
Development of a 3D graphene aerogel and 3D porous graphene/MnO2@polyaniline hybrid film for all-solid-state flexible asymmetric supercapacitors†
Kalyan
Ghosh
a,
Chee Yoon
Yue
 *a,
Md Moniruzzaman
Sk
b,
Rajeeb Kumar
Jena
a and
Shuguang
Bi
a
*a,
Md Moniruzzaman
Sk
b,
Rajeeb Kumar
Jena
a and
Shuguang
Bi
a
aSchool of Mechanical and Aerospace Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798. E-mail: mcyyue@ntu.edu.sg; Fax: +65 6792 4062; Tel: +65 6790 6490
bSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798
First published on 20th November 2017
Abstract
There is an increasing demand for safe, environmentally benign energy storage devices in portable electronic appliances, wearable gadgets, flexible displays, and other personal multimedia devices. In this study, we have fabricated an all-solid-state flexible asymmetric supercapacitor using a novel 3D porous reduced graphene oxide/manganese dioxide@polyaniline (RGO/MnO2@PANI) hybrid film as the positive electrode and a self-assembled 3D pillared graphene aerogel as the negative electrode material with a polyvinyl alcohol/potassium hydroxide (PVA/KOH) gel electrolyte. The flexible composite film was synthesized by vacuum filtration of GO and a MnO2@PANI mixture followed by chemical reduction of the resulting film in a hydrothermal autoclave. The 3D graphene aerogel was synthesized by a hydrothermal route using a solution of the nonionic triblock copolymer Pluronic F-68 as a soft template and vitamin C as a reducing agent. Herein, the Pluronic copolymer played dual roles: first, it enabled the effective dispersion of graphene oxide in water, and second, it assisted the formation of a stable 3D pillared hydrogel assembly. The RGO/MnO2@PANI-based symmetric supercapacitor shows a high energy density of 18.33 W h kg−1 at a power density of 0.388 kW kg−1. An asymmetric supercapacitor (graphene aerogel//RGO/MnO2@PANI), which was fabricated by optimizing the individual electrode materials, exhibited a very high energy density of 38.12 W h kg−1 at a power density of 1.191 kW kg−1 utilizing a large potential window of 1.5 V. Moreover, 3 cells connected in series successfully lit up a red LED for 45 s and displayed similar performance under bending conditions.
1. Introduction
In today's world, energy storage devices play a significant role in sustainable energy output systems. The development of portable, lightweight, flexible, high-performance, and safe power sources is essential to meet the increasing demand for energy storage devices such as electronic paper, wearable electronics, roll-up displays, mobile phones, flexible biosensors, and flexible displays. Supercapacitors1–4 (SC) have attracted significant attention in the field of energy storage devices because their power densities are higher in comparison with those of secondary batteries and their energy densities are higher than those of traditional capacitors. However, to meet the future demand for portable energy devices, the specifications of supercapacitors, such as their size, power and energy densities, and mechanical integrity, should be improved. Earlier SCs basically comprised liquid-based aqueous, organic and ionic electrolytes. These SCs faced two major challenges: first, possible leakage of the corrosive and toxic electrolyte materials and second, difficulties in the manufacture of flexible supercapacitors due to issues with the packaging of the liquid electrolyte. In this regard, a polymer gel electrolyte is more suitable for a solid-state flexible SC.5–8 A flexible solid-state SC has several benefits in comparison with a conventional liquid-based SC.9 Their small size, light weight, ease of handling, avoidance of electrolyte leakage, and wide range of operating temperatures have made flexible solid-state SCs promising for use in flexible and wearable electronics.Carbon-based supercapacitors have stood out as emerging energy storage devices owing to their high power density and long cycling stability.10 Among carbon-based materials, graphene has been attractive, which was mostly due to its large surface area, high conductivity and high flexibility.11,12 A recent approach was followed to prepare graphene films for flexible electrode materials by integrating individual graphene sheets into a macroscopic structure. Dikin et al. prepared free-standing graphene oxide paper using a vacuum filtration method.13 The specific capacitance of pure graphene film was reported to be only 57 F g−1, which is very low, owing to the poor accessibility to ions of the entire surface.14 In contrast, Niu et al. prepared porous paper of reduced graphene oxide and achieved a specific capacitance of 110 F g−1 in a 1 M H2SO4 electrolyte.15 Weng et al.16 synthesized graphene–cellulose paper, which displayed a specific capacitance of 120 F g−1 at 1 mV s−1 in a 1 M H2SO4 electrolyte. Wang et al. prepared graphene film by introducing carbon black as spacers within graphene sheets and achieved a specific capacitance of 138 F g−1 at 10 mV s−1 in a 6 M aqueous KOH electrolyte.17 The limitation of the above systems is that their energy storage mechanism is limited to electrochemical double-layer capacitance. Recently, researchers have been involved in the fabrication of graphene film by incorporating pseudocapacitive materials.7,18,19 In our current work, we will further develop graphene-based materials for solid-state SCs.
Among all transition metal oxides, manganese dioxide (MnO2) is regarded as the most attractive oxide material owing to its high theoretical specific capacitance (1370 F g−1),20 low cost, natural profusion and environmentally benign nature.21,22 Sumboja et al. reported flexible paper of reduced graphene oxide/manganese dioxide that exhibited a specific capacitance of 217 F g−1 at 100 mA g−1 in a 1 M Na2SO4 electrolyte.18 Li et al. fabricated a self-supporting graphene/MnO2 paper, which displayed a specific capacitance of about 256 F g−1 in a 0.1 M Na2SO4 aqueous electrolyte at 500 mA g−1.19 However, the poor conductivity of MnO2 inhibited the easy transportation of ions throughout the graphene matrix. Among numerous CPs such as polyaniline (PANI), polypyrrole (PPy) and polythiophene (PTh), PANI has been regarded as an attractive conductive coating material because of its easy synthesis, environmentally benign nature, and high conductivity. Hence, in our work we incorporated coaxial MnO2@PANI particles into graphene oxide film, which was then reduced by hydrazine vapor.
Coaxial MnO2@PANI particles, which resemble sea urchins, are used as spacers here to create channels in the interior layers of GO film to facilitate the access of ions. The urchin-shaped MnO2@PANI particles also provide high pseudocapacitance by the synergistic effect of MnO2 and PANI. There are two procedural steps that enhance the final film. Firstly, the gas released during the chemical reduction of GO causes significant exfoliation of the crude GO film, which thus makes the film highly porous. Secondly, MnO2@PANI microurchins act as spacers that prevent the GO sheets from collapsing and becoming attached to each other. Hence, the overall capacitance of the electrode could be increased greatly via the contributions of both pseudocapacitive and electrochemical double-layer (EDL) capacitive materials.
The energy density of a supercapacitor cell can be calculated from the equation Ecell = 1/2Csp(ΔV)2,23 where Csp is the specific capacitance of the cell and ΔV is the operating potential window. It is obvious from this equation that the performance of a supercapacitor depends on its capacitance and operating potential window. It is well known that ionic and organic electrolytes can be used to achieve a larger potential window (3 V). However, these electrolytes are very toxic and costly. Another way to expand the potential window comprises the asymmetric combination of the electrode materials, whereby the voltage window can be expanded to reach 2 V.23–25 This approach will be adopted in the current work.
Hence, we propose here the use of a 3D porous reduced graphene oxide/MnO2@PANI (RGO/MnO2@PANI) hybrid film as the positive electrode material in conjunction with graphene as the negative electrode material for the fabrication of an asymmetric supercapacitor. In order to prevent the collapse and restacking of graphene sheets in the negative electrode, we propose to develop graphene sheets that can be self-assembled into a complex 3D macrostructure. The 3D open structure will serve to increase the accessible surface area.
We believe that the self-assembly of graphene into a 3D pillared microstructure using a conventional hydrothermal method is a significant strategy. During hydrothermal treatment, graphene oxide sheets are crosslinked by H-bonds, strong van der Waals forces and π–π interactions to ultimately form a 3D pillared structure.26–28 Xu et al. synthesized self-assembled graphene foam using a hydrothermal method, which exhibited a capacitance of 175 F g−1 in an aqueous electrolyte.29 Zu and Han developed a supramolecular graphene hydrogel by using a Pluronic copolymer.30 By using pyrrole, Zhao et al. prepared 3D graphene foam, which displayed a capacitance of 350 F g−1 at 1.5 A g−1 in an aqueous NaClO4 electrolyte.31 Recently, Jiang et al. fabricated self-assembled 3D reduced graphene oxide by using divalent ions (Ca2+, Ni2+, or Co2+) as linkers.32
However, there is still a lack of self-assembly preparation methods of 3D graphene and its application in supercapacitors based on gel electrolytes. Moreover, the surface area of graphene aerogels can still be increased significantly. We will show that a soft template can be used to build a stable hydrogel column. The soft template, which can be removed easily, will eventually leave pores and voids in the aerogel matrix. Hence, we here propose to prepare a 3D graphene aerogel column with the help of a soft template of a solution of Pluronic F-68, which is a nonionic triblock copolymer comprising poly(ethylene oxide)-block-poly(propylene oxide)-block-poly(ethylene oxide) (PEO-b-PPO-b-PEO), to increase the surface area of the graphene aerogel.
