
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral molecular face-rotating sandwich structures constructed through restricting the phenyl flipping of tetraphenylethylene†
Hang
Qu‡
,
Xiao
Tang‡
,
Xinchang
Wang
 ,
Zhihao
Li
,
Zheyu
Huang
,
Hui
Zhang
,
Zhongqun
Tian
and
Xiaoyu
Cao
*
,
Zhihao
Li
,
Zheyu
Huang
,
Hui
Zhang
,
Zhongqun
Tian
and
Xiaoyu
Cao
*
State Key Laboratory of Physical Chemistry of Solid Surfaces, Key Laboratory of Chemical Biology of Fujian Province, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: xcao@xmu.edu.cn
First published on 14th September 2018
Abstract
Chiral tetraphenylethylene (TPE) derivatives have great potential in chiral recognition and circularly polarized luminescence. However, they were mainly constructed through introducing chiral substituents at the periphery of the TPE moiety, which required additional chemical modifications and limited the variety of chiralities of products. Herein, we constructed a series of chiral face-rotating sandwich structures (FRSs) through restricting the phenyl flipping of TPE without introducing any chiral substituents. In FRSs, the complex arrangements of TPE motifs resulted in a variety of chiralities. We also found that non-covalent repulsive interactions in vertices caused the facial hetero-directionality of FRSs, and the hydrogen bonds between imine bonds and hydroxy groups induced excited-state intramolecular proton transfer (ESIPT) emission of FRSs. In addition, the fluorescence intensity of FRSs decreases with the addition of trifluoroacetic acid. This study provides new insights into the rational design of chiral assemblies from aggregation-induced emission (AIE) active building blocks through restriction of intramolecular rotation (RIR).
Introduction
Tetraphenylethylene (TPE), as an aggregation-induced-emission chromophore (AIEgen),1 has been extensively applied in fluorescent probes,2 optoelectronic materials,3 and bioimaging agents.4 As a pseudo-C4 symmetric building block, TPE has been assembled into covalent-organic frameworks (COFs),5 molecular cages6 and supramolecular fibres.7 Among those TPE-based materials, the chiral ones are particularly interesting because they exhibit extensive functions such as chiral recognition8 and circularly polarized luminescence.9 Generally, chiral TPE derivatives are constructed through introducing chiral substituents at the periphery of the TPE moiety.10 Nevertheless, such strategies required additional chemical modifications on TPE, and the chiral substituents limited the variety of chiralities of products. Interestingly, the propeller-like TPE core can exhibit inherent two-dimensional (2D) chirality in P or M helical configurations11 through the restriction of intramolecular rotation (RIR).12 In addition, the spatial orientation of vinyl bonds (Fig. 1a), either vertical or horizontal, also influences the chirality of assembled structures. Therefore, the complex arrangements of the four TPE motifs (Fig. 1b) could generate numerous chiral diastereoisomers without introducing any chiral substituents. But such a strategy to construct chiral TPE-based assemblies was rarely investigated and challenging due to difficulties in chiral separation.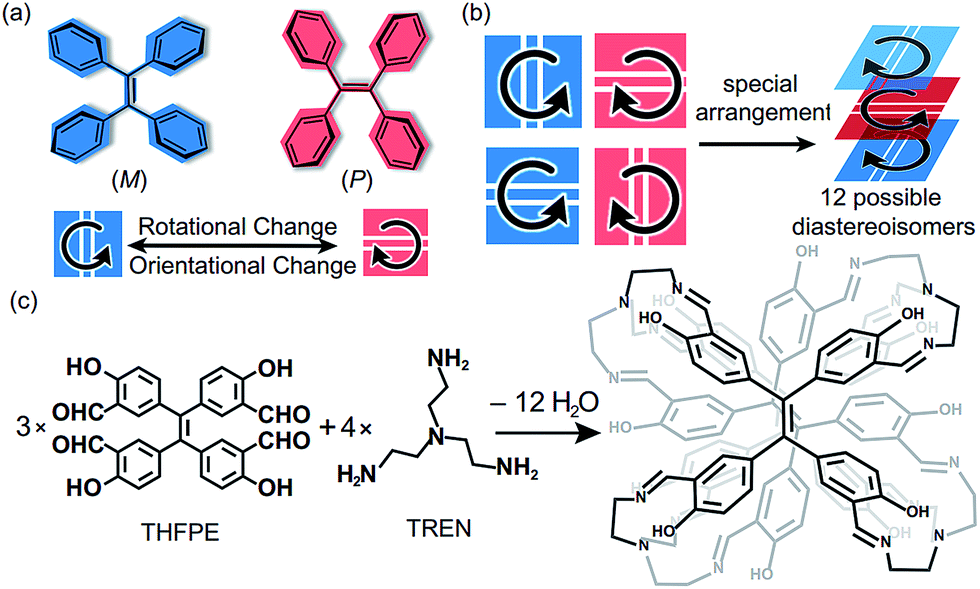 | ||
| Fig. 1 (a) TPE motif exhibits two rotational modes (P or M) of the phenyl groups and two orientational modes (horizontal or vertical) of the vinyl bonds in two dimensions. (b) Four 2D motifs of TPE could generate numerous diastereoisomers through special arrangements. (c) Structural formula of THFPE, TREN and the corresponding sandwich structures formed by dynamic imine chemistry. | ||
