
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA bio-inspired approach to ligand design: folding single-chain peptoids to chelate a multimetallic cluster†
Andy I.
Nguyen
 a,
Ryan K.
Spencer
b,
Christopher L.
Anderson
a and
Ronald N.
Zuckermann
*a
a,
Ryan K.
Spencer
b,
Christopher L.
Anderson
a and
Ronald N.
Zuckermann
*a
aMolecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. E-mail: rnzuckermann@lbl.gov
bDepartment of Chemistry, Department of Chemical Engineering & Materials Science, University of California, Irvine, CA 92697, USA
First published on 15th November 2018
Abstract
Synthesis of biomimetic multimetallic clusters is sought after for applications such as efficient storage of solar energy and utilization of greenhouse gases. However, synthetic efforts are hampered by a dearth of ligands that are developed for multimetallic clusters due to current limitations in rational design and organic synthesis. Peptoids, a synthetic sequence-defined oligomer, enable a biomimetic strategy to rapidly synthesize and optimize large, multifunctional ligands by structural design and combinatorial screening. Here we discover peptoid oligomers (≤7 residues) that fold into a single conformation to provide unprecedented tetra- and hexadentate chelation by carboxylates to a [Co4O4] cubane cluster. The structures of peptoid-bound cubanes were determined by 2D NMR spectroscopy, and their structures reveal key steric and side-chain-to-main chain interactions that work in concert to rigidify the peptoid ligand. This efficient ligand design strategy holds promise for creating new scaffolds for the abiotic synthesis and manipulation of multimetallic clusters.
Introduction
There is growing interest in multimetallic clusters found in enzymes that catalyze important reactions such as water splitting (photosystem II), nitrogen fixation (nitrogenase), and carbon dioxide reduction (carbon monoxide dehydrogenase).1–3 Successful mimicry of these enzymes would result in more stable catalysts, that are readily produced and potentially deployable on an industrial scale to enable solar-to-fuel conversion and minimize man-made climate change.4 To understand and mimic these enzymes requires synthesis of multimetallic clusters in a simplified chemical environment; however, approaches to synthesize chelating ligands needed to stabilize and control cluster formation are underdeveloped. Since a multimetallic cluster has a larger volume than a single metal, its chelating ligand must be significantly larger than that of a single metal, complicating both the design and synthesis.Typical ligand designs use branching motifs with idiosyncratic synthetic routes that can take substantial amounts of time to optimize.5 The few known examples of synthetic ligands that chelate clusters depend on high symmetry, in order to simplify organic synthesis.6–9 An ideal ligand synthesis would permit full access to asymmetry, site-differentiation, and donor diversity, while also remaining easy to synthesize.
An alternative approach to ligand design is to mimic the way nature bind metals (Fig. 1). The linear and modular architecture of polypeptides is an efficient way to modulate size, shape, and function of suitable ligands. Individual metal binding moieties dispersed in a linear sequence are brought into the appropriate geometry through folding of the polypeptide chain. The folding of sequence-defined oligomers is complex, but the modular and efficient synthesis permits the use of combinatorial approaches to seek out properly folded or functional sequences. We aim to develop modular peptidomimetic systems to rapidly create and evaluate large organic ligands for metal cluster chelation. Recently, a short minimalist peptide comprised of natural and mirror-image amino acids was designed to chelate a Fe4S4 cubane, demonstrating an important first step toward non-natural metalloenzymes.10
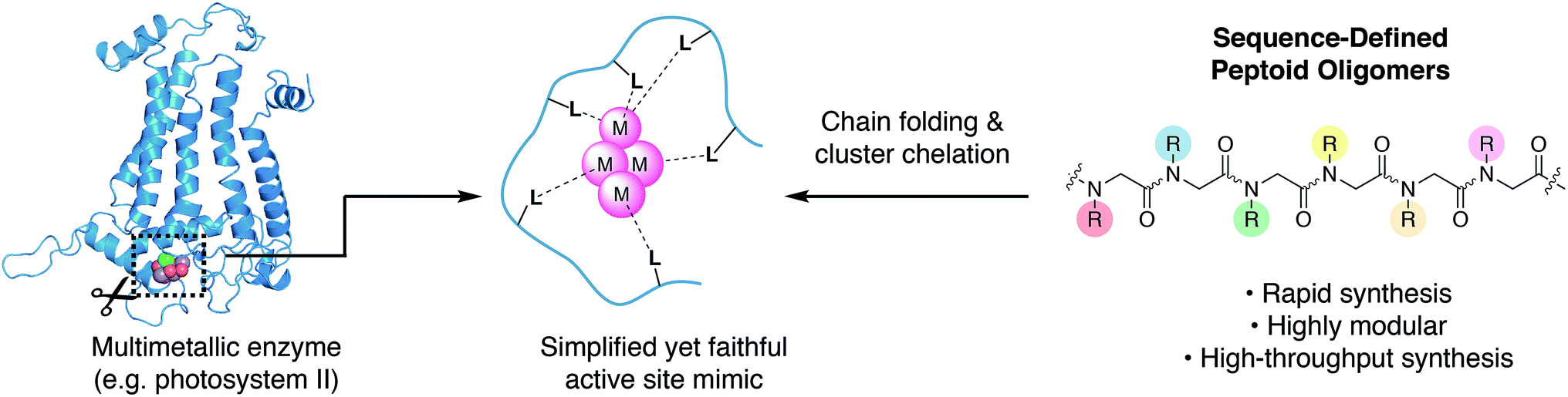 | ||
| Fig. 1 To faithfully reproduce the multimetallic cluster active site in a simplified environment, chelating ligands must be developed to control the coordination and assembly of the cluster. Peptoids have biomimetic structural and synthetic properties that enable discovery of such cluster-chelating ligands. | ||
N-Substituted glycine oligomers, also called “peptoids”, are a class of synthetic peptidomimetics with great potential for ligand design (Fig. 1).11–13 Like polypeptides, they are synthesized in an iterative fashion with their linear monomer sequence precisely defined at each elongation step. However, their synthesis via the solid-phase submonomer method is uniquely rapid and chemically diverse compared to other classes of sequence-defined polymers since the side-chain functional groups are readily introduced from easily obtainable primary amine synthons.14 Furthermore, the properties of peptoids can be efficiently evaluated by high-throughput synthesis and subsequent screening.15 Many laboratories have engineered peptoid side-chains that locally influence the backbone dihedral angles to achieve robust secondary structures such as helices and sheets.16–25 In addition, peptoids can fold into a variety of other secondary structures including ribbons and square-helices.25–29 However, using peptoids as ligands for targeted metal coordination is still has only recently gained momentum,30–39 with very few atomically-defined metallopeptoid structures. To date, most metallopeptoids have mainly been characterized by circular dichroism spectroscopy.40–42 Recently, the first examples of crystal structures of metallopeptoids were reported, however, their solution-phase characterization was limited.31 It is important to note that in cases like this where the molecules have multiple degrees of freedom and multiple pendant functional groups, crystal packing effects can yield solid-state peptoid conformations that do not correlate with their solution-phase structure.19,43 Here we address cluster chelation and peptoid folding in solution, through the design and discovery of conformationally stable peptoids that provide tetradentate to hexadentate chelation to a [Co4O4] cluster via several simple carboxylate moieties. The only previous example of peptoid–metallocluster interaction featured a lanthanide cluster capped with a peptoid monomer.44 NMR spectroscopy was used to elucidate the atomic structures of the peptoid cluster complexes in solution, and single-site mutation studies reveal several non-covalent interactions that contribute to stable peptoid folding.