In this report, we have fabricated an all-solid-state flexible asymmetric supercapacitor (ASC) by utilizing a unique 3D porous RGO/MnO2@PANI hybrid film as the positive electrode and 3D graphene aerogel as the negative electrode material and utilized a polyvinyl alcohol/potassium hydroxide (PVA/KOH) gel electrolyte. The positive electrode material was developed via the preparation of a hybrid film by the vacuum filtration of a mixture of GO and MnO2@PANI, followed by the reduction of the film by hydrazine vapor in a hydrothermal autoclave. The negative electrode material was synthesized by the self-assembly of graphene sheets using a soft-template Pluronic copolymer, where vitamin C was used as a reducing agent, via a one-step hydrothermal reaction. These methods provided a facile, economic and green approach for fabricating a flexible all-solid-state ASC from a 3D porous RGO/MnO2@PANI hybrid film and 3D graphene aerogel.
2. Experimental
2.1 Materials
Potassium permanganate (KMnO4), hydrochloric acid (HCl), sodium dodecylbenzenesulfonate (SDBS), hydrazine monohydrate, aniline, Pluronic F-68 solution (10%), polyvinylidene fluoride (PVDF), dimethylformamide (DMF), polyvinyl alcohol (PVA) and potassium hydroxide (KOH) were purchased from Sigma-Aldrich, Singapore. Graphene oxide (GO) was purchased from Cheap Tubes, and Ni foam was purchased from MTI Corporation. Conductive carbon black was supplied by Age D'or Pte Ltd, and Anodisc filter paper (pore Ø 0.2 μm) was obtained from Newton 101 Pte Ltd.2.2 Materials synthesis
To prepare the graphene aerogel electrode, 80 wt% of the graphene aerogel, 15 wt% of conductive carbon black and 5 wt% of a solution of PVDF in DMF (with respect to the total mass of 2 mg) were mixed to obtain a homogeneous paste. The paste was then embedded in the Ni sheet and dried in an electric oven at 65 °C for 6 h. After drying, a weight of 1 ton was again applied to the Ni sheet/sample to ensure effective binding of the electroactive components in the electrode. Then, a solid-state graphene aerogel SSC cell was fabricated using the PVA/KOH gel electrolyte by following the same procedure as outlined above. The ASC cell was fabricated by assembling the RGO/MnO2@PANI electrode with the graphene aerogel electrode. A schematic representation of the assembly of the solid-state cell is shown in Fig. S1.†
2.3 Materials characterization
The morphology of the samples was analyzed by field emission scanning electron microscopy (FESEM, JEOL JSM-7600) and transmission electron microscopy (TEM, JEOL JEM-2010). X-ray diffraction (XRD) patterns were recorded using a PANalytical X-ray diffractometer with Cu Kα radiation (λ = 0.15418 nm) operated at 40 kV and 40 mA. Thermogravimetric analysis (TGA) was carried out with a TGA Q500 analyzer (Universal V4.5A, TA Instruments). Raman spectroscopy was performed using a Renishaw 1000 system with a He–Ne laser (518 nm) source. Analysis of surface areas and porosity was carried out using an ASAP 2020 analyzer by the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods. The electrical conductivity of the samples was measured using a Keithley 2000 (Keithley Instruments Inc., USA) apparatus utilizing the four-probe method.2.4 Electrochemical measurements
The electrochemical performance was investigated using a galvanostat/potentiostat (Gamry Reference 3000) at room temperature in conventional two-electrode and three-electrode setups. For the three-electrode system, an Ag/AgCl electrode was used as the reference electrode, a Pt wire was used as the counter electrode, a glassy carbon electrode was used as the working electrode and a 6 M aqueous KOH solution was used as the electrolyte. The electrochemical performance was assessed by cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS) and cyclic charge–discharge (CCD) experiments.The specific capacitance (Csp, F g−1) of the supercapacitors was calculated from the experimental CV data using the following relationship:35
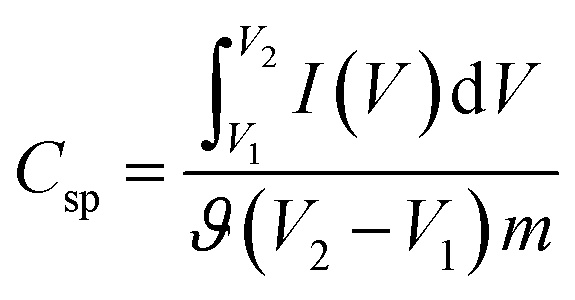 | (1) |
The specific capacitance (Csp, F g−1) of the supercapacitors can also be calculated from the experimental CCD data using the following equation:35
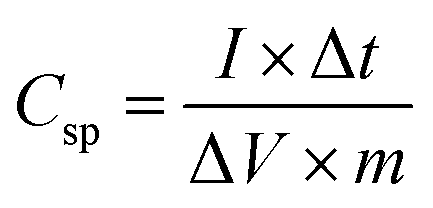 | (2) |
The energy density (Esp, W h kg−1) of the supercapacitors can be calculated from the experimental CCD data according to eqn (3):35
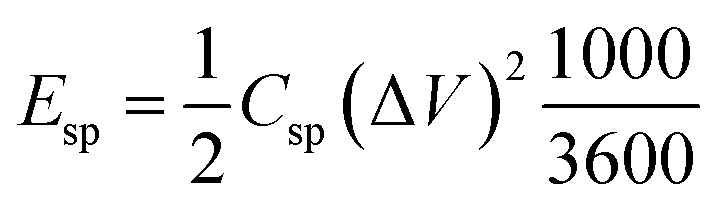 | (3) |
The power density (Psp, kW kg−1) of the supercapacitors can be calculated according to eqn (4):35
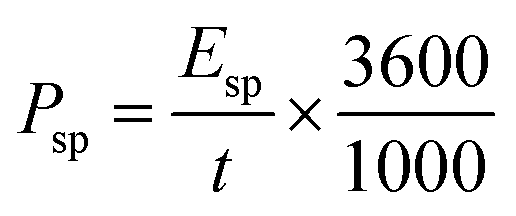 | (4) |
3. Results and discussion
3.1 Negative electrode material
 | ||
| Fig. 1 (a) GO dispersion in deionized water; (b) 3D self-assembled GF20 hydrogel; (c) low-magnification and (d) high-magnification FESEM images of the GF20 graphene aerogel. | ||
Video S1† shows that the cylindrical sample of the 3D hydrogel could absorb water. After freeze-drying, the 3D sample displayed spongy morphology, which was due to the sublimation of ice trapped during freeze-drying. The microstructure of a specimen of the 3D porous graphene aerogel was observed by FESEM, and it can be seen from Fig. 1c that the aerogel had a 3D interconnected porous network structure that comprised numerous micropores and mesopores. The porous structure of the 3D graphene aerogel is apparent in Fig. 1d. Our work revealed that the pore diameter, pore volume and surface area could be adjusted by varying the amount of Pluronic F-68 solution. To make a reasonable comparison, FESEM images of the GF0, GF10, GF20, GF40 and GF50 aerogels are shown in Fig. S2 (ESI).† The surface area and pore size distribution were analyzed by the BET and BJH methods via nitrogen adsorption–desorption. The nitrogen porosimetry data indicated that the Pluronic F-68 solution had a significant impact on the variation of the mesoporous structure of the aerogel. The BET surface area significantly increased when the amount of the F-68 solution was high (volume ratio to GO of 40%). A comparison of the adsorption–desorption plots of the GF0, GF10 and GF40 aerogels is shown in Fig. S3a (ESI).† The BET surface areas of the GF0, GF10, GF20, GF40 and GF50 aerogels were found to be 295, 310, 377, 635 and 638 m2 g−1, respectively. The similarity of the specific surface areas of the GF40 and GF50 specimens indicated that the GF40 aerogel has reached an optimum level of porosity. Fig. S3b (ESI)† shows how the distribution of the incremental pore volume varied with the pore diameter. The pore size distribution and peak pore volume increased with the incorporation of the triblock copolymer to form a highly porous graphene aerogel.
Raman spectroscopic studies were employed to confirm the reduction of GO by vitamin C and the removal of the F-68 copolymer from the aerogel after heat treatment. The Raman spectra of GO, raw-GF20 and GF20 are shown in Fig. 2a. GO displayed two prominent peaks at 1345 and 1586 cm−1, which corresponded to the D and G bands, respectively. The D band is assigned to structural defects and disordered carbon atoms in graphite layers, and the G band corresponds to E2g phonons of sp2 C atoms, which are well documented in the literature.36 Raw-GF20 and GF20 exhibited similar peaks that corresponded to the D and G bands. The ID/IG intensity ratio increased gradually from GO to raw-GF20 and then to GF20, which confirms the reduction of GO by vitamin C. Upon the reduction of GO, more numerous and smaller graphene domains were created, which eventually increased the ID/IG ratio.37–39 The intensity (ID/IG) ratio for raw-GF20 was lower with respect to that for GF20, which would be due to the presence of the copolymer in the graphene matrix. The higher ID/IG ratio of GF20 confirmed that the copolymer was removed after the heat treatment.