Recently, we have constructed a series of molecular face-rotating polyhedra (FRP),13 which display a special form of supramolecular chirality originating from the arrangements of 2D chiral building blocks. In TPE-based FRP,14 we found that the phenyl flipping of TPE faces was significantly restricted and hence FRP exhibited remarkable stability of chirality and fluorescence in diluted solution. To further control the chirality of FRP, we modulated the rotational patterns of faces with several features. On one hand, the subtle difference in building blocks could bring a huge change in the resultant assemblies and their chirality. For instance, Fujita et al. demonstrated that only a 3° difference in the bending angles of two ligands can critically switch achiral Platonic or Archimedean structures into chiral Goldberg polyhedra.15 More recently, our group reported that different positions of aldehyde in triazatruxene (TAT) lead to distinct assembly behaviour and geometry.16 On the other hand, vertices also determined the chirality of FRP. We reported that chiral vertices sufficed to control the facial directionality and the conformations of other achiral vertices in FRP assembled from truxene.17 Nevertheless, how achiral vertices influence the rotational patterns of TPE units remains unclear.
Herein, we assemble the meta-aldehyde TPE (THFPE, Fig. 1c) with tris(2-aminoethyl)amine (TREN) to form a series of chiral face-rotating sandwich structures (FRSs) without introducing any chiral substituents on TPE. The subtle difference in TPE (the positions of aldehyde groups) changes the resultant structures from previously reported molecular cubes14 into FRSs, wherein only four FRSs emerge as products out of twelve possible diastereoisomers. In FRSs, three TPE units exhibit hetero-directional configurations, which are controlled by the repulsive interactions in TREN vertices as indicated by theoretical calculations. In addition, the hydroxyl groups form hydrogen bonds with imine bonds and induce the excited-state intramolecular proton transfer (ESIPT) phenomenon. In addition, the fluorescence intensity of FRSs decreases with the addition of trifluoroacetic acid.
Results and discussion
The facial building block THFPE was prepared from 4,4′-hydroxybenzophenone through multi-step synthesis (Fig. S1†). In the Schiff-base condensation, THFPE (3 equiv.) was mixed with TREN (4 equiv.) and catalytic amounts of trifluoroacetic acid in chloroform at ambient temperature (Fig. 1c). High-resolution mass spectrometry identified the molecular weight to be 1894.21 (Fig. S2†), corresponding to THFPE3TREN4. However, the initial nuclear magnetic resonance (NMR) spectrum showed notable overlap of proton signals in the aromatic region (Fig. S3†), suggesting that more than one stereoisomer existed in the products.High-performance liquid chromatography (HPLC) was employed to separate the mixed products by using a chiral column (Daicel Chiralpak IE) and a mixed solvent as the mobile phase (toluene/chloroform/methanol). After 24 hours of reaction, the HPLC spectrum confirmed that only four fractions eluted in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 ratio (Fig. 2a, top). Further characterization revealed that the four fractions correspond to two pairs of hetero-directional enantiomers, i.e., PMP-1, MPM-1, PMP-2 and MPM-2, which respectively eluted at 17.5, 32.9, 15.5 and 44.7 min. However, as the reaction was heated at 50 °C for five days, the fraction of FRS 2 gradually decreased (Fig. S4†) and eventually disappeared (Fig. 2a, bottom), suggesting that FRS 2 was a kinetic product which finally converted into thermodynamic FRS 1. The reaction yield of THFPE3TREN4 was calculated to be 97.6% according to the full HPLC spectrum (Fig. S5†).