The tetrametallic cobalt oxo cluster, Co4O4(OAc)4py4 (OAc = acetate, py = pyridine)45,46 (1) has garnered attention as a water splitting catalyst in the context of artificial photosynthesis and as a structural mimic for the oxygen-evolving center (OEC) in the active site of photosystem II (PSII).47–50 It also has convenient properties that make it well-suited for combinatorial and structural studies: kinetic stability, established ligand substitution chemistry,51 strong color (useful for colorimetric binding assay), and diamagnetism (useful for NMR spectroscopy). The cluster has a cubane geometry comprised of four cobalt(III) ions and four oxides. Designing a single peptoid chain that can encapsulate this cubane and determining its atomic structure should give general insight on the key elements required for metal cluster chelation, and provide inroads for synthesis of stable biomimetic “cutouts” of multimetallic enzyme cofactors and their ligands outside of the native protein environment (Fig. 1).
Results and discussion
The crystal structure of PSII provides a good starting point for designing a peptoid ligand that can chelate a cubane.52 The cubane subcluster of the OEC is predominantly chelated by carboxylates at the interface of two different protein subunits, D1 and CP43. Examination of the D1 subunit reveals that it provides most of the carboxylate ligands to the cubane subcluster, three of which reside in a small 11-mer C-terminal domain (Fig. 2a). Two faces of the cubane are chelated by an aspartate side-chain (Asp342) and the alanine C-terminus (Ala344). A longer segment (9 residues) connects the second face to the third face via Glu333. Here we set out to recreate the 3-dimensional orientation of the carboxylate moieties in the D1 fragment using a peptoid analogue of minimal length. To break down the complexity of the design, we first created peptoids that bound well to two faces of the cubane, and then built off that sequence to continue onto the third face. We therefore first synthesized the peptoid analogue of the C-terminal trimer fragment, Asp–Leu–Ala–OH, as Fm–Ncm–Nbn–NAla–NH2 (H2A) (Fm = formyl, Ncm = N-carboxymethyl glycine, Nbn = N-benzyl glycine, NAla = N-carboxymethyl-L-alanine) (Fig. 2b) to target two-face binding of a cubane. Note that the isobutyl side-chain of Leu343 was changed to a benzyl moiety, but is not expected to have consequences on the binding behavior since it points away from the cluster.52 The peptoid and peptide have the same number of residues, but because the peptoid side-chains attach to N rather than Cα, the number of atoms between the carboxylates is longer in the peptoid (12 atoms) than in the peptide (11 atoms). Nonetheless, one equivalent of tripeptoid H2A reacted with 1 to produce a complex, with m/z = 1169.12 (Co4O4(OAc)2(A)(py)4H+), consistent with binding of A to 1 and displacement of two acetic acid molecules (1-A) (Fig. S5†). 1H NMR spectroscopy of the purified complex in d4-methanol53 showed four singlets (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5:0.7:1 ratio) belonging to the formyl proton indicating that there are four conformational isomers (Fig. S6†). 1H–13C HSQC showed two distinct classes of NAla methines (1:1.3 ratio) (Fig. S8†). The chemical shifts of the methine, in addition to the nuclear Overhauser effect (NOE) crosspeaks, identify them as cis/trans amide isomers (ω ∼ 0 or 180, respectively) of NAla with a Kcis/trans ∼ 0.8 (Fig. S10†). There is also cis/trans isomerization from the formyl cap, giving a total of 22 conformational isomers. The ω of the second amide is undetermined, but it is either frozen in a single isomer or its conformation is coupled with that of the other residues. This distribution of isomers is consistent with the fact that in general, cis/trans isomerization of tertiary amides is nearly isoenergetic (Kcis/trans ∼ 1).23 The multiple overlapping resonances of these isomers precluded full assignment of the NMR spectrum for 1-A.
:1.5:0.7:1 ratio) belonging to the formyl proton indicating that there are four conformational isomers (Fig. S6†). 1H–13C HSQC showed two distinct classes of NAla methines (1:1.3 ratio) (Fig. S8†). The chemical shifts of the methine, in addition to the nuclear Overhauser effect (NOE) crosspeaks, identify them as cis/trans amide isomers (ω ∼ 0 or 180, respectively) of NAla with a Kcis/trans ∼ 0.8 (Fig. S10†). There is also cis/trans isomerization from the formyl cap, giving a total of 22 conformational isomers. The ω of the second amide is undetermined, but it is either frozen in a single isomer or its conformation is coupled with that of the other residues. This distribution of isomers is consistent with the fact that in general, cis/trans isomerization of tertiary amides is nearly isoenergetic (Kcis/trans ∼ 1).23 The multiple overlapping resonances of these isomers precluded full assignment of the NMR spectrum for 1-A.
