
Thermo-electrochemical cells empowered by selective inclusion of redox-active ions by polysaccharides†
H.
Zhou
a,
T.
Yamada
 *abc and
N.
Kimizuka
ab
*abc and
N.
Kimizuka
ab
aDepartment of Chemistry and Biochemistry, Graduate School of Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: teppei343@gmail.com
bCenter for Molecular Systems, Kyushu University, Japan
cPRESTO, Japan Science and Technology Agency, 4-1-8, Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 4th December 2017
Abstract
Thermo-electrochemical cells (TECs) are a class of thermoelectric materials that offer high thermoelectric voltage (Seebeck coefficient) with potentially lower costs compared to the conventional thermoelectric materials. To maximize the potential of TECs, we show that the Seebeck coefficient of TECs with a redox pair of I−/I3− is enhanced by introducing polymer–ion interactions. Starch and polyvinylpyrrolidone (PVP) are employed as polymeric hosts for I3− ions. The effective concentration of free I3− ions in the cold cell decreases due to their selective inclusion in host polymers, resulting in an increase of the [I−]/[I3−] ratio. Meanwhile in the higher temperature cell, the inclusion of I3− ions by host polymers is less effective and the [I−]/[I3−] ratio is mostly determined by the intrinsic equilibrium without polymers. Consequently, the two electrode cells differing in temperature show a considerable difference in the concentration of I3− ions, which causes a significant increase of the Seebeck coefficient up to 1.5 mV K−1. The performance of polymer TECs can be tuned depending on the polymer–I3− interactions, and starch showed notable performance as compared to PVP, with increased output power by a factor of two.
1. Introduction
Thermo-electrochemical cells (TECs), also described as thermogalvanic cells or thermocells, are one of the thermoelectric generation devices1–7 that are based on reversible redox reactions. TECs generate voltage based on the difference in electrochemical potentials between a pair of electrodes which are thermally stabilized at different temperatures.8,9 The key advantages of TECs are the higher Seebeck coefficients (Se, several mV K−1)10 and lower cost compared to those obtained by conventional thermoelectric (TE) materials (Se, in the order of 0.1 mV K−1).11,12 The first study of TECs was reported by Richards at the end of the 19th century,13 where the thermoelectric voltage was observed between two calomel electrodes kept at different temperatures. Later, his theory was explained by using the Nernst equation.8,9 Since the 1970s, practical use of TECs for thermoelectric conversion has been investigated by Quickenden,3,14,15 Burrow,16 Chum,2 and Ikeshoji.4 In these TECs, an aqueous redox couple of hexacyanoferrate(II/III) was used because of its chemical stability, reversible redox reaction, and high Seebeck coefficient (ca. −1.4 mV K−1). More recently, Katayama,17 Pringle, and Macfarlane1,10,18 studied ionic liquids as electrolytes for TECs. They discovered that a redox couple of tris(bipyridyl)cobalt(II/III) shows a higher Seebeck coefficient of 1.9 mV K−1.10 Kim used mixtures of organic solvent and water, and the Seebeck coefficient of hexacyanoferrate(II/III) increased to 2.9 mV K−1.19 To realize a higher Seebeck coefficient is crucial for the development of efficient thermoelectric generators, thermoelectric cooling (the Peltier effect),20 and thermometric applications, which will have a pervasive effect in a wide range of research areas. Although guiding principles for increasing the Seebeck coefficient of TECs are eagerly anticipated, they are still elusive.21–27 Theoretically, changes in entropy during the redox reactions play essential roles in determining the Seebeck coefficients of TECs.28–31 Meanwhile, conventional approaches to control entropy changes in TECs have been limited to the study of solvent effects, i.e., by changing the degree of solvation of the relevant redox species.To break ground in this area, we have recently developed a simple supramolecular methodology.7 α-Cyclodextrin (α-CD) was introduced into TECs consisting of the redox pair of I−/I3− to control the effective concentration of I3− ions. α-CD shows a higher tendency to encapsulate I3− ions at lower temperatures. It consequently decreases the effective concentration of I3− on the cold electrode side, giving a higher [I−]/[I3−] ratio. Meanwhile, the binding of I3− by α-CD is less effective at higher temperatures, resulting in a decrease in the [I−]/[I3−] ratio in the hot-side cell. The consequent temperature-dependent gradients in the [I−]/[I3−] ratio generated additional voltage according to the Nernst equation.32,33 The Seebeck coefficient of this CD-based TEC reached 1.45 mV K−1.7 The drawback of the CD-based TECs is the precipitation of the I3−/α-CD complex by addition of a supporting electrolyte, which hindered their stable performance for a sustained period of time. Apparently, it is imperative to develop new host–guest inclusion complexes that do not precipitate in the presence of supporting electrolytes, which would also serve to generalize the concept of supramolecular TECs.