:2:2:1 ratio (Fig. 2a, top). Further characterization revealed that the four fractions correspond to two pairs of hetero-directional enantiomers, i.e., PMP-1, MPM-1, PMP-2 and MPM-2, which respectively eluted at 17.5, 32.9, 15.5 and 44.7 min. However, as the reaction was heated at 50 °C for five days, the fraction of FRS 2 gradually decreased (Fig. S4†) and eventually disappeared (Fig. 2a, bottom), suggesting that FRS 2 was a kinetic product which finally converted into thermodynamic FRS 1. The reaction yield of THFPE3TREN4 was calculated to be 97.6% according to the full HPLC spectrum (Fig. S5†).
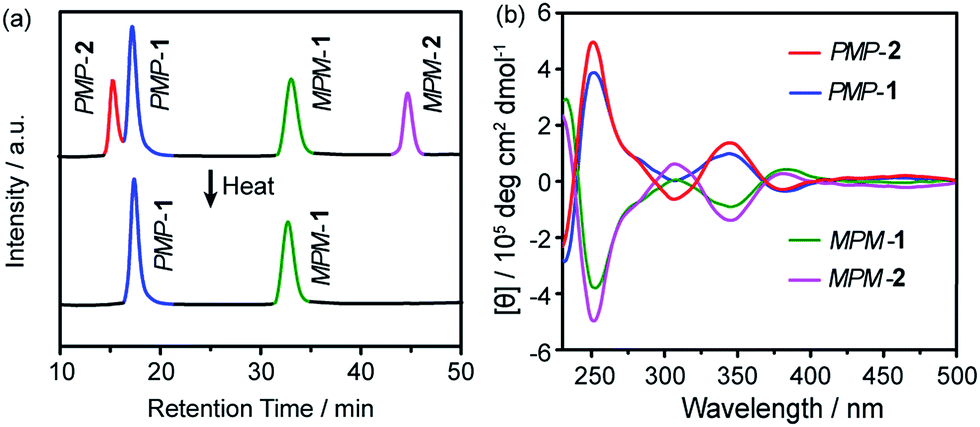 | ||
| Fig. 2 Chiral-HPLC and CD analyses of 1 and 2. (a) Chiral HPLC spectra of the kinetic products (top) synthesised at ambient temperature revealing two pairs of diastereoisomers, (i.e., MPM-1 and PMP-1, PMP-2 and MPM-2) that changed into the thermodynamic products (MPM-1 and PMP-1) when heated at 50 °C for five days (bottom). (b) CD spectra of PMP-2, MPM-1, MPM-1 and MPM-2 (10 μM) in dichloromethane. | ||
Slow evaporation of FRSs in chloroform was allowed to afford prismatic crystals after a month. Although we used enantiopure fractions to grow crystals, single-crystal X-ray diffraction analysis revealed a pair of enantiomers co-crystallized in a unit cell, indicating racemization of FRSs 1 or 2 during the crystallization process. Further time-dependent chiral-HPLC analyses revealed that only 2% of MPM-1 or MPM-2 can change into their enantiomers after five days in solution (Fig. S6†), indicating that the racemization of each enantiopure fraction was quite slow in solution. All crystal structures of FRSs bear a tetra-capped hetero-directional sandwich structure, wherein one TPE unit was stuck between the other two TPE units with a different rotational configuration and TREN occupied four vertices to link three TPE units.
The crystal structure of PMP-1 is C2 symmetric according to the central axis of each TPE face (Fig. 3a). The M face in the middle is 5.29 Å from two P faces (Fig. 3b). The vinyl orientations of P faces are parallel whereas they form a 51° angle with the vinyl bond of the middle M face (Fig. 3a, inset). In addition, three phenyl rings linked by the same TREN have highly twisted conformations, wherein two phenyl rings from two P faces are nearly parallel and they are perpendicular to that from the M face as shown in Fig. S7.† Similar to PMP-1, PMP-2 has only a subtle difference in the vinyl orientations of two P faces (Fig. 3c): they are noncoplanar perpendicular to each other and form a 45° angle with the vinyl bond of the middle M face (Fig. 3d and more structural details are in Fig. S7†).