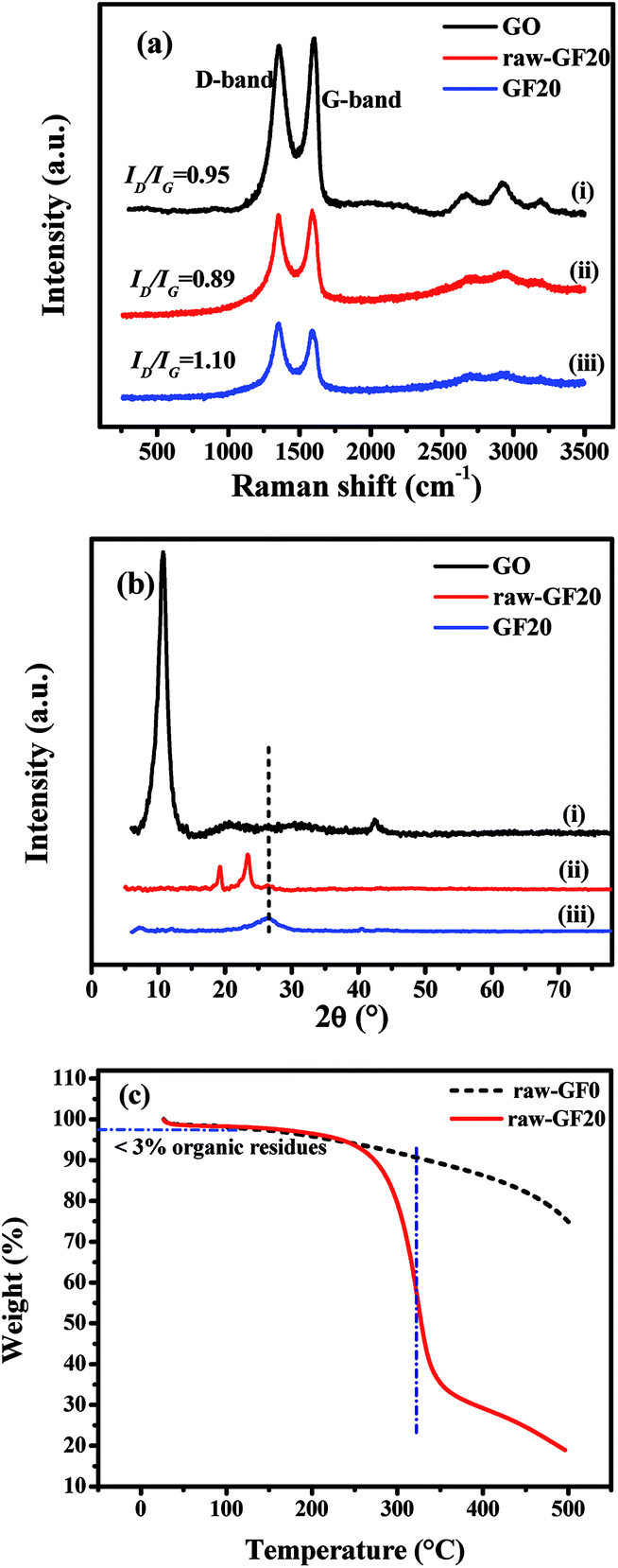 | ||
| Fig. 2 (a) Raman spectra and (b) XRD patterns of (i) GO, (ii) raw-GF20, and (iii) GF20 aerogel; (c) TGA of raw-GF0 and raw-GF20. | ||
The X-ray diffraction patterns of the crystal phases of GO, raw-GF20 and GF20 are shown in Fig. 2b. In Fig. 2b(i), the sharp peak at 2θ = 10.8° corresponds to the (001) plane of GO with a d-spacing of 0.215 Å. Raw-GF20 (Fig. 2b(ii)) displayed peaks at 2θ = 19.8°, 23.4° and 26.5°. The sharp peaks at 19.8° and 23.4° correspond to the crystalline structure of PEO chains with interlayer d-spacings of 3.74 and 3.83 Å, respectively.40 The characteristic broad peak at 26.5° corresponds to graphene with a d-spacing of 3.41 Å. Fig. 2b(iii) shows the XRD pattern of GF20, and the single broad peak at 26.4° is typical of graphene sheets with a d-spacing of 3.25 Å. The peak intensity ratio was higher for GF20 in comparison with raw-GF20. No other peak corresponding to PEO chains was observed, which confirms that the F-68 copolymer was removed after heat treatment of the template.
The thermal stability of raw aerogels (freeze-dried samples) was determined by TGA at a heating rate of 10 °C min−1 to a temperature of 500 °C in an oxygen atmosphere. It can be seen from Fig. 2c that raw-GF20 displayed a weight loss of ∼3% in the initial stage owing to the evaporation of adsorbed water and organic residues. The major weight loss at around 310 °C was associated with the combustion of Pluronic copolymer. The weight loss remained stable at ∼19% when the temperature reached 500 °C. The raw-GF0 aerogel exhibited a mass loss of about 25% at 500 °C. The mass loss was due to the loss of organic residues and functional groups from the pure graphene aerogel.
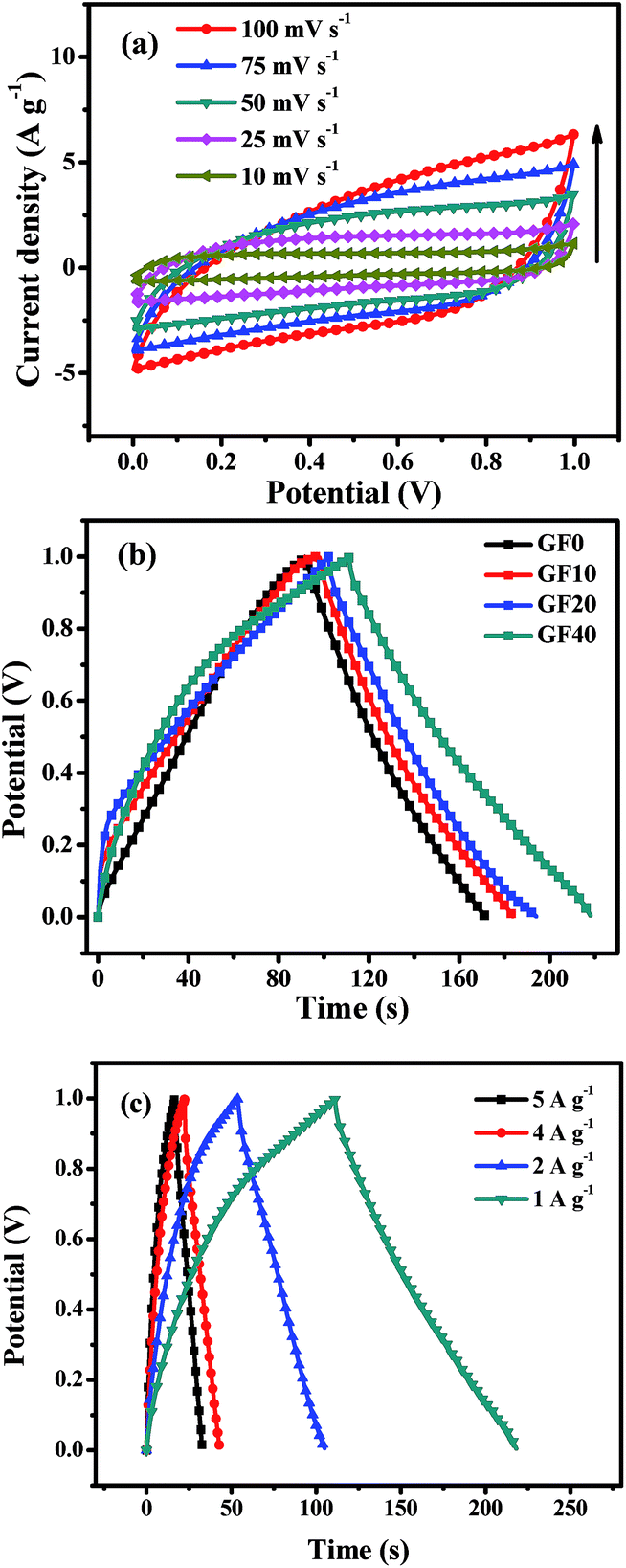 | ||
| Fig. 3 (a) CV curves of GF20 SSC for scan rates of 10–100 mV s−1; (b) CCD curves of GF0, GF10, GF20 and GF40 SSCs at 1 A g−1; and (c) CCD curves at different current densities of the GF40 SSC. | ||
A comparison of the CCD curves for SSCs based on the GF0, GF10, GF20 and GF40 aerogels is shown in Fig. 3b. The specific capacitance (Csp) of SSCs based on GF0, GF10, GF20 and GF40 was found to be 81, 87, 95 and 111 F g−1 at 1 A g−1, respectively. These results are in accordance with the basic EDL mechanism, whereby a larger surface area produces higher values of capacitance and energy density. Hence, we selected the GF40 aerogel for further study in an asymmetric supercapacitor.
A CCD study of a GF40//GF40 SSC was carried out at different current densities of 5, 4, 2, and 1 A g−1, as shown in Fig. 3c. It can be seen that the charging and discharging times decreased with an increase in current density. The Csp value and energy density of the GF40//GF40 SSC were calculated from the discharge curve using eqn (2) and (3). The Csp value of the GF40//GF40 SSC was found to be 87, 91, 107, and 111 F g−1 at current densities of 5, 4, 2 and 1 A g−1, respectively. The energy density was found to be 15.41 W h kg−1 at a power density of 0.523 kW kg−1 and 12.08 W h kg−1 at a high power density of 2.967 kW kg−1. It is apparent that the highly porous structure of the aerogel induced by the copolymer was responsible for the high energy density and high capacitance of the GF40//GF40 SSC.