 | ||
| Fig. 2 (a) Selected amino acid residues from the D1 subunit (blue) that bind to the Mn4Ca oxygen-evolving complex (OEC). (b) Design of the peptoid analogues based on D1. (c) Ensemble of the five lowest energy NMR structures of 1-2B (pyridine ligands omitted for clarity). (d) The lowest energy structure of 1-2B. | ||
To remove the amide isomerization at each terminus, NAla was changed to Ala-OH (simulating cis-NAla since it places the 2-carboxyethyl functionality cis with respect to the carbonyl O atom) and formyl was replaced with p-toluenesulfonyl (Ts). Reaction of one and two equivalents of Ts–Ncm–Nbn–Ala–OH (H2B) with 1 produced the mono-peptoid complex, Co4O4(OAc)2(B)(py)4 (1-B), and bis-peptoid complex, Co4O4(B)2(py)4 (1-2B), respectively, demonstrating that termini modification did not significantly affect binding ability. The expected masses for 1-B and 1-2B were observed (m/z = 1238.06 and 1623.24, respectively; Fig. S11 and S13†), and their 1H NMR spectra in d4-methanol showed only one conformer (Fig. S12 and S14†). In contrast, the free peptoid shows two rotamers due to isomerization about the amide bond between Nbn and Ncm (Fig. S2†). While the spectra for 1-B and 1-2B show similar features, the higher symmetry of 1-2B allowed a more facile assignment. The NMR spectrum of 1-2B is consistent with a C2 symmetric molecule, evidenced by two sets of equally intense para-pyridine protons and a singular set of peptoid resonances. This C2 point group symmetry implies that the two chains wrap around the equatorial plane of the cubane in a “head-to-tail” or antiparallel fashion. In contrast, parallel alignment of the peptoid chains around the cubane would result in C1 symmetry that would be distinguishable by NMR spectroscopy. Methylene protons of the peptoid backbone and side-chains are diastereotopic with geminal protons separated up to 1.5 ppm (Fig. S12 and S14†), suggestive of a rigid fold. In addition to mass spectrometry and NMR data, evidence that the [Co4O4] core remains intact upon peptoid coordination comes from solution electrochemistry data, which is a known metric that is sensitive to the [Co4O4] coordination environment.51 For context, the [Co4O4] core in compound 1 exhibits a characteristic reversible redox event in the cyclic voltammogram (CV) corresponding to the [CoIII4O4]4+/[CoIVCoIII3O4]5+ couple (0.35 V vs. Fc+/Fc in methanol, Fig. S33†), and this redox couple has been previously shown to be also sensitive to the ligand environment around the cubane.51 Compound 1-2B exhibits a nearly identical feature at 0.34 V in the CV (Fig. S33†), suggesting that the cubane core is preserved and surrounded in a highly similar ligand environment to that of 1.
Multiple NOE crosspeaks were used as distance restraints for structural determination (Fig. S34, S35, S42†) by replica-exchange molecular dynamics (REMD) to obtain low energy structures consistent with the NMR data (Fig. 2c). In particular, a strong NOE crosspeak between the backbone methylene protons of Ncm and Nbn indicates a less than 3 Å separation, which is diagnostic of a cis-amide linkage between those two residues. The determined NMR structure is consistent with expectations, with the two μ2-carboxylate donors per peptoid interacting with three cobalt ions, for an overall tetradentate chelation per peptoid. Analysis of the backbone dihedral angles, phi (ϕ) and psi (ψ), gives insight into the validity of the structure (Fig. 3a). Satisfyingly, dihedral combinations (ϕ, ψ) of the backbone reside in the low energy regions of the cis-peptoid Ramachandran plot (Fig. 3b).27,54
 | ||
| Fig. 3 (a) The ϕ and ψ dihedrals of a peptoid. (b) Backbone dihedral combinations (ϕ, ψ) of the lowest energy NMR structure for 1-2B (triangles) overlaid on the cis-peptoid Ramachandran free energy plot.27 | ||
It is noteworthy that Ncm adopts a cis-amide, whereas PSII's Asp–Leu–Ala–OH sequence is all trans; perhaps the shorter Cα–Cα distance in a cis-amide backbone is needed to account for the longer linkage (12 atoms) between the two carboxylate groups in the peptoid (vide supra). Titration of a monodentate competitor, d4-acetic acid into 1-2B, monitored by NMR spectroscopy, gave stepwise stability constants of log(Ka1) = 2.0 ± 0.6 and log(Ka2) = 0.9 ± 0.1, and overall stability log(β) = 2.9 ± 0.7, demonstrating a stabilization from the chelate effect (Fig. S30–S32†).
Next, we attempted to extend the sequence of 1-2B to allow carboxylate binding to the third face within a single ligand. Similar to the tripeptide fragment that binds the OEC, the N-terminus of [Co4O4]-bound B is pointed outward and slightly away from the third face of the cluster such that more than one peptoid monomer would be needed to connect to the third face in a μ2-fashion. Preliminary geometry-optimized 3-dimensional computer models suggested that a three peptoid monomer chain should be long enough to link the Ncm–Nbn–Ala–OH segment to a carboxylate residue, Nce (N-2-carboxyethyl glycine), bound on the third face. It was unclear whether there would be a preference for cis or trans amides in this loop (as either appeared reasonable), and concern that a loop that is too flexible would diminish binding.
Thus, we employed a combinatorial approach for discovering classes of loop sequences that would result in high-affinity cluster-binding peptoids.32,33,55 The “split-and-pool” strategy,56 was used to synthesize a “one-bead-one-compound” library of 125 resin-bound sequences varied at the three loop positions (Scheme S2†). In this method, the ensemble of beads contains all possible sequence combinations, but a single bead contains only a homogeneous population of a unique sequence. The peptoids were covalently attached to a TentaGel macrobead support by a labile methionine linkage that can be cleaved using cyanogen bromide, which is orthogonal to the synthesis and screening conditions (see ESI† for details). The monomer basis set comprised of two hydrogen bond donors, Nae (N-2-aminoethyl glycine) and Nhe (N-2-hydroxyethyl glycine), a chiral Nrpe (N-(R)-1-phenylethyl glycine), a strictly trans-enforcing residue Nanis (N-4-methoxyphenyl glycine), and a neutral residue, sarcosine (N-methyl glycine). The peptoid library was synthesized by solid-phase synthesis, and side-chains were deprotected (without cleavage from the resin) by trifluoroacetic acid. The [Co4O4] unit was complexed onto the resin-bound peptoids by ligand exchange with 1.