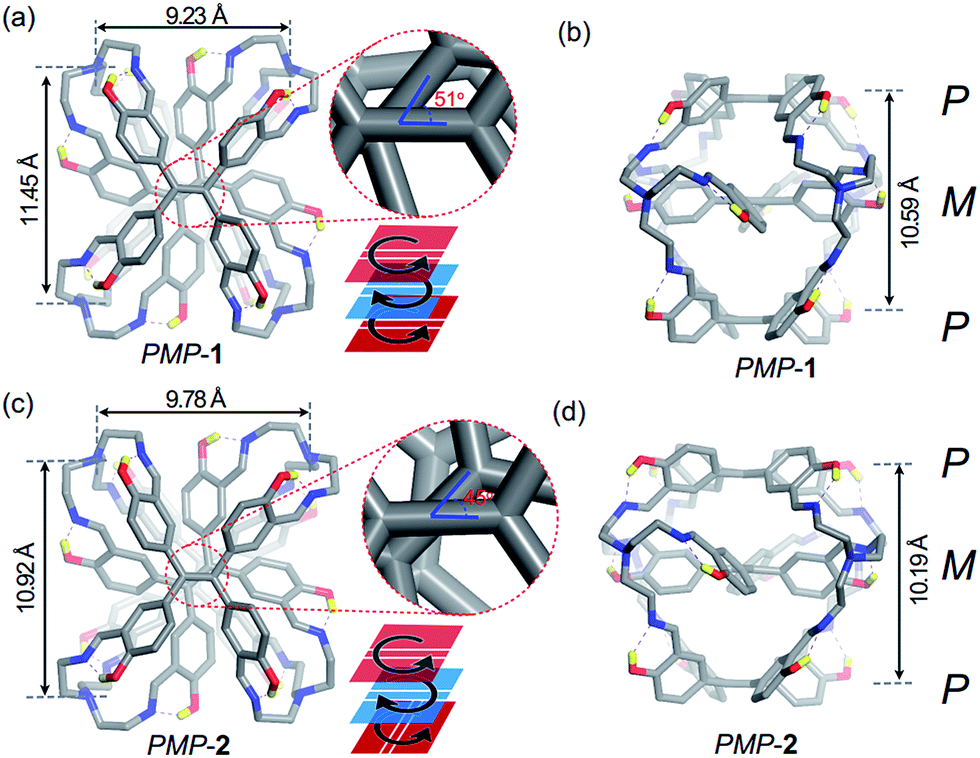 | ||
| Fig. 3 Single-crystal structures of PMP-1 and PMP-2. Front and side views of the sandwich structures of PMP-1 (a and b) and PMP-2 (c and d). Carbon atoms are shown in grey, nitrogen atoms are shown in blue, oxygen atoms are shown in red, and hydrogen atoms (yellow) are omitted for clarity except those with hydrogen bonds. | ||
The imine bonds in FRSs form hydrogen bonds (O–H⋯N) with hydroxyl groups and are conjugated with phenyl rings, resulting in complete coplanarity with the adjacent phenyl rings. In our previous work, the imine bonds and the adjacent phenyl rings, without the assistance of hydrogen bonds, have a 7°dihedral angle.14 To investigate the role of hydrogen-bonding interactions, we substituted the hydroxyl groups in THFPE with ethoxy groups (TOMPE, Fig. S8a†) to replace hydrogen bonds with imine bonds. TOMPE was reacted with TREN under the same conditions and mass spectrometry revealed that the condensation reaction only produced some uncharacterized oligomers (Fig. S8c†), indicating that the hydrogen bonds assisted in the imine formation and subsequently stabilized the resultant structures.