3.2 Positive electrode material
 | ||
| Fig. 4 (a) Low- and (b) high-magnification FESEM micrographs of MnO2@PANI [inset: TEM image of MnO2@PANI]; (c and f) normal images of RGO/M@P25 film; FESEM images of edge of cross-section of RGO/M@P25 film at (d) lower magnification and (e, g and h) higher magnification. | ||
Using our technique, we obtained a 3D RGO/M@P25 porous film (Fig. 4c) after the reduction of the GO/M@P25 film by hydrazine vapor in the hydrothermal autoclave. The GO sheet contained epoxy, hydroxyl and carboxyl groups in its basal plane and edges, which promoted the anchoring of MnO2@PANI nanostructures onto the surfaces of GO sheets. The advantage of our technique is that the MnO2@PANI that is produced acts as a spacer between graphene sheets, which thus prevents the sheets from aggregating. Moreover, during the reduction process, gaseous products (H2O(g) and CO2(g)) are formed inside the GO layers.15 The released gases serve to exfoliate the RGO film and make it porous. The content (wt%) of the RGO/M@P25 film was determined from mass calculations for the MnO2@PANI coaxial nanoparticles and RGO/MnO2@PANI film. The RGO/M@P25 film contained ∼75 wt% RGO and ∼25% MnO2@PANI.
The top view of the hybrid film shown in Fig. 4c shows curved surface regions where exfoliation had occurred owing to the entrapment of gas bubbles within the film. A side view FESEM image of a cross-section of the RGO/M@P25 film is shown in Fig. 4d. The magnified image in Fig. 4g confirms that the films were decorated with many pores and channels. The magnified side view images of the cross-section of the film in Fig. 4e and h clearly highlight the exfoliation of RGO and the presence of an even distribution of MnO2@PANI in the RGO matrix. Fig. 4f shows that the RGO/M@P25 film remained intact after bending and folding, which thus indicates its remarkable flexibility. It is clear from Fig. 5a and b that the GO/M@P25 film expanded by a factor of ∼3.5 after the reduction step. This high level of exfoliation and the MnO2@PANI spacer particles enabled a greater surface area to be accessible for the diffusion of ions, which led to higher charge transfer and higher capacitance. It was found that the flexibility of the film decreased when the amount of MnO2@PANI spacer particles increased.
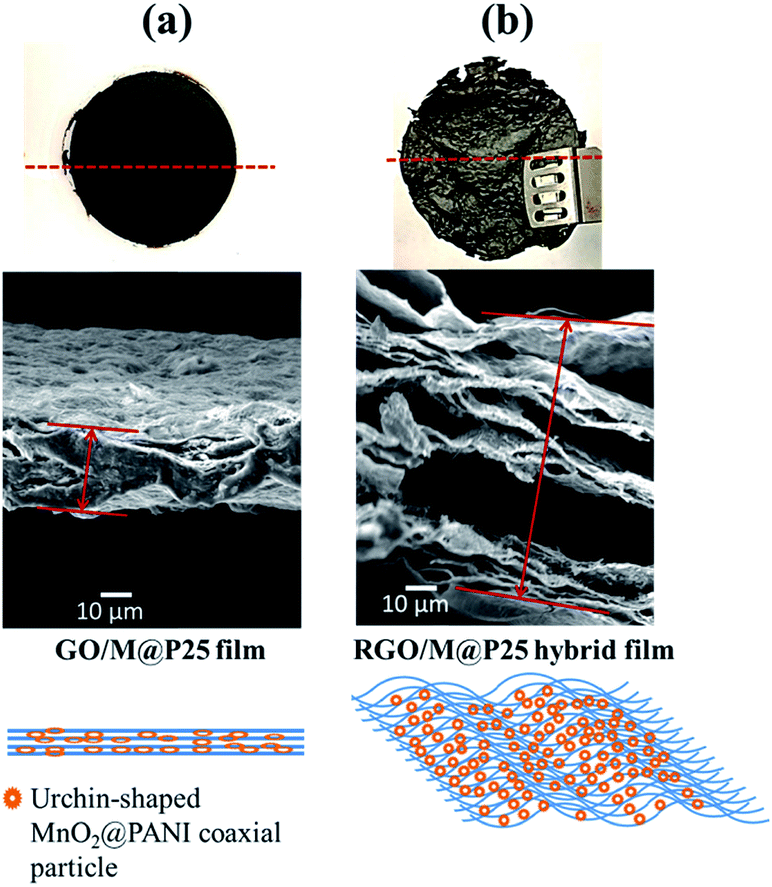 | ||
| Fig. 5 Schematic of exfoliation of (a) the GO/M@P25 film and (b) the RGO/M@P25 film. | ||
The structures of the MnO2@PANI-based graphene film and RGO/M@P25 film were evaluated by comparing their XRD patterns (see Fig. 6a). It can be seen from Fig. 6a(i) that MnO2@PANI exhibited 2θ peaks at 12.7°, 18.1°, 28.8°, 37.5°, 42.1°, 49.9°, 56.7°, 60.3° and 69.7°, which corresponded to the diffraction peaks of the (110), (200), (310), (211), (301), (411), (600), (521) and (541) crystal planes, respectively. These data closely match the standard data for α-MnO2 in JCPDS no. 44-0141.41 The diffraction peaks for the RGO/M@P25 film are shown in Fig. 6a(ii). The peaks that appeared at 2θ = 11.6°, 28.8°, 37.9°, 42.5°, 46.9°, 47.9°, 56.3° and 60.4° matched the expected diffraction peaks of the crystalline phase of α-MnO2. However, the peak intensity decreased owing to the complex nature of the ternary composite system. For the RGO/M@P25 film, a new broad peak appeared at 2θ = ∼25°, which corresponded to the crystal plane of RGO and confirmed the reduction of GO to RGO.42,43
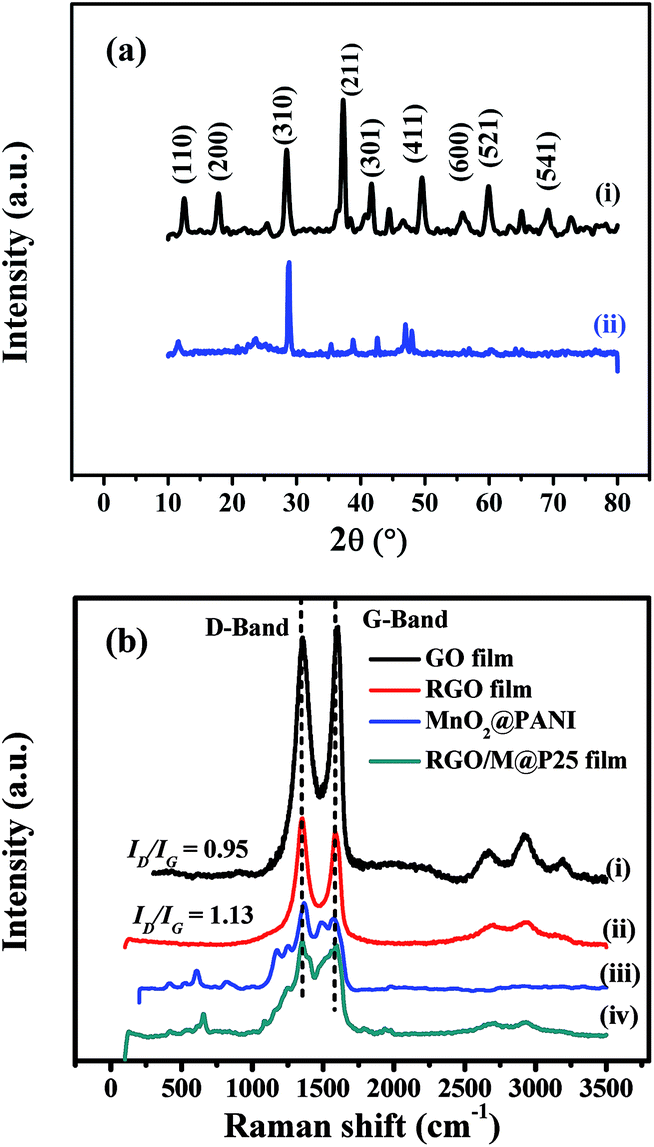 | ||
| Fig. 6 (a) XRD patterns of (i) MnO2@PANI and (ii) RGO/M@P25 film; (b) Raman spectra of (i) GO film, (ii) RGO film, (iii) MnO2@PANI and (iv) RGO/M@P25 film. | ||
Raman spectroscopy was used to confirm the reduction of GO to RGO and the presence of interactions between MnO2@PANI and RGO in the RGO/M@P25 hybrid film. Fig. 6b(i) and (ii) show the Raman spectra of the GO film and RGO film, which contained peaks that corresponded to the D band and G band.36 The ID/IG intensity ratio increased when GO was transformed into RGO owing to the formation of more numerous but smaller graphene networks.37–39 This increase in the ID/IG ratio confirmed that GO was successfully reduced by hydrazine vapor. The Raman spectra of MnO2@PANI and the RGO/M@P25 film are shown in Fig. 6b(iii) and (iv), respectively. MnO2@PANI displayed all the characteristic peaks of MnO2 and PANI, as mentioned in our previous work.33 The Raman spectrum of the RGO/M@P25 film exhibited peaks that corresponded to the characteristic D band and G band of graphene, as well as peaks that corresponded to MnO2@PANI, which overlapped in some regions. Moreover, there was a change in peak intensities for the RGO/M@P25 film with respect to those for the MnO2@PANI composite and the RGO film, which indicated that there were interactions between MnO2@PANI and the RGO film.