Since the [Co4O4] unit is a dark green color, direct colorimetric analysis was used to identify hit sequences. Subjecting the resin-bound clusters to increasing concentrations of acetic acid at 50 °C for 20 h and selecting for the remaining green-colored beads allowed identification high-affinity sequences. As a benchmark, the resin-bound tripeptoid, Ts–Ncm–Nbn–NAla, showed complete loss of green color at 1.0 M acetic acid (Fig. S3†). While most beads in the heptameric library lost color, ∼25% of the beads retained color up to 3.0 M acetic acid (Fig. S3†). The peptoids belonging to the colored beads were cleaved and sequenced with tandem MS/MS (see ESI† for details).57 A hit sequence, Ts–Nce–Nae–Nhe–Nrpe–Ncm–Nbn–NAla, appeared multiple times and was chosen for further analysis. The hit was resynthesized as the C-terminal acid derivative, Ts–Nce–Nae–Nhe–Nrpe–Ncm–Nbn–Ala–OH (H3C), in order to prevent amide isomerization at the termini (vide supra). Note that due to trifluoroacetic acid used in HPLC purification procedures, Nae is in the ammonium form with a trifluoroacetate counterion.
Peptoid H3C reacts with 1 in a 1:1 ratio to form the expected complex 1-C, with expected m/z= 1669.27 (Co4O4(OAc)(C)(py)4+) and 835.14 (Co4O4(OAc)(C)(py)4H2+) (Fig. S17†). The 1H NMR spectrum of purified 1-C in d4-methanol exhibits ∼51% of a single conformation species having sharp resonances, and the remaining less folded conformer exhibiting very broad resonances (Fig. S18†). Backbone methylene protons of the single conformation species are diastereotopic with large separation consistent with rigid folding. This species also exhibits a nearly identical redox couple to that of 1 and 1-2B at 0.36 V that suggests the [Co4O4] core remains intact (Fig. S33†).
The NMR resonances were fully assigned (Fig. S21†), and many NOE crosspeaks were resolved for structure determination (Fig. S36–S38†). Notably, the appearance of medium strength NOE crosspeaks between i, i + 1 and i, i − 1 backbone methylene protons correspond to cis amides for all peptoid residues. The side-chain methylene protons of Nce and Nae are also diasterotopic, and show a J-coupling pattern consistent with a gauche relationship of the non-hydrogen substituents (Fig. S39†). While the side-chain of Nce bound to the cluster should adopt a rigid rotational state, the rigidity of the Nae side-chain is more surprising and suggests that it may be involved in a strong non-covalent interaction. Several crosspeaks between the loop residues and the pyridine ligands of the cluster are also observed. NMR-constrained REMD was used to elucidate the structure of 1-C (Fig. 4 and S43†). All backbone dihedral angles fall into the stable regions of the peptoid Ramachandran plot (Fig. 5). Travelling from the C to N terminus, the chain begins to dip far below the equatorial plane of the cubane after Ncm. The chiral Nrpe4 residue initiates a turn, directing the backbone, via sterics, to steer around the corner of the cubane (Fig. 6a). Nhe3 adopts similar dihedrals as Nrpe4, beginning a partial helical stretch that exits back up to the cubane equatorial plane at Nae2. The chirality of the backbone is reversed at