All TREN vertices in FRSs influence the stacking of TPE because they exhibited clockwise or anti-clockwise configurations (Fig. S9a†) and strong non-covalent repulsive interactions inside TREN. In a previous report, Bein et al. demonstrated that the TPE entities in highly crystalline COFs are energetically favourable for homo-directional stacking rather than alternating stacking of P or M configurations to form successive layers.5b However, in FRSs, TPE units take the alternating stacking of P or M configurations. We attributed this unusual stacking in FRSs to two types of non-covalent repulsive interactions in TREN vertices. The first type is repulsive interactions among the three alkyl chains of TREN (Fig. S9c†), and the second one is repulsive interactions of atoms in the same alkyl chain (Fig. S9a,† top). In further density functional theory (DFT) calculations, we constructed a structure of PPP-1 through substituting the middle M face into the P face in PMP-1 (Fig. S9b†). The energy of PPP-1 was much higher than that of PMP-1. In contrast to the anti-clockwise rotational configurations of TREN in PMP-1, TREN in the optimized structure of PPP-1 has an extremely distorted configuration. This suggested that the first type of repulsive interactions led to an anti-clockwise rotational configuration of TREN. We also varied the torsional angle (θ, Fig. S9†) in the N–C–C–N gauche conformation of PMP-1 from 0° to 180° in one alkyl chain to construct the potential energy surfaces (PESs). In PESs, due to the second type of repulsive forces in one alkyl chain, the energy of PMP-1 was the lowest when θ was 65° (Fig. S9d†), which is consistent with the crystal structure of PMP-1. Therefore, the second type of repulsive interactions restricted the movement of alkyl chains and thus transferred the chirality from rotational conformations of TREN to TPE entities, resulting in facial hetero-directionality of FRSs.
Similarly, the crystal structures of MPM-1 or MPM-2 were the enantiomers of PMP-1 or PMP-2 as shown in Fig. S10,† respectively. In addition, the theoretical calculations also confirmed that the energy of thermodynamic product 1 was 67.01 kJ mol−1 lower than that of kinetic product 2.
The circular dichroism (CD) spectra of PMP-1 and MPM-1 showed mirror-image Cotton effect signals at 250 nm (molar ellipticity of 5.1 × 105 deg cm2 dmol−1, Fig. 2b), which belong to the absorption region of TPE, and thus confirmed that phenyl flipping was restricted in diluted solution. MPM-2 and PMP-2 also displayed mirror-like Cotton effect signals at the similar intensity of FRSs 1 as shown in Fig. 2b. More importantly, their CD spectral signals at 250 nm were dominated by the major rotational faces of TPE units, that is, the number of M or P faces in FRSs. For instance, FRSs with more P faces exhibited negative signals at 250 nm, which is consistent with previous work by our group.14 All four CD spectra of FRSs well matched the (ZINDO/S)-predicted CD spectra (Fig. S11†), confirming the absolute configuration of each diastereoisomer.
The 1H-NMR spectrum of THFPE only displayed one peak for protons in the aldehyde groups (Fig. S12†), indicating high freedom of phenyl rings in solution. In contrast, the 1H-NMR spectrum of PMP-1 exhibited complex proton signals of imine bonds because the restriction of phenyl flipping in PMP-1 lowers the symmetry of THFPE (Fig. S14†). The 2D NMR spectra allowed us to assign all the protons in PMP-1 (details in ESI†), which exhibited six peaks for protons in 12 imine bonds (Fig. S16†). Therefore, those protons of PMP-1 are individually interchangeable through C2-symmetric operations, which is consistent with its single-crystal structure. Particularly, two protons of methylene in TREN, namely He and He* (Fig. S17†), are in a distinct chemical environment and split into two individual peaks, confirming that the non-covalent repulsive interactions restricted the movement of flexible TREN in solution. In addition, the nuclear Overhauser effect (NOE) crosspeaks between He and Hd reflected the E conformation of imine bonds and the structural rigidity of PMP-1 (Fig. S17c†).
Compared with the fluorescence of THFPE and some similar compounds,18 FRSs 1 and 2 exhibited an emission peak at 550 nm with a large Stokes shift (∼200 nm) in diluted solution (Fig. 4b). In addition, the fluorescence quantum yields of FRSs 1 and 2 were measured to be 2.08% and 1.56% in toluene, respectively. The emission at 550 nm can be rationalized by restriction of phenyl rotation of TPE units and the ESIPT phenomenon induced by the intramolecular hydrogen bonds (Fig. 4a). ESIPT emission involves a four-level photophysical cycle,19 in which the enol–keto tautomerism from E* to cis-K* occurs upon photoexcitation and then cis-K returns to the initial enol–imine form through ground state intramolecular proton transfer (GSIPT). However, there are two competitive processes for cis-K* relaxation: radiative decay to the ground state (cis-K) and rotation-induced non-radiative decay to trans-K. Generally, E/Z isomerization from cis-K* to trans-K is forbidden on aggregation and it will increase the intensity of ESIPT emission.20 In FRSs, the rigidity of TREN vertices can also prevent the E/Z isomerization of cis-K* and strong ESIPT emission of FRSs can be observed in diluted solution. In addition, the ESIPT emission of FRSs can be tuned by pH and solvent, which can remarkably block the enol–keto tautomerism in the excited state.21 By adding TFA into FRSs 1 in chloroform incrementally, the fluorescence intensity decreased dramatically at 550 nm (Fig. 4c), whereas the fluorescence can recover to the initial intensity by using trimethylamine to neutralize TFA (Fig. S19c†). As expected, increasing the solvent polarity can also switch off emission at 550 nm with a red shift (Fig. S19b†).