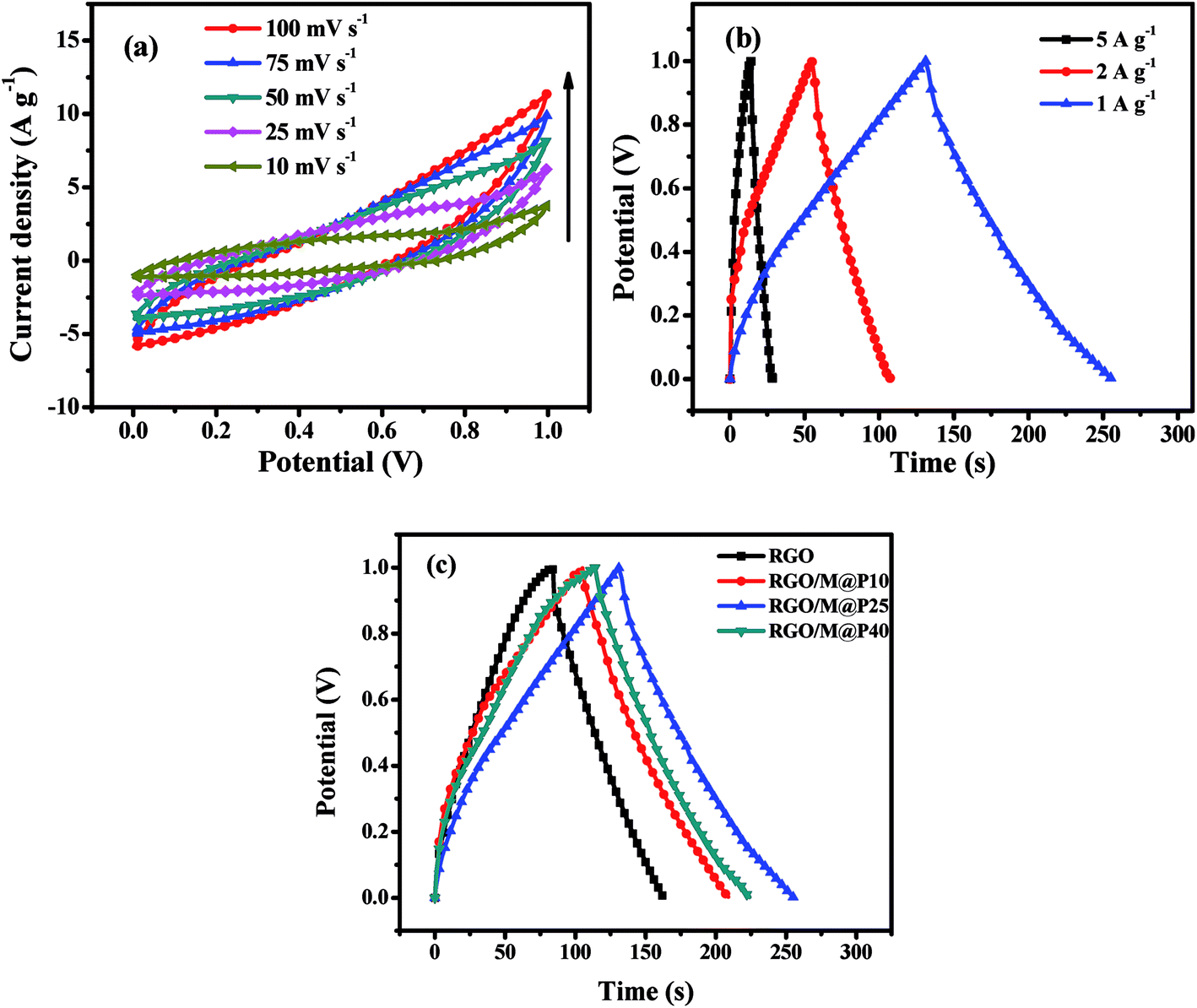 | ||
| Fig. 7 (a) CV plots of RGO/M@P25 SSC at different scan rates; (b) CCD plots of RGO/M@P25 SSC at different current densities; (c) comparison of CCD plots of RGO, RGO/M@P10, RGO/M@P25 and RGO/M@P40 SSCs at 1 A g−1 current density. | ||
A cyclic charge–discharge study of the RGO/M@P25 SSC was performed at a constant current within the voltage window of 0–1.0 V at current densities of 5, 2 and 1 A g−1, as shown in Fig. 7b. The Csp values of the RGO/M@P25 SSC at current densities of 5, 2 and 1 A g−1 were found to be 80, 103 and 125 F g−1, respectively. The energy density was found to be 18.33 W h kg−1 at a power density of 0.388 kW kg−1. At low current densities, the increase in the Csp value was small, which was probably due to the slower charge–discharge of electrolyte ions. In general, the Csp values followed the same trend as was observed from the CV study.
CCD studies were carried out for the RGO, RGO/M@P10, RGO/M@P25 and RGO/M@P40 hybrid films to determine which had the best performance (see Fig. 7c). The RGO, RGO/M@P10, RGO/M@P25 and RGO/M@P40 SSCs were found to have Csp values of 82, 104, 125 and 110 F g−1, respectively. It is evident that the Csp values increased when the contents of the pseudocapacitive materials MnO2 and PANI increased. The conductive PANI layer on the MnO2 spikes facilitated ion transport from the coaxial microurchin composite to the RGO matrix, which provided high double-layer capacitance. This served to increase the overall capacitance owing to the synergistic effect of pseudocapacitive and EDL capacitive materials. Moreover, the 3D porous graphene film provided a larger surface area for the ready access of electrolyte ions, which ultimately increased the capacitance.
It can be seen that the MnO2@PANI particles played a dual role. Firstly, they acted as spacers between the graphene layers to prevent stacking from occurring. Secondly, they contributed to the high pseudocapacitance of the composite. Although the Csp value increased with an increase in the MnO2@PANI content, the RGO/M@P40 sample exhibited a lower capacitance than the RGO/M@P25 sample, which was counterintuitive. However, detailed studies of the sample morphology revealed that at higher ratios of MnO2@PANI the microurchins tended to agglomerate, which inhibited their even distribution in the graphene matrix. This was the reason why the gravimetric capacitance could not increase further. Thus, for the asymmetric device we selected the RGO/M@P25 porous film, as it had the highest capacitance and energy density.
3.3 Asymmetric hybrid supercapacitor
A flexible all-solid-state asymmetric supercapacitor (ASC) was fabricated using the RGO/M@P25 composite film as the positive electrode, GF40 as the negative electrode and PVA/KOH as the gel electrolyte. A schematic diagram of the solid-state ASC is shown in Fig. 8a. To determine the best working potential window, separate CV studies in a three-electrode system were first carried out for the GF40 and RGO/M@P25 electrode materials, respectively, as shown in Fig. 8b. It is apparent that the best working range for the negative electrode was 0 to −0.8 V, whereas that for the positive electrode was 0 to +1.0 V. Hence, the maximum potential window for the asymmetric supercapacitor cell can reach 1.8 V. To achieve a potential window of 1.8 V, it is essential to balance the charges stored at the positive and negative electrodes.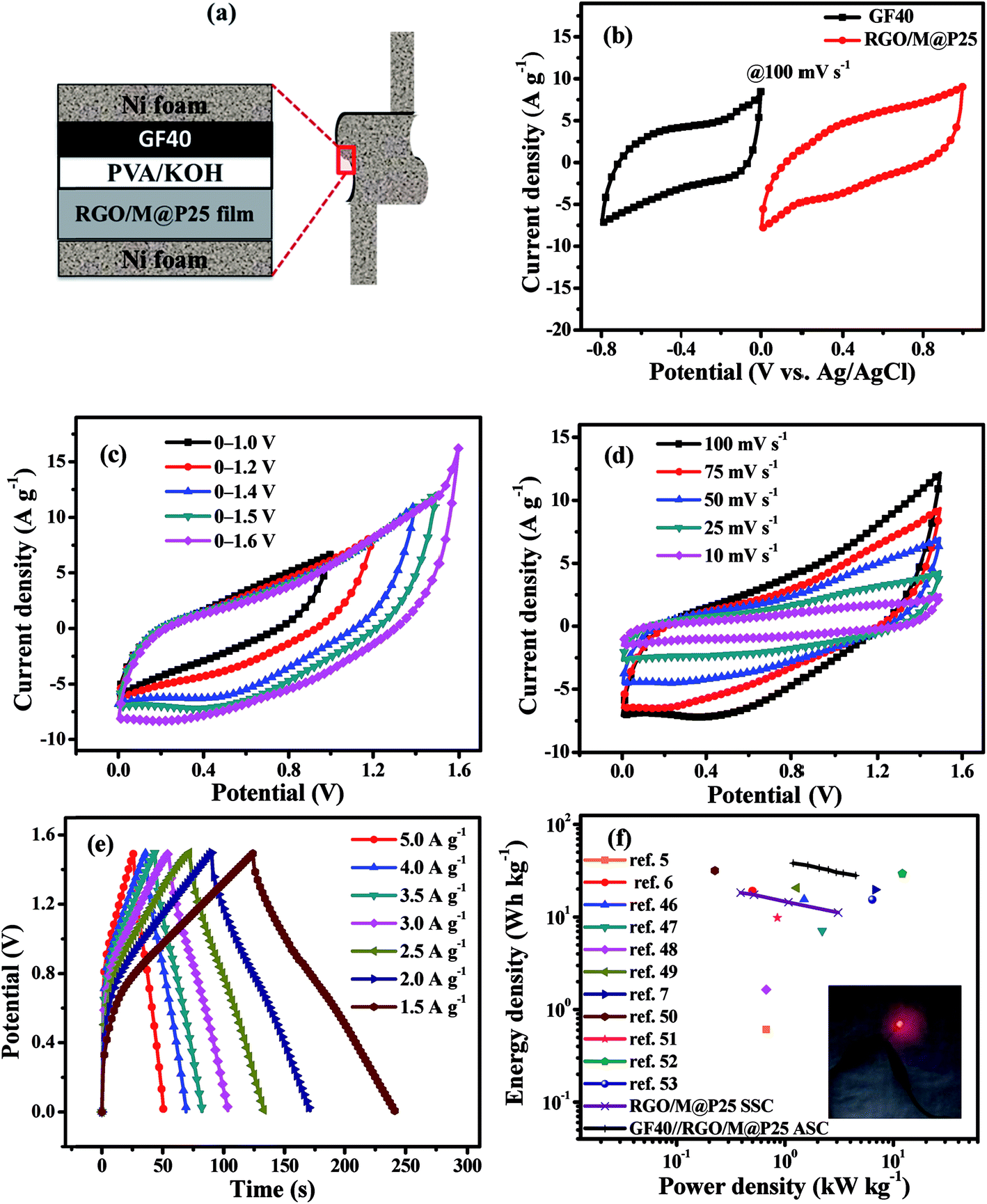 | ||
| Fig. 8 (a) Schematic of flexible solid-state GF40//RGO/M@P25 ASC; (b) CV curves for RGO/M@P25 and GF40 materials in a three-electrode system at 100 mV s−1; (c) CV curves for different voltage windows at a scan rate of 100 mV s−1, (d) CV curves at different scan rates of 10–100 mV s−1 and (e) CCD curves at different current densities for the GF40//RGO/M@P25 ASC; (f) Ragone plots for RGO/M@P25 SSC and GF40//RGO/M@P25 ASC with reference values [inset: a red LED was powered by the ASC]. | ||
The charge stored in an electrode can be calculated by the following equation:23
| Q = Cspe × ΔV × m | (5) |
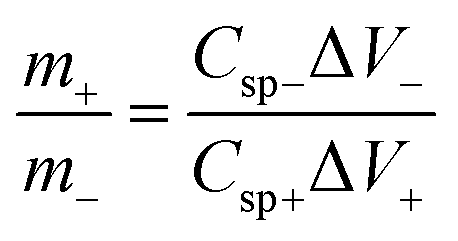 | (6) |
To determine the optimal working window of the solid-state GF40//RGO/M@P25 ASC, CV studies were carried out in the ranges of 0–1.0, 0–1.2, 0–1.4, 0–1.5 and 0–1.6 V at 100 mV s−1, as shown in Fig. 8c. It is apparent that the ASC had a stable potential window in the range of 0–1.5 V. Next, the CV studies were conducted at scan rates of 10 to 100 mV s−1 within the potential range of 0–1.5 V (see Fig. 8d). It can be seen from Fig. 8d that the potential window remained stable at all scan rates. The current density was found to increase with an increase in the scan rate.