In this paper, we report the use of water-soluble polymers as host materials for enhancing the performance of the I−/I3− TEC system (Fig. 1). Starch and poly-N-vinyl-2-pyrrolidone (PVP) were investigated as the host polymers. The Seebeck coefficient of the I−/I3− TEC increased from 0.86 to 1.5 mV K−1 (with starch) and 1.2 mV K−1 (with PVP). The thermoelectric power of the single starch–I−/I3− TEC reached 3.0 μW at the temperature difference of 33 K. Importantly, the output power lasted for more than six hours without deterioration, which successfully overcame the drawback of the previously reported α-CD–I3− complex system. It is to note that starch, an inexpensive biopolymer that consists of amylose and branched amylopectin,34,35 plays a remarkable role in achieving the outstanding advance for the long-lived, high-performance TECs which opens a field of green-sustainable biopolymer devices.
 | ||
| Fig. 1 A schematic of a working TEC with a host polymer which binds triiodide (I3−, purple sphere) at the cold side and releases it at the hot side. Oxidation of iodide, and reduction of triiodide take place at the cold and hot side, respectively. | ||
2. Results and discussion
2.1 Seebeck coefficient of the starch–I−/I3− TEC
As described, the main component of potato starch is amylose and amylopectin.34,35 It is widely known that amylose binds to iodine and change its color to dark blue,36–38 which is ascribed to the supramolecular complexation of iodine in the amylose helix as the I5− ion.39 Schneider and co-workers studied the cooperative binding of iodine, and reported that an iodine in the amylose is stabilized by the neighboring iodine.40 Szejtli and Pfannemüller reported that the higher degree of polymerization (DP) of amylose increases the iodine-binding capacity and causes a red shift of the maximum absorption wavelength (λmax).41,42 Yasunaga and co-workers performed a quantitative analysis of the temperature dependence of the binding constant between amylose and iodine.43 They reported that the binding constant of iodine to amylose (DP = 42) is 1.6 × 104 M−1 at 10 °C, while it decreases at higher temperatures (e.g., 3.9 × 103 M−1 at 30 °C).According to the relationship between DP and λmax reported by Banks,44 the average DP of the amylose used in this study was estimated as 42 (λmax = 555 nm, Fig. 2a). In Fig. 2b, the absorbance of amylose–iodine complexes decreased with increasing temperature while that of unbound triiodide (λmax = 289 nm and 352 nm) showed an increase. These results are in good agreement with the report by Yasunaga, and confirm that the binding constant of amylose is decreased by elevating temperature.43,45
 | ||
| Fig. 2 (a) UV-vis spectra of the starch–iodine complex at various starch concentrations. Initial concentrations of KI and I2 were 6.0 and 0.15 mM, respectively. Absorption peaks of triiodide (290 and 350 nm) and the starch–iodine complex (550 nm) are clearly observed. (b) Temperature dependence of the peak absorbance for the starch–iodine complex (552 nm) and I3− (352 and 289 nm) in the temperature range from 1 to 35 °C at a scan rate of 0.5 °C min−1. The solid and dashed lines correspond to the plots with increasing and decreasing temperature, respectively. A reversible change was observed. Initial concentrations of starch, KI and I2 were 3.6, 6.0 and 0.15 mM, respectively. (c and d) Plots of open-circuit voltage (Voc) at various temperature differences (ΔT) for (c) Sample A ([KI]0 = 12.5 mM, [I2]0 = 2.5 mM), and (d) Sample B ([KI]0 = 100 mM, [I2]0 = 2.5 mM). Experimental errors are within the length of each mark side. The temperature of the cold side is kept at 10 °C. (e) Plots of the Seebeck coefficient at various concentrations of starch. Error bars show the deviation from the value evaluated from the fitting lines in (c) and (d). | ||
Triiodide (I3−) is formed in equilibrium with iodide (I−) and iodine (I2):46,47
| I− + I2 ⇄ I3− | (1) |
Ramette and Sandford determined the formation constant of triiodide in water to be 748 M−1 at 25 °C.48 We confirmed the formation constant by UV-vis spectroscopy (Fig. S5 in the ESI†). Since the concentration of I− correlates with the concentration of I3− as in eqn (1), we prepared two different samples that contain different concentrations of I−, abbreviated as Samples A and B (Sample A, [I−]0 = 12.5 mM; Sample B, [I−]0 = 100 mM). Starch was added to these samples and the Seebeck coefficient was evaluated from the slope of Voc–ΔT plots as shown in Fig. 2c and d. The pristine Seebeck coefficients of Samples A (Se = 0.86 mV K−1) and B (Se = 0.64 mV K−1) are different because of the difference in the concentration of I−.