Nae2, creating a ∼90° bend that positions the chain parallel with the cubane face, allowing Nce1 to coordinate the cubane. The backbone can sample both positive and negative stable (ϕ, ψ) regions due to the prochiral nature of peptoids (Fig. 5). This flexibility seems advantageous in allowing such a short oligomer to “freely” mold into the most suitable fold. The cis-amides of the backbone are reinforced in two ways – (1) Nae forms a 7-membered H-bond to the i − 1 carbonyl (average N⋯O distance of 3.2 ± 0.3 Å, average N–H–O angle of 122.3° ± 13.6°), and (2) the steric bulk of Nrpe favors a cis-amide in order to minimize allylic strain (Fig. 6a).
 | ||
| Fig. 4 (a) Primary sequence of peptoid H3C. (b) Overlay of the five lowest energy NMR structures, with pyridine and acetate removed for clarity. (c) Peptoid backbone view only. (d) A selected low energy NMR structure. | ||
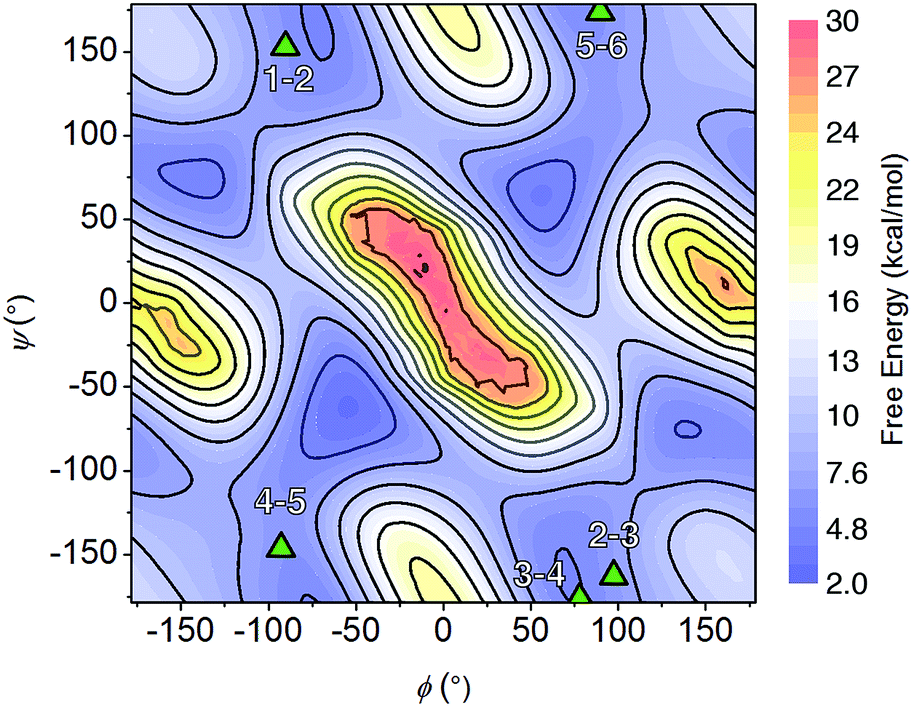 | ||
| Fig. 5 Backbone dihedral combinations (ϕ, ψ) of the lowest energy NMR structure for 1-C (triangles) overlaid on the cis-peptoid Ramachandran free energy plot.27 The residue numbers are indicated. | ||
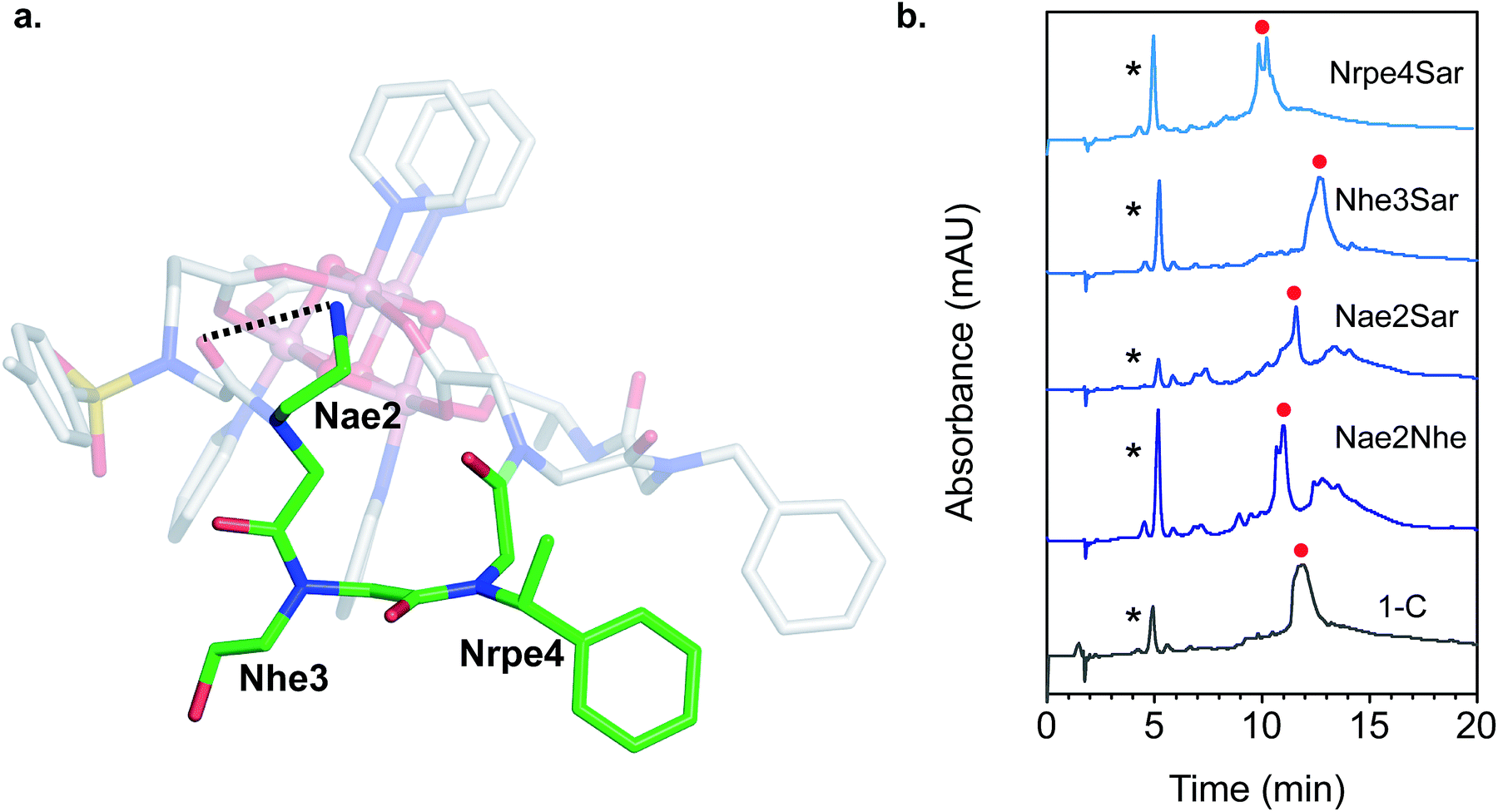 | ||
| Fig. 6 (a) Close-up view of the loop region of 1-C. The loop residues are highlighted with green carbon atoms and labeled. The dashed line indicates the H-bond donor–acceptor distance. (b) LCMS of the crude reaction mixture between 1 and the mutated loop derivatives of H3C (both 0.5 mM) in methanol after 20 h at 50 °C. The asterisk (*) indicates residual 1, and the red circle indicates peaks with masses corresponding to the hexadentate chelated cluster. Mobile phase: 5–95% gradient of acetonitrile/water with 10 mM NH4OAc/HOAc (pH ∼ 5.2) buffer. | ||
Single mutation of each of the three loop residues to sarcosine,58 analogous to alanine scanning59 in peptides, was used to probe the structure–function relationship of each residue (Fig. 6b). Liquid chromatography mass spectrometry (LCMS) of the crude reaction mixture of 1 with H3C exhibited a large major peak corresponding to the desired complex, 1-C. Removing the cationic H-bond donor via a Nae2Sar mutation was highly detrimental, producing significant amounts of side products, including partially chelated and a dimeric cluster complex. Even a conservative mutation to an isosteric, weaker H-bond donor, Nae2Nhe, yielded similar side products. However, chelation ability is unaffected by an Nhe3Sar mutation. NMR characterization of the purified cluster-bound Nhe3Sar mutant (1-C-Nhe3Sar) shows a conformation with many characteristic NOE features of 1-C, indicating an identical fold (Fig. S24, S27, S40, S41†). This suggests that the third residue may be useful as a site for future substitution or conjugation. An Nrpe4Sar mutation does not produce as many side products, but gives two identical-mass species having different retention times, suggestive of isomers (possibly isomers about the ω or Φ dihedral angle between residues 3–4). The results of this sarcosine scan are in line with the NMR structure of 1-C – the H-bond from Nae and steric constraints of Nrpe are critical in decreasing backbone flexibility leading to better binding.