The charge–discharge curves (CCD) of the solid-state GF40//RGO/M@P25 ASC within the potential range of 0–1.5 V at current densities of 5, 4, 3.5, 3, 2.5, 2, and 1.5 A g−1 are shown in Fig. 8e. It was found that a high Csp value of 122 F g−1 was achieved when the current density was 1.5 A g−1. A Ragone plot, which shows the relationship between the power density and energy density, was plotted for both the SSCs and the ASC, as shown in Fig. 8f. It can be seen that at the same power density, the GF40//RGO/M@P25 ASC exhibited a higher energy density than the RGO/M@P25//RGO/M@P25 SSC and the GF40//GF40 SSC. The highest energy density was found to be 38.12 W h kg−1 at a power density of 1.191 kW kg−1. The ASC cell maintained a high energy density of 28.12 W h kg−1 even at a power density of 4.490 kW kg−1.
Electrochemical impedance spectroscopy (EIS) tests were carried out in the frequency range of 100 kHz to 1 Hz for all the symmetric and asymmetric supercapacitors. Fig. 9a shows Nyquist plots for the GF40//GF40 and RGO/M@P25//RGO/M@P25 SSCs and the GF40//RGO/M@P25 ASC. The inset in Fig. 9a illustrates a typical Nyquist plot to facilitate the analysis of different parts of the curve. The intercept with the Zreal axis at point A represents the equivalent series resistance Rs, which includes the contact resistance between the current collector and the electrode material, the intrinsic resistance of the active material and the ionic resistance of the electrolyte.44 In the medium-frequency region (denoted as B), the diameter of the semicircle indicates the charge transfer resistance (Rct). In the low-frequency region (denoted as C), the portion of the curve with a slope of 45° corresponds to the Warburg resistance (RW), which is a measure of the diffusion of electrolyte ions towards the electrodes. A higher slope of the line indicates a higher diffusion rate and a lower RW value.44,45 The Rs values of the GF40//GF40 and RGO/M@P25//RGO/M@P25 SSCs were found to be 0.47 and 11.0 Ω, respectively. In comparison, the GF40//RGO/M@P25 ASC had an Rs value of 7.38 Ω. The higher Rs value for the RGO/M@P25 SSC was due to the high intrinsic resistance and contact resistance of the porous substrate. The low Rs value for the GF40 aerogel was consistent with its high conductivity and also indicated that strong bonding occurred with the Ni foam. The presence of the pseudocapacitive MnO2 and PANI materials caused the RGO/M@P25 sample to have a higher Warburg resistance. The GF40 aerogel, which exhibited EDL capacitive behavior, had the highest slope in the low-frequency region. The slope for the ASC was between those of the positive and negative electrode materials.
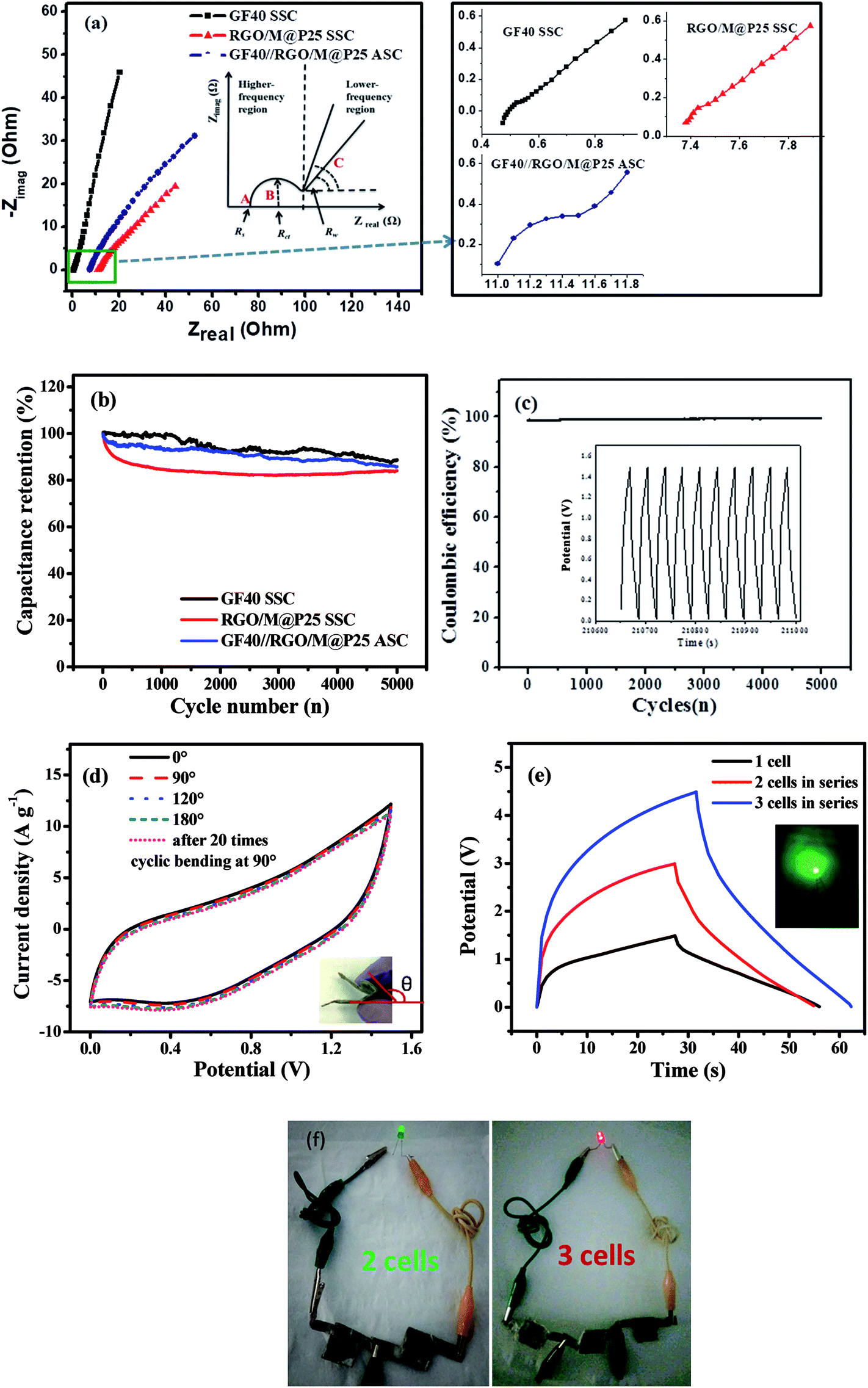 | ||
| Fig. 9 (a) Nyquist plots for GF40, the RGO/M@P25 SSC and the GF40//RGO/M@P25 ASC [the inset shows a schematic diagram explaining the Nyquist plot. Magnifications of the high-frequency region for the SSC and ASC are shown in the right-hand panel]; (b) cycling stability performance over 5000 cycles and (c) coulombic efficiency for the GF40//RGO/M@P25 ASC over 5000 cycles [the inset shows the charge–discharge behavior over the last 10 cycles]; (d) CV curves for the ASC in different bending states [inset: optical image showing the bent sample]; (e) CCD curves for GF40//RGO/M@P25 where two and three ASC cells were connected in series [inset: green LED powered by the ASC]; (f) images of green and red LEDs lighted up by 2 and 3 ASC cells connected in series, respectively. | ||
Cycling stability tests were carried out for both the SSCs and the ASC at a current density of 5 A g−1 over 5000 cycles. It can be seen in Fig. 9b that the percentage capacitance retention after cycling was high for the SSCs and ASC. The GF40//GF40 SSC exhibited a high capacitance retention of 88.6%. The high cycling stability of this SSC can be explained by the pure EDL capacitive behavior of the 3D porous foam, in which the capacitance was derived exclusively from the adsorption–desorption mechanism of ions on the surface of the porous foam. The RGO/M@P25//RGO/M@P25 SSC displayed a capacitance retention of 83.9%, whereas the GF40//RGO/M@P25 ASC exhibited a capacitance retention of 85.8%. In the SSCs and ASC degradation of the urchin-shaped MnO2@PANI during charge–discharge cycling was mitigated by the RGO film matrix that covered and protected the MnO2@PANI phase. The GF40//RGO/M@P25 ASC achieved a stable coulombic efficiency of ∼98% up to 5000 cycles (see Fig. 9c). This is especially evident from the inset in Fig. 9c, which illustrates the last 10 cycles of the charge–discharge study for the GF40//RGO/M@P25 ASC.