After the addition of starch into the electrolyte, an increase in the Seebeck coefficient is observed for both Samples A and B (Fig. 2e). The concentration of I3− unbound to starch is amenable to change depending on the temperature difference between the hot- and cold-side cells; the higher the difference, the larger the concentration gap of I3− species and it generates additional voltage. It is noteworthy that the increase in the Seebeck coefficient showed saturation for both Samples A and B beyond the starch concentration of 60 mM, indicating that the concentration difference of I3− between the hot- and cold-half-cells reached a constant at sufficiently high host polymer concentrations. Compared to the previous report, Se shows a rather gradual change with the addition of starch, which could be associated with the deviation of the chain length of starch. Similar to concentration cells, the difference of concentration evokes voltage according to the Nernst equation. It is to note that in conventional concentration cells, the difference of concentration was introduced by the continuous supply of redox species, while in the TEC the concentration difference was evoked by the temperature difference and host–guest interactions. Hence, it could be regarded as a new type of Seebeck effect. The Seebeck coefficient shown in Fig. 2e is a combination of conventional TECs and host–guest interactions.
To elucidate the effect of I3− inclusion on the redox reaction, cyclic voltammetry (CV) was conducted for the starch–iodine solutions. Fig. 3a shows the temperature dependence of CV curves obtained at various scan rates, where details are given in Fig. S6.† The reduction of I3− was effectively suppressed by the addition of starch at low temperature, and starch prevents the electron hopping of the captured I3−. The CV of the aqueous solution containing potassium iodide (12.5 mM), iodine (2.5 mM) and potassium chloride (0.2 M) displayed two peaks, peaks (i) and (ii). The literature reported that the oxidation of iodide to iodine occurs in the following two steps:49–51
| 3I− ⇄ I3− + 2e− | (2) |
| 2I3− ⇄ 3I2 + 2e− | (3) |
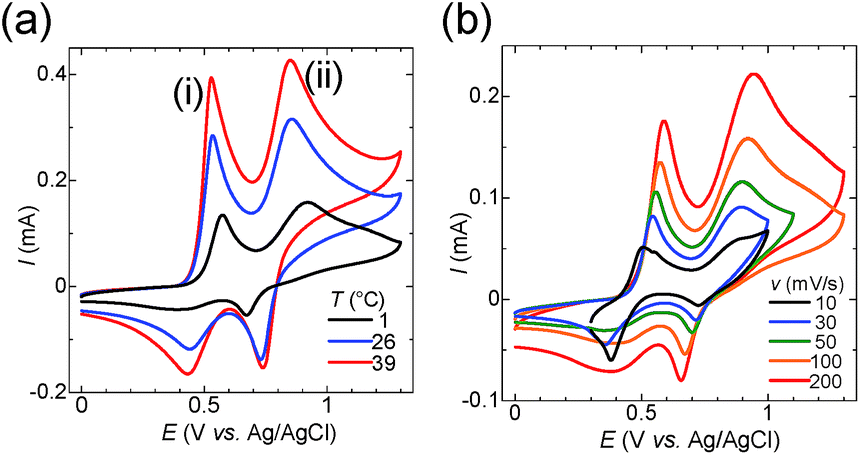 | ||
| Fig. 3 (a) Variable-temperature cyclic voltammograms of the starch–I−/I3− solution ([KI]0 = 12.5 mM, [I2]0 = 2.5 mM, [starch] = 60 mM, [KCl] = 0.2 M) at a scan rate of 100 mV s−1. (b) Cyclic voltammograms of the starch–I−/I3− solution at 1 °C at various scan rates. | ||
The peak potentials of eqn (2) and (3) depend on the solvent, and the two peaks coalesce in water into one peak.52 In the presence of chloride ions, iodide can be further oxidized to I+ in the form of the [ICl2]− complex.53,54
| I− + 2Cl− ⇄ [ICl2]− + 2e− | (4) |
Peak (ii) disappears in the absence of potassium chloride (see Fig. S7†). Therefore, peak (i) corresponds to eqn (2) and (3), and peak (ii) corresponds to eqn (4). The peak current for the reduction of I3− at 0.4 V diminished with decreasing temperature (Fig. 3a). However, a sharp reduction peak appeared at 1 °C when the scan rate was lower than 30 mV s−1 (Fig. 3b). These results show that the I3− anion is electrochemically inactivated as a result of the encapsulation by starch. The I3− anion binds to starch according to the host–guest interaction in equilibrium. Thus it is released slowly by the consumption of I3− during the reduction process, which was observed as a recovered reduction peak of free I3− in the CV at a slow scan rate. Meanwhile, as the reduction peak of [ICl2]− at 0.7 V is not affected by the addition of starch, the interaction between starch and [ICl2]− is negligible. To summarize, I3− was successfully encapsulated into starch at the cold side of the TEC test cells, and released at the hot side, and this increased the voltage of TECs by a factor of two.