Conclusions
We show that the synthetic efficiency of peptoids enables rapid strategies to overcome the highly challenging design of chelating ligands for multimetallic clusters. By exploiting the versatile solid-phase syntheses of sequence-defined oligomers, cluster-binding peptoids were discovered and optimized using both structure-based design and combinatorial screening. The better binding properties of the optimized sequences, which exhibit less aggregation and more quantitative chelation, result from stronger non-covalent interactions within the peptoid chain that decreases conformational entropy. The peptoid allows for binding to the metal cluster in an anisotropic fashion, where every residue of the peptoid is modifiable, allowing for site-differentiation. Looking forward, the techniques established in this work can be extended to target the chelation of the fourth acetate face, or even the pyridine faces. Preliminary studies with an non-optimized 11-mer peptoid, H4D, made by extending peptoid C's sequence with another three-residue loop (based of the loop discovered for H3C) and an Nce residue, exhibits the correct m/z value for octadentate binding by displacement of all four acetates from 1 to form Co4O4(D)(py)4 (Fig. S29†). Although this species is predominant in the mass spectrum, a noticeable amount of hexadentate species (partially chelated) is present. Nonetheless, it demonstrates that the peptoid sequence can be systematically and easily extended to increase denticity. Also notable is that these compounds are the first conformationally stable metallopeptoids to be structurally characterized in solution phase, an important milestone in understanding metallopeptoid dynamics to create a new class of stable, protein-mimetic metal catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. We thank Dr John Edison for providing the raw data for the Ramachandran plot.Notes and references
- J. Yano and V. Yachandra, Mn4Ca Cluster in Photosynthesis: Where and How Water is Oxidized to Dioxygen, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Mechanism of Nitrogen Fixation by Nitrogenase: The Next Stage, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Structure, Function, and Mechanism of the Nickel Metalloenzymes, CO Dehydrogenase, and Acetyl-CoA Synthase, Chem. Rev., 2014, 114, 4149–4174 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Powering the planet: chemical challenges in solar energy utilization, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- A. L. Gavrilova and B. Bosnich, Principles of Mononucleating and Binucleating Ligand Design, Chem. Rev., 2004, 104, 349–384 CrossRef CAS PubMed.
- T. D. P. Stack and R. H. Holm, Subsite-differentiated analogs of biological [4Fe-4S]2+ clusters: synthesis, solution and solid-state structures, and subsite-specific reactions, J. Am. Chem. Soc., 1988, 110, 2484–2494 CrossRef CAS.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, A Synthetic Model of the Mn3Ca Subsite of the Oxygen-Evolving Complex in Photosystem II, Science, 2011, 333, 733–736 CrossRef CAS PubMed.
- Q. Zhao and T. A. Betley, Synthesis and Redox Properties of Triiron Complexes Featuring Strong Fe–Fe Interactions, Angew. Chem., Int. Ed., 2011, 50, 709–712 CrossRef CAS PubMed.
- G. L. Guillet, F. T. Sloane, D. M. Ermert, M. W. Calkins, M. K. Peprah, E. S. Knowles, E. Čižmár, K. A. Abboud, M. W. Meisel and L. J. Murray, Preorganized assembly of three iron(II) or manganese(II) β-diketiminate complexes using a cyclophane ligand, Chem. Commun., 2013, 49, 6635–6637 RSC.
- J. D. Kim, D. H. Pike, A. M. Tyryshkin, G. V. T. Swapna, H. Raanan, G. T. Montelione, V. Nanda and P. G. Falkowski, Minimal Heterochiral de Novo Designed 4Fe–4S Binding Peptide Capable of Robust Electron Transfer, J. Am. Chem. Soc., 2018, 140(36), 11210–11213 CrossRef CAS PubMed.
- A. S. Knight, E. Y. Zhou, M. B. Francis and R. N. Zuckermann, Sequence Programmable Peptoid Polymers for Diverse Materials Applications, Adv. Mater., 2015, 27, 5665–5691 CrossRef CAS PubMed.
- N. Gangloff, J. Ulbricht, T. Lorson, H. Schlaad and R. Luxenhofer, Peptoids and Polypeptoids at the Frontier of Supra- and Macromolecular Engineering, Chem. Rev., 2016, 116, 1753–1802 CrossRef CAS PubMed.
- K. Kirshenbaum, A. E. Barron, R. A. Goldsmith, P. Armand, E. K. Bradley, K. T. V. Truong, K. A. Dill, F. E. Cohen and R. N. Zuckermann, Sequence-specific polypeptoids: a diverse family of heteropolymers with stable secondary structure, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4303–4308 CrossRef CAS.
- R. N. Zuckermann, J. M. Kerr, S. B. H. Kent and W. H. Moos, Efficient method for the preparation of peptoids [oligo(N-substituted glycines)] by submonomer solid-phase synthesis, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- R. N. Zuckermann and T. Kodadek, Peptoids as potential therapeutics, Curr. Opin. Mol. Ther., 2009, 11, 299–307 CAS.
- C. Caumes, O. Roy, S. Faure and C. Taillefumier, The Click Triazolium Peptoid Side Chain: A Strong cis-Amide Inducer Enabling Chemical Diversity, J. Am. Chem. Soc., 2012, 134, 9553–9556 CrossRef CAS PubMed.
- P. Armand, K. Kirshenbaum, R. A. Goldsmith, S. Farr-Jones, A. E. Barron, K. T. V. Truong, K. A. Dill, D. F. Mierke, F. E. Cohen, R. N. Zuckermann and E. K. Bradley, NMR determination of the major solution conformation of a peptoid pentamer with chiral side chains, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4309–4314 CrossRef CAS.
- N. H. Shah, G. L. Butterfoss, K. Nguyen, B. Yoo, R. Bonneau, D. L. Rabenstein and K. Kirshenbaum, Oligo(N-aryl glycines): A New Twist on Structured Peptoids, J. Am. Chem. Soc., 2008, 130, 16622–16632 CrossRef CAS PubMed.
- C. W. Wu, K. Kirshenbaum, T. J. Sanborn, J. A. Patch, K. Huang, K. A. Dill, R. N. Zuckermann and A. E. Barron, Structural and Spectroscopic Studies of Peptoid Oligomers with α-Chiral Aliphatic Side Chains, J. Am. Chem. Soc., 2003, 125, 13525–13530 CrossRef CAS PubMed.