The ASC device could withstand bending to different angles without being physically damaged or experiencing impairments in its electromechanical properties. This was confirmed by CV profiles obtained when the ASC was held at different angles, as shown in Fig. 9d. It is evident that there was little or no deviation in the CV curves of the ASC at different bending angles. This was observed even after the ASC was subjected 20 times to reversible bending between 0° and 90°. Thus, the ASC can be utilized in a bent condition without any loss of performance. Photographs of the ASC device in different bending states are shown in Fig. S4 (ESI).†
To assess the performance of the ASC cell in a practical application, two and three ASC cells were connected in series and charged to potential windows of 0–3.0 and 0–4.5 V, respectively. The charge–discharge profiles at a constant current of 20 mA of the cells that were connected in series are shown in Fig. 9e. Three cell devices that were connected in series lit up a red light-emitting diode (1.8 V red LED) successfully for 45 s. Photographs of two cells and three cells in series lighting up green and red LEDs are shown in Fig. 9f.
To determine the leakage current and self-discharge behavior, our GF40//RGO/M@P25 ASC cell was initially charged from 0 to 1.0 V at 0.5 A g−1 current density. Then, a potentiostatic test was conducted by keeping the voltage unchanged at 1.0 V for 2 h whilst the current flowing through the device was measured. The current flowing in the stabilized device represents the leakage current. A plot of current versus time is shown below in Fig. 10a. It can be seen that the current decayed rapidly at the beginning and was stabilized at 120 μA after 2 h. Thus, the leakage current of the ASC cell is 120 μA.
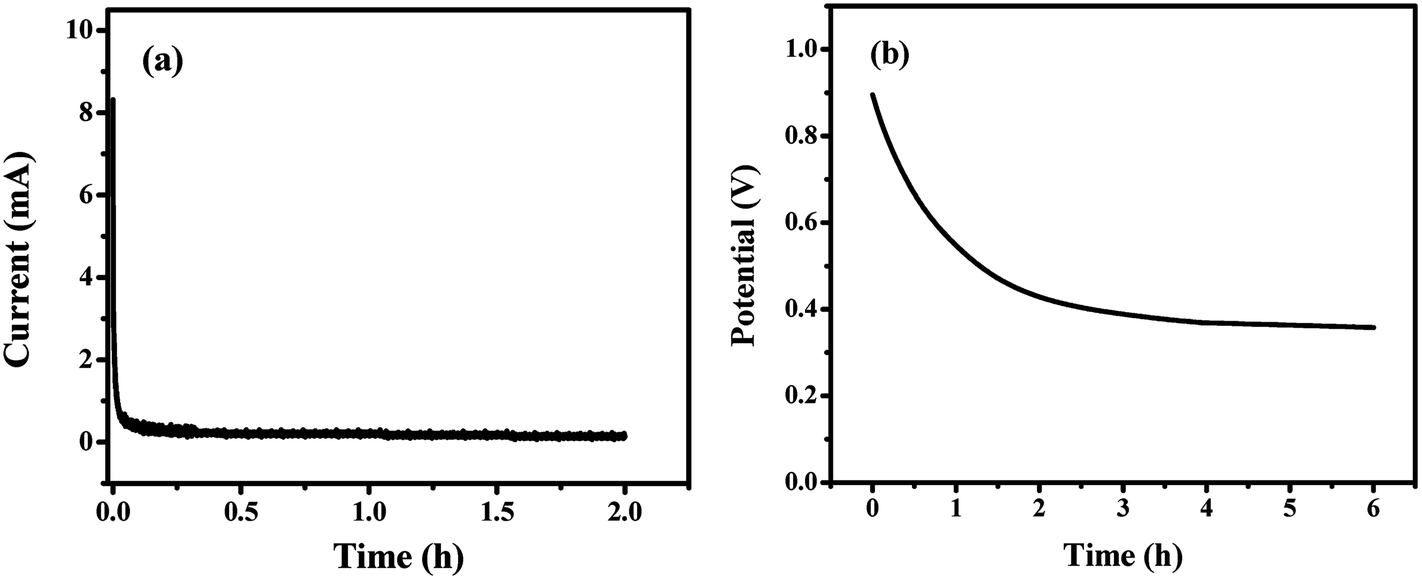 | ||
| Fig. 10 (a) Leakage current and (b) self-discharge tests of the GF40//RGO/M@P25 ASC cell, which was initially charged to 1.0 V at a 0.5 A g−1 current density. | ||
The self-discharge behavior of the device was then determined using an open-circuit potential test. It can be seen from the results in Fig. 10b that the potential decayed gradually during the first hour and was stabilized at a constant value after 4 hours. About 40% of the potential was retained after 6 hours.
Comparisons of the energy density, power density and cycling stability of all-solid-state symmetric and asymmetric supercapacitors from the literature are presented in Tables 1 and 2, respectively. It is evident that our synthesized GF40//RGO/M@P25 ASC displayed higher energy density and power density with high cycling performance in comparison with other ASCs mentioned in the literature.
| Materials | Electrolyte | Energy density (W h kg−1) | Power density (kW kg−1) | Cycling stability (% capacitance retention) | Ref. |
|---|---|---|---|---|---|
| Graphene hydrogel film | H2SO4/PVA gel | 0.61 | 0.67 | 91.6 (after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles) 000 cycles) |
5 |
| RCF/MnO2/PEDOT | PVA/KCl gel | 19.17 | 0.500 | 83 (after 10000 cycles) |
6 |
| CNTs/bacterial nanocellulose paper | PS-PEO-PS/[EMIM] [NTf2] | 15.5 | 1.500 | 99.5 (after 5000 cycles) | 46 |
| PANI/CNT | H2SO4/PVA gel | 7.1 | 2.2 | 91.9 (after 1000 cycles) | 47 |
| Graphene@carbon cloth | H2SO4/PVA gel | 1.64 | 0.670 | — | 48 |
| Polypyrrole nanofiber/RGO | PVA/H3PO4 gel | 20.6 | 1.280 | 79 (after 1000 cycles) | 49 |
| RGO/MnO2@PANI (RGO/M@P25) film | PVA/KOH gel | 18.33 | 0.388 | 83.9 (after 5000 cycles) | Our work |
| Graphene aerogel (GF40) | PVA/KOH gel | 15.41 | 0.523 | 88.6 (after 5000 cycles) | Our work |
| Materials | Electrolyte | Energy density (W h kg−1) | Power density (kW kg−1) | Cycling stability (% capacitance retention) | Ref. |
|---|---|---|---|---|---|
| Graphene (IL-CMG)//RuO2–IL-CMG | H2SO4/PVA gel | 19.7 | 6.800 | 95 (after 2000 cycles) | 7 |
| N-doped graphene/layered MnO2//AC | PVA/LiCl gel | 31.67 | 0.222 | 90.5 (after 1500 cycles) | 50 |
| MnO2@poly(3,4-ethylenedioxythiophene)//poly(3,4-ethylenedioxythiophene) | LiClO4/PMMA gel | 9.8 | 0.850 | 86 (after 1250 cycles) | 51 |
| GNR//GNR-MnO2 | PAAK/KCl gel | 29.4 | 12.1 | 88 (after 5000 cycles) | 52 |
| CNTs/MnO2//CNTs/PANI | Na2SO4/PVP gel | 24.8 | 0.120 | — | 35 |
| TiN@GNS//Fe2N@GNS | PVA/LiCl gel | 15.4 | 6.4 | ∼98 (after 20000 cycles) |
53 |
| MnO2-ERGO//CNT-ERGO | PAAK/KCl gel | 31.8 | 0.4536 | 84.4 (after 10000 cycles) |
54 |
| GF40//RGO/M@P25 | PVA/KOH gel | 38.12 | 1.191 | 85.8 (after 5000 cycles) | Our work |
4. Conclusions
We have fabricated a novel all-solid-state flexible asymmetric supercapacitor (ASC) that comprised a 3D graphene-based composite, whereby high energy density was achieved by enlarging the operational potential window. This novel ASC system utilized a novel 3D porous reduced graphene oxide/manganese dioxide@polyaniline (RGO/MnO2@PANI) hybrid film as the positive electrode and a self-assembled 3D pillared graphene aerogel as the negative electrode material. The ASC, which used PVA/KOH as the gel electrolyte, operated in the wide potential window of 0–1.5 V. The self-assembled free-standing 3D graphene aerogel was synthesized using Pluronic copolymer as a template. A special feature of the positive electrode material was the presence of microurchin MnO2@PANI particles, which acted as spacers to mitigate agglomeration of the 3D RGO layers to maximize the surface area for ion transfer. The MnO2@PANI particles also contributed to the pseudocapacitive performance of the cell. The asymmetric supercapacitor exhibited a very high energy density of 38.12 W h kg−1 at a power density of 1.191 kW kg−1 and stable cycling stability with capacitance retention of 85.8% after 5000 cycles. The solid-state asymmetric supercapacitor exhibited high flexibility upon bending and can be employed in various portable devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Nanyang Technological University, Singapore for financial support and the provision of research equipment.References
- B. Conway, Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications, New York, 1999 Search PubMed.