2.2 Seebeck coefficient of the PVP–I−/I3− TEC
The encapsulation of iodine into PVP in aqueous solution is also widely known and used for antiseptics.55 The interaction between I3− and PVP is explained by the formation of hydrogen bonds with two pyrrolidone residues.56Upon addition of PVP(M.W. 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) into the I3− solution, a broad absorbance peak from 360 to 500 nm appeared in UV spectra (Fig. 4a),57 which reflects the complexation of I3− with PVP. The absorption peak decreases with elevating temperature, because the binding constant between I3− and PVP decreases at higher temperature. The Seebeck coefficients of Samples A and B also increased with addition of PVP as shown in Fig. 4b and c, respectively. Similar to the case of starch, the Seebeck coefficient reached a plateau at higher PVP concentrations (Fig. 4e). As discussed in the previous section, PVP concentrations above 36 mM were required to create the concentration gap of triiodide between the heated and cooled half-cells.
000) into the I3− solution, a broad absorbance peak from 360 to 500 nm appeared in UV spectra (Fig. 4a),57 which reflects the complexation of I3− with PVP. The absorption peak decreases with elevating temperature, because the binding constant between I3− and PVP decreases at higher temperature. The Seebeck coefficients of Samples A and B also increased with addition of PVP as shown in Fig. 4b and c, respectively. Similar to the case of starch, the Seebeck coefficient reached a plateau at higher PVP concentrations (Fig. 4e). As discussed in the previous section, PVP concentrations above 36 mM were required to create the concentration gap of triiodide between the heated and cooled half-cells.
 | ||
| Fig. 4 (a) UV-vis spectra of the PVP–I3− complex. Absorption peaks at ca. 360 nm correspond to the PVP–I3− complex, and the absorbance of the peak decreases with increasing temperature, which suggests that more PVP–I3− complex is formed at a lower temperature. Initial concentrations of PVP, KI and I2 were 2.2, 6.0 and 0.15 mM, respectively. (b, c, and d) Plots of Voc and ΔT between heated and cooled sides of the TEC. (b) Sample A: [KI]0 = 12.5 mM, [I2]0 = 2.5 mM; (c and d) Sample B: [KI]0 = 100 mM, [I2]0 = 2.5 mM. Molecular weight of PVP: (b and c) 40000, and (d) 1300000. The temperature of the cold side is kept at 10 °C. Error bares not shown are within the size of marks. Seebeck coefficient, i.e. slope of the fitting line, increases with the increasing concentration of PVP. (e) Change of Seebeck coefficients by addition of various concentrations of PVP. (f) Cyclic voltammograms of the PVP (M.W. 40k)–I−/I3− solution ([KI]0 = 12.5 mM, [I2]0 = 2.5 mM, [PVP] = 0–36 mM, [KNO3] = 0.1 M) at a scan rate of 50 mV s−1 at 20 °C. | ||
CV of the aqueous solution of PVP (M.W. 40000, 0–36 mM), potassium iodide (12.5 mM), iodine (2.5 mM), and potassium nitrate (0.1 M) was performed and the results are summarized in Fig. 4f (see Fig. S8 and S9† for details). Potassium nitrate was used here as the supporting electrolyte instead of potassium chloride, because potassium chloride caused precipitation of PVP. The reduction current of triiodide was suppressed by the addition of 36 mM of PVP, which is the sufficient concentration to maximize the Seebeck coefficient (Fig. 4e). Naturally, when the concentration of PVP is low (9 mM), a considerable amount of free triiodide still exists in the solution and the reduction current of triiodide is not suppressed. A small oxidation peak is present at 0.55 V, which is due to the oxidation of triiodide to iodine (eqn (3), see Fig. S6† for the detailed discussion). Interestingly, the peak vanishes at 36 mM of PVP, because triiodide ions are stabilized by PVP, and further oxidation to iodine is suppressed.