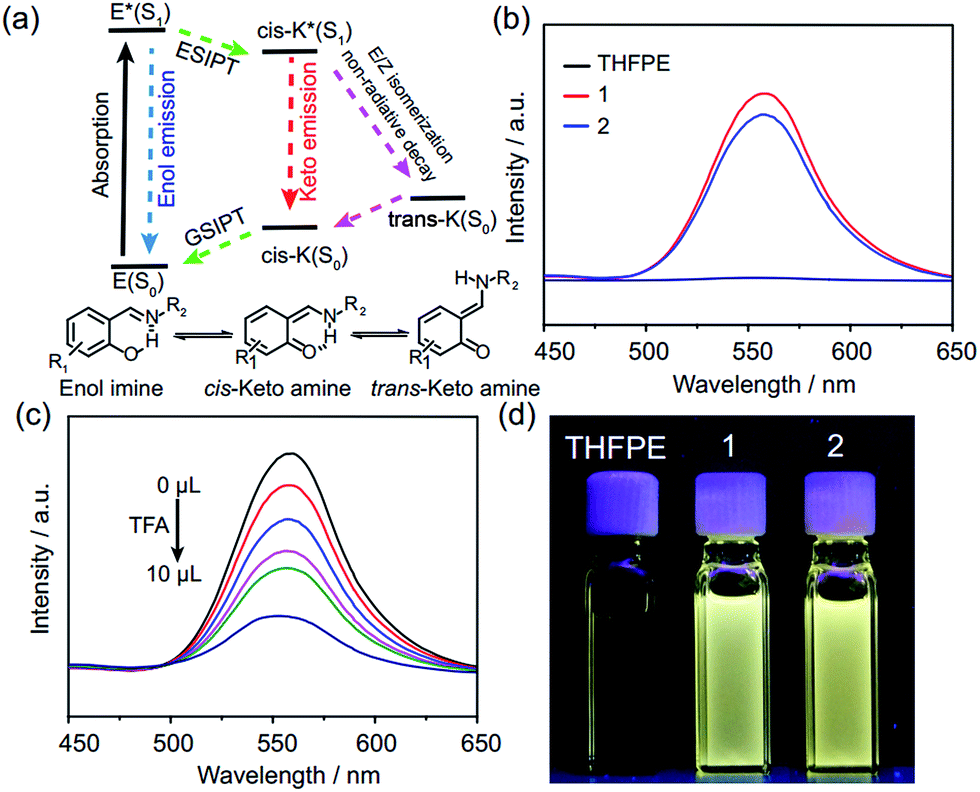 | ||
| Fig. 4 (a) The ESIPT mechanism of enol-imine compounds through a four-level photo-cycle. (b) Fluorescence spectra of THFPE (30 μM, black), FRSs 1 (10 μM, red) and 2 (10 μM, blue) in chloroform, excited at 360 nm. (c) pH-dependent fluorescence spectra of FRS 1 in chloroform. (d) Fluorescence images of THFPE (1.2 mM, left), 1 (400 μM, middle) and 2 (400 μM, right) under 365 nm UV-irradiation. | ||
Conclusions
To conclude, we have constructed a series of FRSs with emergent chirality and fluorescence through restricting the phenyl flipping of TPE units in diluted solution. Due to the non-covalent repulsive interactions, TREN vertices exhibited rotational configurations and transmitted their chirality to the TPE entities, resulting in facial hetero-directionality of FRSs. In addition, the hydrogen bonds between imine bonds and hydroxy groups induced ESIPT properties, which can be tuned by pH and solvents. This study provides a strategy to construct chiral supramolecular assemblies from AIEgens by restricting intramolecular rotation, which can be applied in luminescent materials, molecular recognition or asymmetric catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the support from the NSFC (No. 21722304, 91427304, 21573181, and 91227111), the 973 Program (No. 2015CB856505), the Top-Notch Young Talents Program of China and the Fundamental Research Funds for the Central Universities of China (No. 20720160050). We thank Wei Zhang, Hao Li, Haitao Feng, and Jonathan Nitschke for helpful discussions. We also thank Linlin Yang, Wenting Deng, Kai Wu and Jijun Jiang for assistance in experiments.References
- (a) D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed; (b) J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- (a) M. Gao and B. Z. Tang, ACS Sens., 2017, 2, 1382–1399 CrossRef CAS PubMed; (b) W. Chen, Q. Li, W. Zheng, F. Hu, G. Zhang, Z. Wang, D. Zhang and X. Jiang, Angew. Chem., Int. Ed., 2014, 53, 13734–13739 CrossRef CAS PubMed.