- O. Roy, C. Caumes, Y. Esvan, C. Didierjean, S. Faure and C. Taillefumier, The tert-Butyl Side Chain: A Powerful Means to Lock Peptoid Amide Bonds in the Cis Conformation, Org. Lett., 2013, 15, 2246–2249 CrossRef CAS PubMed.
- O. Roy, G. Dumonteil, S. Faure, L. Jouffret, A. Kriznik and C. Taillefumier, Homogeneous and Robust Polyproline Type I Helices from Peptoids with Nonaromatic α-Chiral Side Chains, J. Am. Chem. Soc., 2017, 139, 13533–13540 CrossRef CAS PubMed.
- B. C. Gorske and H. E. Blackwell, Tuning Peptoid Secondary Structure with Pentafluoroaromatic Functionality: A New Design Paradigm for the Construction of Discretely Folded Peptoid Structures, J. Am. Chem. Soc., 2006, 128, 14378–14387 CrossRef CAS PubMed.
- B. C. Gorske, J. R. Stringer, B. L. Bastian, S. A. Fowler and H. E. Blackwell, New Strategies for the Design of Folded Peptoids Revealed by a Survey of Noncovalent Interactions in Model Systems, J. Am. Chem. Soc., 2009, 131, 16555–16567 CrossRef CAS PubMed.
- B. C. Gorske, B. L. Bastian, G. D. Geske and H. E. Blackwell, Local and Tunable n → π* Interactions Regulate Amide Isomerism in the Peptoid Backbone, J. Am. Chem. Soc., 2007, 129, 8928–8929 CrossRef CAS PubMed.
- B. C. Gorske, E. M. Mumford, C. G. Gerrity and I. Ko, A Peptoid Square Helix via Synergistic Control of Backbone Dihedral Angles, J. Am. Chem. Soc., 2017, 139, 8070–8073 CrossRef CAS PubMed.
- B. C. Hudson, A. Battigelli, M. D. Connolly, J. Edison, R. K. Spencer, S. Whitelam, R. N. Zuckermann and A. K. Paravastu, Evidence for cis Amide Bonds in Peptoid Nanosheets, J. Phys. Chem. Lett., 2018, 9, 2574–2578 CrossRef CAS PubMed.
- J. R. Edison, R. K. Spencer, G. L. Butterfoss, B. C. Hudson, A. I. Hochbaum, A. K. Paravastu, R. N. Zuckermann and S. Whitelam, Conformations of peptoids in nanosheets result from the interplay of backbone energetics and intermolecular interactions, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5647–5651 CrossRef CAS PubMed.
- D. R. Greer, M. A. Stolberg, J. Kundu, R. K. Spencer, T. Pascal, D. Prendergast, N. P. Balsara and R. N. Zuckermann, Universal Relationship between Molecular Structure and Crystal Structure in Peptoid Polymers and Prevalence of the cis Backbone Conformation, J. Am. Chem. Soc., 2018, 140, 827–833 CrossRef CAS PubMed.
- J. A. Crapster, I. A. Guzei and H. E. Blackwell, A Peptoid Ribbon Secondary Structure, Angew. Chem., Int. Ed., 2013, 52, 5079–5084 CrossRef CAS PubMed.
- G. Maayan, M. D. Ward and K. Kirshenbaum, Metallopeptoids, Chem. Commun., 2008, 56–58 Search PubMed.
- T. Ghosh, N. Fridman, M. Kosa and G. Maayan, Self-Assembled Cyclic Structures from Copper(II) Peptoids, Angew. Chem., Int. Ed., 2018, 57, 7703–7708 CrossRef CAS PubMed.
- A. S. Knight, E. Y. Zhou and M. B. Francis, Development of peptoid-based ligands for the removal of cadmium from biological media, Chem. Sci., 2015, 6, 4042–4048 RSC.
- A. S. Knight, E. Y. Zhou, J. G. Pelton and M. B. Francis, Selective Chromium(VI) Ligands Identified Using Combinatorial Peptoid Libraries, J. Am. Chem. Soc., 2013, 135, 17488–17493 CrossRef CAS PubMed.
- B. F. Parker, A. S. Knight, S. Vukovic, J. Arnold and M. B. Francis, A Peptoid-Based Combinatorial and Computational Approach to Developing Ligands for Uranyl Sequestration from Seawater, Ind. Eng. Chem. Res., 2016, 55, 4187–4194 CrossRef CAS.
- S. Young and S. C. Young, A Systematic Review of Antiamyloidogenic and Metal-Chelating Peptoids: Two Structural Motifs for the Treatment of Alzheimer's Disease, Molecules, 2018, 23, 296 CrossRef PubMed.
- I. Izzo, G. Ianniello, C. De Cola, B. Nardone, L. Erra, G. Vaughan, C. Tedesco and F. De Riccardis, Structural Effects of Proline Substitution and Metal Binding on Hexameric Cyclic Peptoids, Org. Lett., 2013, 15, 598–601 CrossRef CAS PubMed.
- M. Baskin and G. Maayan, A rationally designed metal-binding helical peptoid for selective recognition processes, Chem. Sci., 2016, 7, 2809–2820 RSC.
- A. D'Amato, R. Volpe, M. C. Vaccaro, S. Terracciano, I. Bruno, M. Tosolini, C. Tedesco, G. Pierri, P. Tecilla, C. Costabile, G. Della Sala, I. Izzo and F. De Riccardis, Cyclic Peptoids as Mycotoxin Mimics: An Exploration of Their Structural and Biological Properties, J. Org. Chem., 2017, 82, 8848–8863 CrossRef PubMed.
- A. D'Amato, G. Pierri, C. Costabile, G. Della Sala, C. Tedesco, I. Izzo and F. De Riccardis, Cyclic Peptoids as Topological Templates: Synthesis via Central to Conformational Chirality Induction, Org. Lett., 2018, 20, 640–643 CrossRef PubMed.
- M. Baskin and G. Maayan, Chiral Cu(II), Co(II) and Ni(II) complexes based on 2,2′-bipyridine modified peptoids, Dalton Trans., 2018, 47, 10767–10774 RSC.
- M. Baskin, L. Panz and G. Maayan, Versatile ruthenium complexes based on 2,2′-bipyridine modified peptoids, Chem. Commun., 2016, 52, 10350–10353 RSC.
- L. Zborovsky, A. Smolyakova, M. Baskin and G. Maayan, A Pure Polyproline Type I-like Peptoid Helix by Metal Coordination, Chem.–Eur. J., 2018, 24, 1159–1167 CrossRef CAS PubMed.
- G. L. Butterfoss, B. Yoo, J. N. Jaworski, I. Chorny, K. A. Dill, R. N. Zuckermann, R. Bonneau, K. Kirshenbaum and V. A. Voelz, De novo structure prediction and experimental characterization of folded peptoid oligomers, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 14320–14325 CrossRef CAS PubMed.
- D. T. Thielemann, A. T. Wagner, E. Rösch, D. K. Kölmel, J. G. Heck, B. Rudat, M. Neumaier, C. Feldmann, U. Schepers, S. Bräse and P. W. Roesky, Luminescent Cell-Penetrating Pentadecanuclear Lanthanide Clusters, J. Am. Chem. Soc., 2013, 135, 7454–7457 CrossRef CAS PubMed.
- J. K. Beattie, J. A. Klepetko, A. F. Masters and P. Turner, The chemistry of cobalt acetate. VIII. New members of the family of oxo-centred trimers, [Co3(μ3-O)(μ-O2CCH3)5-p(μ-OR)pL5]2+ (R = H, alkyl, L = ligand, p = 0–4). The preparation and characterisation of the trimeric tetrakis(μ-acetato)-(μ-hydroxo)-μ3-oxo-pentakis(pyridine)-tri-cobalt(III) hexafluorophosphate, [Co3(μ3-O)(μ-O2CCH3)4(μ-OH)(C5H5N)5][PF6]2, and the preparation and crystal structure of the trimeric tris(μ-acetato)-(μ-hydroxo)-(μ-methoxo)-μ3-oxo-pentakis(pyridine)-tri-cobalt(III) hexafluorophosphate·methanol·water solvate [Co3(μ3-O)(μ-O2CCH3)3(μ-OH)(μ-OCH3)(C5H5N)5][PF6]2·CH3OH·0.25H2O, Polyhedron, 2003, 22, 947–965 CrossRef CAS.
- R. Chakrabarty, S. J. Bora and B. K. Das, Synthesis, Structure, Spectral and Electrochemical Properties, and Catalytic Use of Cobalt(III)-Oxo Cubane Clusters, Inorg. Chem., 2007, 46, 9450–9462 CrossRef CAS PubMed.
- A. I. Nguyen, M. S. Ziegler, P. Oña-Burgos, M. Sturzbecher-Hohne, W. Kim, D. E. Bellone and T. D. Tilley, Mechanistic Investigations of Water Oxidation by a Molecular Cobalt Oxide Analogue: Evidence for a Highly Oxidized Intermediate and Exclusive Terminal Oxo Participation, J. Am. Chem. Soc., 2015, 137, 12865–12872 CrossRef CAS PubMed.
- P. F. Smith, L. Hunt, A. B. Laursen, V. Sagar, S. Kaushik, K. U. D. Calvinho, G. Marotta, E. Mosconi, F. De Angelis and G. C. Dismukes, Water Oxidation by the [Co4O4(OAc)4(py)4]+ Cubium is Initiated by OH− Addition, J. Am. Chem. Soc., 2015, 137, 15460–15468 CrossRef CAS PubMed.
- L. Olshansky, R. Huerta-Lavorie, A. I. Nguyen, J. Vallapurackal, A. Furst, T. D. Tilley and A. S. Borovik, Artificial Metalloproteins Containing Co4O4 Cubane Active Sites, J. Am. Chem. Soc., 2018, 140, 2739–2742 CrossRef CAS PubMed.
- R. G. Hadt, D. Hayes, C. N. Brodsky, A. M. Ullman, D. M. Casa, M. H. Upton, D. G. Nocera and L. X. Chen, X-ray Spectroscopic Characterization of Co(IV) and Metal–Metal Interactions in Co4O4: Electronic Structure Contributions to the Formation of High-Valent States Relevant to the Oxygen Evolution Reaction, J. Am. Chem. Soc., 2016, 138, 11017–11030 CrossRef CAS PubMed.
- A. I. Nguyen, J. Wang, D. S. Levine, M. S. Ziegler and T. D. Tilley, Synthetic control and empirical prediction of redox potentials for Co4O4 cubanes over a 1.4 V range: implications for catalyst design and evaluation of high-valent intermediates in water oxidation, Chem. Sci., 2017, 8, 4274–4284 RSC.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9A, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- d 4-methanol was chosen for its ability to dissolve the peptoid-cluster complexes in high concentrations.
- G. L. Butterfoss, P. D. Renfrew, B. Kuhlman, K. Kirshenbaum and R. Bonneau, A Preliminary Survey of the Peptoid Folding Landscape, J. Am. Chem. Soc., 2009, 131, 16798–16807 CrossRef CAS PubMed.
- D. M. Nalband, B. P. Warner, N. H. Zahler and K. Kirshenbaum, Rapid identification of metal-binding peptoid oligomers by on-resin X-ray fluorescence screening, Pept. Sci., 2014, 102, 407–415 CrossRef CAS PubMed.
- K. S. Lam, M. Lebl and V. Krchňák, The “One-Bead-One-Compound” Combinatorial Library Method, Chem. Rev., 1997, 97, 411–448 CrossRef CAS PubMed.
- W. Heerma, C. Versluis, C. G. de Koster, J. a. W. Kruijtzer, I. Zigrovic and R. M. J. Liskamp, Comparing Mass Spectrometric Characteristics of Peptides and Peptoids, Rapid Commun. Mass Spectrom., 1996, 10, 459–464 CrossRef CAS PubMed.
- M. Park, M. Wetzler, T. S. Jardetzky and A. E. Barron, A Readily Applicable Strategy to Convert Peptides to Peptoid-based Therapeutics, PLoS One, 2013, 8, e58874 CrossRef CAS PubMed.
- K. L. Morrison and G. A. Weiss, Combinatorial alanine-scanning, Curr. Opin. Chem. Biol., 2001, 5, 302–307 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04240c |
| This journal is © The Royal Society of Chemistry 2018 |