The Seebeck coefficient of the I−/I3− TEC was evaluated also by using a higher molecular weight PVP (Mw, 1300k) as the host polymer (Sample B, Fig. 4d). The Seebeck coefficient increased with the increase in PVP concentration, and the rate of increment was identical to the case when lower molecular weight PVP (Mw, 40k) was employed. It is noteworthy that the Seebeck coefficient decreased at the concentration of 72 mM for PVP (Mw, 1300k), probably because the release of triiodide from the higher molecular weight polymer chain becomes more difficult at elevated concentration of the host polymer.
2.3 Thermal efficiency of the starch–I−/I3− TEC
Since the starch–I−/I3− TEC showed a higher Seebeck coefficient without precipitation, a further investigation was carried out on its power-generation ability. Potassium chloride was added to the solution as a supporting electrolyte.By the addition of potassium chloride, the Seebeck coefficient showed an increase from 1.37 to 1.53 mV K−1 at the starch concentration of 60 mM (Fig. S10†). This slight increase in the Seebeck coefficient implies that the higher concentration of potassium cations promoted the formation of hydrogen bonding between two hydroxyl groups of the neighboring glucose unit in an amylose helix,58 which could affect the binding constant between amylose and iodine. The effect of chloride anions on Voc was also evaluated for elucidating the effect of eqn (4), which was found to be negligible (Fig. S11†).
The I–V curve was then evaluated for Sample A with and without starch (60 mM), where ΔT = 34 K (Fig. 5a, left axis and S12a†). The current decreased linearly with the increase of the external voltage (Vex), which indicates that the TEC has ohmic resistance. The output power was evaluated from the I–V curve by using the relationship P = I × V (Fig. 5a, right axis and S12b†). The maximum power (Pmax) of the TEC with starch is higher by a factor of two as compared to that without starch. This is ascribed to the enhanced Voc and Seebeck coefficient by the inclusion of I3− in the host polymer.
 | ||
| Fig. 5 (a) Current–voltage and power–voltage curves of the I−/I3− TEC. ([KI]0 = 12.5 mM, [I2]0 = 2.5 mM, [starch] = 60 mM, [KCl] = 2 M). The temperature of the cold and hot side was kept at 3 and 37 °C, respectively. (b) Time course of the maximum output power (Pmax) of the TEC. ΔT was kept at 33 ± 1 K for the cell with [KCl] = 2 M, and 26 ± 1 K for [KCl] = 0.2 M, throughout the measurement. | ||
2.4 Long-term thermoelectric power generation
The current and power of TECs were evaluated over the course of time (Fig. 5b and S13†). TECs consume I3− and I− at hot and cold sides, respectively, and the diffusion of these redox species toward opposite electrodes occurs. As a result, TECs run over six hours. The Pmax of the I−/I3− TEC gradually decreased during four hours of running (Fig. 5b). UV-vis spectroscopy revealed that the concentration of I3− decreased to one quarter of the initial value after four hours (Fig. S13†). This is because of the high volatility of iodine, which is produced from the decomposition of triiodide. On the other hand, the Pmax of the starch–I−/I3− TEC settled at a constant value even after three hours, which indicates that triiodide is significantly stabilized in the amylose helices.The drop in the Pmax of the starch–I−/I3− TEC ([KCl] = 2 M) during the first two hours in Fig. 5b is presumably because of the slow mass transfer of redox species or the starch–I3− complex. When the redox reaction occurs in the I−/I3− TEC, the concentration of the counter redox species (I− on the heated side and I3− on the cooled side) increases, which decreases the Voc and the current.14 These counter redox species diffuse to the other side (I− to the cold side, I3−/starch complex to the hot side) and maintain a constant Voc and current. When the rate of the redox reaction is higher than that of mass transfer, the current and the reaction rate decrease until it becomes equal to the rate of mass transfer. Indeed, when the rate of the redox reaction is lower than that of mass transfer (starch–I−/I3− TEC, [KCl] = 0.2 M), the Pmax is kept at a constant value for six hours.
3. Conclusion
The complexation of redox ions by host polymers significantly increases the Seebeck coefficient of TECs. The principle of this strategy—creating a concentration difference of redox species by taking advantage of the temperature-dependent host–guest complexation—is now generalized. The relative concentration of the hosts to the guests is responsible for the change of the Seebeck coefficient, and the design of host molecules is the key to the performance of TECs. Starch showed a high binding constant and increased the Seebeck coefficient of TECs up to 1.5 mV K−1. Remarkably, inclusion of triiodide in starch significantly enhanced the stability, which underlines the use of polymeric hosts for TECs. To summarize, addition of PVP increases the Seebeck coefficient, although not as effective as that with the addition of starch. This result demonstrates that the hosts for triiodide are not limited to polysaccharides, such as cyclodextrins and starch. Various hosts can be applied for supramolecular TECs when they have hydrophobic interactions with triiodide in aqueous solution.The polymers used in the present work are inexpensive, water soluble and can be transformed to gel membranes. Although several papers reported that polymers or gel electrolytes can be applied for thermocells, they can only act as ionic conductive matrices.59,60 Our strategy could pave the way to improve the Seebeck coefficient by using polymers as host materials for redox species. We presume that these green-sustainable TECs would be suitable for future device applications which work at atmospheric temperatures.
4. Experimental
KI and refined potato starch were purchased from Wako Pure Chemicals Inc. I2, KCl, PVP (molecular weight = 40000), and PVP (molecular weight = 1300000) were purchased from Chameleon Analytical Reagent, Nacalai Tesque, Kishida Chemical, and Acros Organics, respectively. All reagents were used without further purification.
4.1 Electrolyte solution of polymer and I−/I3−
KI (2.49 g, 15 mmol) and I2 (1.27 g, 5 mmol) were dissolved in Milli-Q water (100 mL) and used as a stock solution (Solution 1, [KI]0 = 150 mM, [I2]0 = 50 mM). An aqueous solution of starch (0.3 M) was produced by dissolving refined potato starch (2.43 g, 15 mmol in glucose units) in 50 mL of hot water (70 °C). The Solution 1 (2.5 mL), 0.1 M KIaq (2.5 mL) and 0.3 M starchaq (10 mL) were mixed and diluted with water to make total of 50 mL of the final solution ([KI]0 = 12.5 mM, [I2]0 = 2.5 mM, [starch]0 = 60 mM, where subscript zero represents the initial state). KCl was added before the final dilution when it was necessary. An aqueous solution of KI/I2 and PVP was prepared according to a similar procedure. These samples are categorized into two solutions according to the initial concentration of KI and I2: Sample A ([KI]0 = 12.5 mM, [I2]0 = 2.5 mM) and Sample B ([KI]0 = 100 mM, [I2]0 = 2.5 mM). Details are summarized in the ESI.†4.2 Evaluation of the Seebeck coefficient
Sample A was put into an H-shaped glass tube (H-cell, Fig. S1 and S2 in the ESI†), and Sample B was put into a miniature glass vial (mini-cell, Fig. S3†), and a temperature difference (ΔT = 0–50 K) was applied to both the samples in the cells. The temperature inside the cell was monitored using a digital thermometer (TM201, AS ONE, Japan). Platinum wires were soaked in concentrated sulfuric acid (97%, Nacalai Tesque, Japan) and rinsed with pure water before use as electrodes (1 mm in diameter and ca. 1 cm in depth). The open-circuit voltage (Voc) between the heated and cooled electrodes was measured using a Source Meter, KEITHLEY 2401, after the values became stable. The Seebeck coefficient (Se) was evaluated from the open-circuit voltage (Voc) and temperature difference (ΔT) as follows:1,6,9 | (5) |
Prior to the other measurements, we evaluated the Voc of the same sample with an H-cell and a mini-cell, and confirmed that the resultant values were identical (Fig. S4†).
4.3 Electrochemical measurements
Voltammetric experiments were carried out with an Electrochemical Analyzer, ALS608E (ALS, Japan), using a standard three-electrode arrangement. A glassy carbon electrode with an inner diameter of 3 mm (ALS, Japan) was used as the working electrode, a calibrated aqueous Ag/AgCl with 3 M NaCl (ALS, Japan) as the reference electrode, and a Pt wire as the counter electrode. The carbon electrode was polished with aqueous alumina slurries with a nominal diameter of 0.05 μm (BAS, Japan) on a clean polishing cloth (Buehler, USA) before use.4.4 Spectroscopic analysis
All spectroscopic experiments were carried out with a UV/Vis spectrophotometer, V-670 (JASCO, Japan), optical path length = 1 mm.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by PRESTO from JST, Grant Number JPMJPR141D. This study was also supported by JSPS KAKENHI Grant Numbers JP16H06513, 17H03046, 26708007, 26600026, and 25220805.Notes and references
- T. J. Abraham, D. R. MacFarlane and J. M. Pringle, Chem. Commun., 2011, 47, 6260–6262 RSC.
- H. L. Chum and R. A. Osteryoung, Rep. SERI/TR-332416, 1980, vol. 1.
- T. I. Quickenden and Y. Mua, J. Electrochem. Soc., 1995, 142, 3985–3994 CrossRef CAS.
- T. Ikeshoji, Bull. Chem. Soc. Jpn., 1987, 60, 1505–1514 CrossRef CAS.
- H. Im, T. Kim, H. Song, J. Choi, J. S. Park, R. Ovalle-Robles, H. D. Yang, K. D. Kihm, R. H. Baughman, H. H. Lee, T. J. Kang and Y. H. Kim, Nat. Commun., 2016, 7, 10600 CrossRef CAS PubMed.
- W. B. Chang, C. M. Evans, B. C. Popere, B. M. Russ, J. Liu, J. Newman and R. A. Segalman, ACS Macro Lett., 2016, 5, 94–98 CrossRef CAS.
- H. Zhou, T. Yamada and N. Kimizuka, J. Am. Chem. Soc., 2016, 138, 10502–10507 CrossRef CAS PubMed.
- Y. V. Kuzminskii, V. A. Zasukha and G. Y. Kuzminskaya, J. Power Sources, 1994, 52, 231–242 CrossRef CAS.
- P. F. Salazar, S. Kumar and B. A. Cola, J. Appl. Electrochem., 2014, 44, 325–336 CrossRef CAS.
- T. J. Abraham, D. R. MacFarlane and J. M. Pringle, Energy Environ. Sci., 2013, 6, 2639–2645 CAS.
- B. Poudel, Q. Hao, Y. Ma, Y. Lan, A. Minnich, B. Yu, X. Yan, D. Wang, A. Muto, D. Vashaee, X. Chen, J. Liu, M. Dresselhaus, G. Chen and Z. Ren, Science, 2008, 320, 634–638 CrossRef CAS PubMed.
- K. Biswas, J. He, I. Blum, C.-I. Wu, T. Hogan, D. Seidman, V. Dravid and M. G. Kanatzidis, Nature, 2012, 489, 414–418 CrossRef CAS PubMed.
- T. W. Richards, Z. Phys. Chem., 1897, 24, 39–54 Search PubMed.
- T. I. Quickenden and C. F. Vernon, Sol. Energy, 1986, 36, 63–72 CrossRef CAS.
- Y. Mua and T. I. Quickenden, J. Electrochem. Soc., 1996, 143, 2558 CrossRef CAS.
- B. Burrows, J. Electrochem. Soc., 1976, 123, 154–159 CrossRef CAS.
- T. Migita, N. Tachikawa, Y. Katayama and T. Miura, Electrochemistry, 2009, 77, 639–641 CrossRef CAS.
- T. Abraham, N. Tachikawa, D. Macfarlane and J. Pringle, Phys. Chem. Chem. Phys., 2014, 16, 2527–2532 RSC.
- T. Kim, J. S. Lee, G. Lee, H. Yoon, J. Yoon, T. J. Kang and Y. H. Kim, Nano Energy, 2017, 31, 160–167 CrossRef CAS.
- F. J. Disalvo, Science, 1999, 285, 703–706 CrossRef CAS PubMed.
- T. M. Tritt, Science, 1999, 283, 804–805 CrossRef CAS.
- G. A. Slack and M. A. Hussain, J. Appl. Phys., 1991, 70, 2694 CrossRef CAS.
- G. A. Slack and V. G. Tsoukala, J. Appl. Phys., 1994, 76, 1665 CrossRef CAS.
- L. D. Hicks and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 727–731 CrossRef.
- L. D. Hicks and M. S. Dresselhaus, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 631–634 Search PubMed.
- G. J. Snyder and E. S. Toberer, Nat. Mater., 2008, 7, 105–114 CrossRef CAS PubMed.
- O. Bubnova, Z. U. Khan, A. Malti, S. Braun, M. Fahlman, M. Berggren and X. Crispin, Nat. Mater., 2011, 10, 429–433 CrossRef CAS PubMed.
- S. Sahami and M. J. Weaver, J. Electroanal. Chem., 1981, 122, 155–170 CrossRef CAS.
- J. Hupp and M. Weaver, Inorg. Chem., 1984, 23, 3639–3644 CrossRef CAS.
- E. L. Yee, R. J. Cave, K. L. Guyer, P. D. Tyma and M. J. Weaver, J. Am. Chem. Soc., 1979, 101, 1131–1137 CrossRef CAS.
- S. Sahami and M. J. Weaver, J. Electroanal. Chem., 1981, 122, 171–181 CrossRef CAS.
- J. E. Anderson and Y. B. Graves, J. Electrochem. Soc., 1981, 128, 294–300 CrossRef CAS.
- H.-I. Yoo and S.-J. Yang, J. Electrochem. Soc., 2001, 148, A642 CrossRef CAS.
- J. J. M. Swinkels, Starke, 1985, 37, 1–5 CrossRef CAS.
- R. G. F. Visser, L. C. J. M. Suurs, P. M. Bruinenberg, I. Bleeker and E. Jacobsen, Starke, 1997, 49, 438–443 CrossRef CAS.
- F. L. Bates, D. French and R. E. Rundle, J. Am. Chem. Soc., 1943, 65, 142–148 CrossRef CAS.
- R. S. Stein and R. E. Rundle, J. Chem. Phys., 1948, 16, 195–206 CrossRef CAS.
- R. Bersohn and I. Isenberg, J. Chem. Phys., 1961, 35, 1640–1643 CrossRef CAS.
- W. Saenger, Naturwissenschaften, 1984, 71, 31–36 CrossRef CAS.
- F. W. Schneider, C. Cronan and S. K. Podder, J. Phys. Chem., 1968, 72, 4563 CrossRef CAS.
- V. B. Pfannemüller, H. Mayerhofer and R. C. Schulz, Makromol. Chem., 1969, 121, 147–158 CrossRef.
- J. Szejtli, M. Richter and S. Augustat, Biopolymers, 1967, 5, 5–16 CrossRef CAS.
- M. Yamamoto, T. Sano and T. Yasunaga, Bull. Chem. Soc. Jpn., 1982, 55, 1886–1889 CrossRef CAS.
- W. Banks, C. T. Greenwood and K. M. Khan, Carbohydr. Res., 1971, 17, 25–33 CrossRef CAS.
- M. Yamamoto, S. Harada, T. Sano, T. Yasunaga and N. Tatsumoto, Biopolymers, 1984, 23, 2083–2096 CrossRef CAS.
- G. Jones and B. B. Kaplan, J. Am. Chem. Soc., 1928, 50, 1845–1861 CrossRef CAS.
- L. Katzin and E. Gebert, J. Am. Chem. Soc., 1955, 77, 5814–5819 CrossRef CAS.
- R. W. Ramette and R. W. Sandford, J. Am. Chem. Soc., 1965, 87, 5001–5005 CrossRef CAS.
- A. I. Popov and D. H. Geske, J. Am. Chem. Soc., 1958, 80, 1340–1352 CrossRef CAS.
- R. T. Iwamoto, Anal. Chem., 1959, 31, 955 CrossRef CAS.
- C. L. Bentley, A. M. Bond, A. F. Hollenkamp, P. J. Mahon and J. Zhang, J. Phys. Chem. C, 2015, 119, 22392–22403 CAS.
- R. T. Iwamoto, Anal. Chem., 1959, 31, 955 CrossRef CAS.
- I. M. Kolthoff and J. Jordan, J. Am. Chem. Soc., 1953, 75, 1571–1575 CrossRef CAS.
- C. L. Bentley, A. M. Bond, A. F. Hollenkamp, P. J. Mahon and J. Zhang, Anal. Chem., 2016, 88, 1915–1921 CrossRef CAS PubMed.
- H. Rackur, J. Hosp. Infect., 1985, 6, 13–23 CrossRef PubMed.
- J. L. Zamora, Am. J. Surg., 1986, 151, 400–406 CrossRef CAS PubMed.
- G. Oster and E. H. Immergut, J. Am. Chem. Soc., 1954, 76, 1393–1396 CrossRef CAS.
- J.-L. Jane, Starke, 1993, 45, 161–166 CrossRef CAS.
- J. Wu, J. J. Black and L. Aldous, Electrochim. Acta, 2017, 225, 482–492 CrossRef CAS.
- L. Jin, G. W. Greene, D. R. MacFarlane and J. M. Pringle, ACS Energy Lett., 2016, 1, 654–658 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00470b |
| This journal is © The Royal Society of Chemistry 2018 |