- (a) J. Huang, N. Sun, Y. Dong, R. Tang, P. Lu, P. Cai, Q. Li, D. Ma, J. Qin and Z. Li, Adv. Funct. Mater., 2013, 23, 2329–2337 CrossRef CAS; (b) Y. Liu, C. Mu, K. Jiang, J. Zhao, Y. Li, L. Zhang, Z. Li, J. Y. Lai, H. Hu, T. Ma, R. Hu, D. Yu, X. Huang, B. Z. Tang and H. Yan, Adv. Mater., 2015, 27, 1015–1020 CrossRef CAS PubMed; (c) Z. Y. Yang, Z. H. Chi, Z. Mao, Y. Zhang, S. W. Liu, J. Zhao, M. Aldred and Z. G. Chi, Mater. Chem. Front., 2018, 2, 861–890 RSC.
- (a) D. Ding, D. Mao, K. Li, X. Wang, W. Qin, R. Liu, D. S. Chiam, N. Tomczak, Z. Yang, B. Z. Tang, D. Kong and B. Liu, ACS Nano, 2014, 8, 12620–12631 CrossRef CAS PubMed; (b) Y. Hong, L. Meng, S. Chen, C. W. T. Leung, L.-T. Da, M. Faisal, D.-A. Silva, J. Liu, J. W. Y. Lam, X. Huang and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 1680–1689 CrossRef CAS PubMed; (c) G. Yu, D. Wu, Y. Li, Z. Zhang, L. Shao, J. Zhou, Q. Hu, G. Tang and F. Huang, Chem. Sci., 2016, 7, 3017–3024 RSC; (d) C. W. T. Leung, Y. Hong, S. Chen, E. Zhao, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 62–65 CrossRef CAS PubMed.
- (a) S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. Jiang, J. Am. Chem. Soc., 2016, 138, 5797–5800 CrossRef CAS PubMed; (b) L. Ascherl, T. Sick, J. T. Margraf, S. H. Lapidus, M. Calik, C. Hettstedt, K. Karaghiosoff, M. Döblinger, T. Clark, K. W. Chapman, F. Auras and T. Bein, Nat. Chem., 2016, 8, 310–316 CrossRef CAS.
- (a) L. Yang, X. Jing, C. He, Z. Chang and C. Duan, Chemistry, 2016, 22, 18107–18114 CrossRef CAS PubMed; (b) X. Yan, T. R. Cook, P. Wang, F. Huang and P. J. Stang, Nat. Chem., 2015, 7, 342–348 CrossRef CAS PubMed; (c) C. Zhang, Z. Wang, L. Tan, T. L. Zhai, S. Wang, B. Tan, Y. S. Zheng, X. L. Yang and H. B. Xu, Angew. Chem., Int. Ed., 2015, 54, 9244–9248 CrossRef CAS PubMed.
- (a) L. J. Chen, Y. Y. Ren, N. W. Wu, B. Sun, J. Q. Ma, L. Zhang, H. Tan, M. Liu, X. Li and H. B. Yang, J. Am. Chem. Soc., 2015, 137, 11725–11735 CrossRef CAS PubMed; (b) A. Schultz, S. Diele, S. Laschat and M. Nimtz, Adv. Funct. Mater., 2001, 11, 441–446 CrossRef CAS; (c) H. Q. Peng, X. Y. Zheng, T. Han, R. Kwok, J. Lam, X. H. Huang and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 10150–10156 CrossRef CAS PubMed.