- M. M. Sk and C. Y. Yue, J. Mater. Chem. A, 2014, 2, 2830–2838 CAS.
- M. M. Sk, C. Y. Yue, K. Ghosh and R. K. Jena, J. Power Sources, 2016, 308, 121–140 CrossRef CAS.
- R. Jena, C. Yue, M. Sk and K. Ghosh, Carbon, 2017, 115, 175–187 CrossRef CAS.
- Y. Xu, Z. Lin, X. Huang, Y. Liu, Y. Huang and X. Duan, ACS Nano, 2013, 7, 4042–4049 CrossRef CAS PubMed.
- X. Hu, W. Xiong, W. Wang, S. Qin, H. Cheng, Y. Zeng, B. Wang and Z. Zhu, ACS Sustainable Chem. Eng., 2016, 4, 1201–1211 CrossRef CAS.
- B. G. Choi, S.-J. Chang, H.-W. Kang, C. P. Park, H. J. Kim, W. H. Hong, S. Lee and Y. S. Huh, Nanoscale, 2012, 4, 4983–4988 RSC.
- Z. Zhang, F. Xiao, J. Xiao and S. Wang, J. Mater. Chem. A, 2015, 3, 11817–11823 CAS.
- Y. Fu, X. Cai, H. Wu, Z. Lv, S. Hou, M. Peng, X. Yu and D. Zou, Adv. Mater., 2012, 24, 5713–5718 CrossRef CAS PubMed.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC.
- C. Liu, Z. Yu, D. Neff, A. Zhamu and B. Z. Jang, Nano Lett., 2010, 10, 4863–4868 CrossRef CAS PubMed.
- Z. Zhang, F. Xiao and S. Wang, J. Mater. Chem. A, 2015, 3, 11215–11223 CAS.
- D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS PubMed.
- Q. Wu, Y. Xu, Z. Yao, A. Liu and G. Shi, ACS Nano, 2010, 4, 1963–1970 CrossRef CAS PubMed.
- Z. Niu, J. Chen, H. H. Hng, J. Ma and X. Chen, Adv. Mater., 2012, 24, 4144–4150 CrossRef CAS PubMed.
- Z. Weng, Y. Su, D.-W. Wang, F. Li, J. Du and H.-M. Cheng, Adv. Energy Mater., 2011, 1, 917–922 CrossRef CAS.
- G. Wang, X. Sun, F. Lu, H. Sun, M. Yu, W. Jiang, C. Liu and J. Lian, Small, 2012, 8, 452–459 CrossRef CAS PubMed.
- A. Sumboja, C. Y. Foo, X. Wang and P. S. Lee, Adv. Mater., 2013, 25, 2809–2815 CrossRef CAS PubMed.
- Z. Li, Y. Mi, X. Liu, S. Liu, S. Yang and J. Wang, J. Mater. Chem., 2011, 21, 14706–14711 RSC.
- X. Feng, Z. Yan, N. Chen, Y. Zhang, Y. Ma, X. Liu, Q. Fan, L. Wang and W. Huang, J. Mater. Chem. A, 2013, 1, 12818–12825 CAS.
- W. Wei, X. Cui, W. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40, 1697–1721 RSC.
- Z. Zhang, K. Chi, F. Xiao and S. Wang, J. Mater. Chem. A, 2015, 3, 12828–12835 CAS.
- J. Yan, Z. Fan, W. Sun, G. Ning, T. Wei, Q. Zhang, R. Zhang, L. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632–2641 CrossRef CAS.
- X. Lu, M. Yu, G. Wang, T. Zhai, S. Xie, Y. Ling, Y. Tong and Y. Li, Adv. Mater., 2013, 25, 267–272 CrossRef CAS PubMed.
- P.-C. Chen, G. Shen, Y. Shi, H. Chen and C. Zhou, ACS Nano, 2010, 4, 4403–4411 CrossRef CAS PubMed.
- L. Qiu, J. Z. Liu, S. L. Y. Chang, Y. Wu and D. Li, Nat. Commun., 2012, 3, 1241 CrossRef PubMed.
- L. Zhang and G. Shi, J. Phys. Chem. C, 2011, 115, 17206–17212 CAS.
- Z. Zhang, F. Xiao, Y. Guo, S. Wang and Y. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 2227–2233 CAS.
- Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324–4330 CrossRef CAS PubMed.
- S.-Z. Zu and B.-H. Han, J. Phys. Chem. C, 2009, 113, 13651–13657 CAS.
- Y. Zhao, J. Liu, Y. Hu, H. Cheng, C. Hu, C. Jiang, L. Jiang, A. Cao and L. Qu, Adv. Mater., 2013, 25, 591–595 CrossRef CAS PubMed.
- X. Jiang, Y. Ma, J. Li, Q. Fan and W. Huang, J. Phys. Chem. C, 2010, 114, 22462–22465 CAS.
- K. Ghosh, C. Y. Yue, M. M. Sk and R. K. Jena, ACS Appl. Mater. Interfaces, 2017, 9, 15350–15363 CAS.
- A. M. Khattak, H. Yin, Z. A. Ghazi, B. Liang, A. Iqbal, N. A. Khan, Y. Gao, L. Li and Z. Tang, RSC Adv., 2016, 6, 58994–59000 RSC.
- Y. Jin, H. Chen, M. Chen, N. Liu and Q. Li, ACS Appl. Mater. Interfaces, 2013, 5, 3408–3416 CAS.
- M. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. Cancado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276–1290 RSC.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- Z. Ji, X. Shen, M. Li, H. Zhou, G. Zhu and K. Chen, Nanotechnology, 2013, 24, 115603 CrossRef PubMed.
- H. Gao, F. Xiao, C. B. Ching and H. Duan, ACS Appl. Mater. Interfaces, 2012, 4, 2801–2810 CAS.
- A. R. Polu and H.-W. Rhee, Adv. Polym. Technol., 2017, 36, 145–151 CrossRef CAS.
- B. Li, G. Rong, Y. Xie, L. Huang and C. Feng, Inorg. Chem., 2006, 45, 6404–6410 CrossRef CAS PubMed.
- X. Qiao, S. Liao, C. You and R. Chen, Catalysts, 2015, 5, 981–991 CrossRef CAS.
- Z.-H. Liu, Z.-M. Wang, X. Yang and K. Ooi, Langmuir, 2002, 18, 4926–4932 CrossRef CAS.
- M. M. Sk and C. Y. Yue, RSC Adv., 2014, 4, 19908–19915 RSC.
- M. M. Sk, C. Y. Yue and R. K. Jena, RSC Adv., 2014, 4, 5188–5197 RSC.
- Y. J. Kang, S.-J. Chun, S.-S. Lee, B.-Y. Kim, J. H. Kim, H. Chung, S.-Y. Lee and W. Kim, ACS Nano, 2012, 6, 6400–6406 CrossRef CAS PubMed.
- C. Meng, C. Liu, L. Chen, C. Hu and S. Fan, Nano Lett., 2010, 10, 4025–4031 CrossRef CAS PubMed.
- S. Wang, B. Pei, X. Zhao and R. A. Dryfe, Nano Energy, 2013, 2, 530–536 CrossRef CAS.
- S. Li, C. Zhao, K. Shu, C. Wang, Z. Guo, G. G. Wallace and H. Liu, Carbon, 2014, 79, 554–562 CrossRef CAS.
- Y. Liu, X. Miao, J. Fang, X. Zhang, S. Chen, W. Li, W. Feng, Y. Chen, W. Wang and Y. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 5251–5260 CAS.
- J. Duay, E. Gillette, R. Liu and S. B. Lee, Phys. Chem. Chem. Phys., 2012, 14, 3329–3337 RSC.
- M. Liu, W. W. Tjiu, J. Pan, C. Zhang, W. Gao and T. Liu, Nanoscale, 2014, 6, 4233–4242 RSC.
- C. Zhu, P. Yang, D. Chao, X. Wang, X. Zhang, S. Chen, B. K. Tay, H. Huang, H. Zhang and W. Mai, Adv. Mater., 2015, 27, 4566–4571 CrossRef CAS PubMed.
- Z. Zhang, F. Xiao, L. Qian, J. Xiao, S. Wang and Y. Liu, Adv. Energy Mater., 2014, 4, 1400064 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00433h |
| This journal is © The Royal Society of Chemistry 2018 |