- (a) N. N. Liu, S. Song, D. M. Li and Y. S. Zheng, Chem. Commun., 2012, 48, 4908–4910 RSC; (b) H.-T. Feng, X. Zhang and Y.-S. Zheng, J. Org. Chem., 2015, 80, 8096–8101 CrossRef CAS PubMed.
- (a) Q. Ye, D. Zhu, L. Xu, X. Lu and Q. Lu, J. Mater. Chem. C, 2016, 4, 1497–1503 RSC; (b) F. Y. Song, Z. Xu, Q. S. Zhang, Z. Zhao, H. K. Zhang, W. J. Zhao, Z. J. Qiu, C. X. Qi, H. Zhang, H. Sung, I. Williams, J. Lam, Z. J. Zhao, A. J. Qin, D. G. Ma and B. Z. Tang, Adv. Funct. Mater., 2018, 28, 1800051–1800063 CrossRef.
- (a) H. Li, X. Zheng, H. Su, J. W. Lam, K. Sing Wong, S. Xue, X. Huang, X. Huang, B. S. Li and B. Z. Tang, Sci. Rep., 2016, 6, 19277–19286 CrossRef PubMed; (b) D. D. L. Anuradha, M. Al Kobaisi and S. V. Bhosale, Sci. Rep., 2015, 5, 15652–15663 CrossRef PubMed; (c) H. Shi, R. T. K. Kwok, J. Liu, B. Xing, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 17972–17981 CrossRef CAS PubMed.
- (a) J. B. Xiong, H. T. Feng, J. P. Sun, W. Z. Xie, D. Yang, M. Liu and Y. S. Zheng, J. Am. Chem. Soc., 2016, 138, 11469–11472 CrossRef CAS PubMed; (b) L. Ding, L. Lin, C. Liu, H. Li, A. Qin, Y. Liu, L. Song, H. Zhang, B. Z. Tang and Y. Zhao, New J. Chem., 2011, 35, 1781 RSC.
- N. L. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Lam and B. Z. Tang, Chemistry, 2014, 20, 15349–15353 CrossRef CAS PubMed.
- X. Wang, Y. Wang, H. Yang, H. Fang, R. Chen, Y. Sun, N. Zheng, K. Tan, X. Lu, Z. Tian and X. Cao, Nat. Commun., 2016, 7, 12469 CrossRef PubMed.
- H. Qu, Y. Wang, Z. Li, X. Wang, H. Fang, Z. Tian and X. Cao, J. Am. Chem. Soc., 2017, 139, 18142–18145 CrossRef CAS PubMed.
- D. Fujita, Y. Ueda, S. Sato, N. Mizuno, T. Kumasaka and M. Fujita, Nature, 2016, 540, 563–566 CrossRef CAS.
- P. Zhang, X. Wang, W. Xuan, P. Peng, Z. Li, R. Lu, S. Wu, Z. Tian and X. Cao, Chem. Commun., 2018, 54, 4685–4688 RSC.
- Y. Wang, H. Fang, I. Tranca, H. Qu, X. Wang, A. J. Markvoort, Z. Tian and X. Cao, Nat. Commun., 2018, 9, 488–496 CrossRef PubMed.
- (a) H. T. Feng and Y. S. Zheng, Chem.–Eur. J., 2014, 20, 195–201 CrossRef CAS PubMed; (b) H. T. Feng, S. Song, Y. C. Chen, C. H. Shen and Y. S. Zheng, J. Mater. Chem. C, 2014, 2, 2353–2359 RSC; (c) J. B. Xiong, Y. X. Yuan, L. Wang, J. P. Sun, W. G. Qiao, H. C. Zhang, M. Duan, H. W. Han, S. Zhang and Y. S. Zheng, Org. Lett., 2018, 20, 373–376 CrossRef CAS PubMed.
- (a) J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC; (b) V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169–202 RSC.
- R. Hu, S. Li, Y. Zeng, J. Chen, S. Wang, Y. Li and G. Yang, Phys. Chem. Chem. Phys., 2011, 13, 2044–2051 RSC.
- (a) P. Jagadesan, G. Eder and P. McGrier, J. Mater. Chem. C, 2017, 5, 5676–5679 RSC; (b) Y. C. Chen, X. X. Liu, H. Huang, W. W. Wu and Y. S. Zheng, Sci. China: Chem., 2010, 53, 569–575 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1856409 and 1856413. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03404d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |