
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing surfaces of atmospherically relevant organic particles by easy ambient sonic-spray ionization mass spectrometry (EASI-MS)†
L. M.
Wingen
 and
B. J.
Finlayson-Pitts
*
and
B. J.
Finlayson-Pitts
*
Department of Chemistry, University of California Irvine, Irvine, CA 92697-2025, USA. E-mail: bjfinlay@uci.edu; Tel: +1-949-824-7670
First published on 9th November 2018
Abstract
Both ambient and laboratory-generated particles can have a surface composition different from the bulk, but there are currently few analytical techniques available to probe these differences. Easy ambient sonic-spray ionization mass spectrometry (EASI-MS) was applied to solid, laboratory-generated particles with core–shell morphologies formed from a variety of dicarboxylic acids. The soft ionization facilitated parent peak detection for the two compounds, from which the depth probed could be determined from the relative signal intensities. Two different configurations of a custom-made nebulizer are reported that yield different probe depths. In the “orthogonal mode,” with the nebulizer ∼10 centimeters away from the particle stream and at a 90° angle to the MS inlet, evaporation of the nebulizer droplets forms ions before interaction with the particles. The probe depth for orthogonal mode EASI-MS is shown to be 2–4 nm in these particle systems. In the “droplet mode”, the nebulizer and particle streams are in close proximity to each other and the MS inlet so that the particles interact with charged liquid droplets. This configuration resulted in full dissolution of the particles and gives particle composition similar to that from collection on filters and extraction of the particles (bulk). These studies establish that EASI-MS is a promising technique for probing the chemical structures of inhomogeneous airborne organic particles.
Introduction
A major focus of atmospheric chemistry research is the formation, growth, composition and properties of secondary organic aerosol particles (SOA).1–8 These particles are formed in air from the gas-phase oxidation of larger organic compounds such as monoterpenes and sesquiterpenes. A number of analytical approaches have been used to examine their composition both in laboratory studies and in field measurements such as collection on filters or impactors with subsequent analysis by chromatographic methods coupled with mass spectrometry.9–13 Particle mass spectrometry techniques11–17 using thermal desorption18–21 or laser ablation22–27 have been applied to particles on-the-fly, with no need for collection or other sample preparation. While these have provided a wealth of data, they generally give information only on bulk particle composition, often without molecular speciation. In a few selected cases, depth profiling has been possible to distinguish the surface from the bulk.28–32Probing such heterogeneity is important for understanding gas–particle interactions that determine particle growth and heterogeneous chemistry. For purely organic particles formed from the oxidation of gas-phase precursors, the general assumption has been that they are homogeneous mixtures. However, there are few definitive data to support this, and some reasons to expect it may not always be the case. For example, there is evidence that nucleation to form new particles during oxidation of gas phase organic compounds involves extremely low volatility organic compounds (ELVOC),33–35 and that subsequent growth of the core involves uptake of relatively more volatile reaction products.36–39 For particles of high viscosity38–48 where diffusion is slow,49–51 this heterogeneity may be preserved.52 Reorganization of surface layers may also occur to minimize surface free energy, resulting in a surface that differs from the bulk. The nature of this surface, e.g. the polarity and presence of hydrogen bonding moieties, will determine growth and ability to take up water and ultimately act as cloud condensation nuclei (CCN).3,53–56 Additionally, the surface composition of ambient particles will play a role in their interactions with biological surfaces, such as the respiratory system, skin, and other membranes.57–59
The development and application of ambient ionization mass spectrometry techniques has increased dramatically in the last decade,60–71 providing rapid, real-time information on composition without the need for extensive sample preparation. Such techniques have been applied to a wide variety of samples, including pharmaceuticals, foods, fuels, cosmetics, and perfumes, and their use in forensic analysis of samples as disparate as wood and paper currency has proven very useful.71 They have also been used to follow reactions in solutions on surfaces in real time.72 A number of these ionization techniques, including ASAP (atmospheric solids analysis probe),73–76 DART (direct analysis in real time),29,30,39,77–80 DESI (desorption electrospray ionization)16,61,62,81,82 and EESI (extractive electrospray ionization)71,83–92 have been applied to atmospheric particles and model systems. However, it is not always clear how much of the particle is being probed, which may depend not only on the technique but also on the nature of the particles and the specific experimental conditions used.90,91
An ambient ionization technique based on sonic spray ionization (SSI)93,94 has found increasing use. Easy ambient sonic-spray ionization mass spectrometry (EASI-MS) forms ions from a solvent stream that is mechanically disturbed using a flow of N2 at supersonic speeds.69,71,95–97 This disruption forms droplets with uneven distributions of positive and negative charges which can then be transferred to an analyte.98 No external energy such as voltage or heating is required. As a result, there is essentially no background signal or reactions of the analyte occurring in the source as is the case for some of the other ambient ionization techniques.
We report here the application of EASI-MS to a stream of airborne organic particles generated in a core–shell structure with a core of one compound and a shell of a different compound of known thickness. The EASI-MS signatures of the two compounds as a function of the shell-to-core ratio compared to those from bulk analysis of the particles provides insight into how deeply EASI-MS probes into such particles. Malonic acid coatings on cores of succinic, glutaric, and pimelic acids were studied. Dicarboxylic acids have been identified in a variety of urban, rural, remote, and marine locations.99–112 They are present in both primary particles, such as combustion emissions, and secondary particles formed during VOC oxidation in air and hence their measurement in surface and bulk is of great interest.
Experimental methods
As described in detail below, poly- or monodisperse core particles were generated by atomization of a dicarboxylic acid solution, diffusion dried, and either left uncoated or coated with a second diacid before characterization by size and number. Particles were then directed to the EASI nebulizer and mass spectrometer for analysis. For comparison, in separate experiments, bulk analysis was performed on filter-collected particles dissolved in the EASI solvent.Solvent spray ionization nebulizer
Fig. 1 shows a schematic of the nebulizer used to carry out EASI-MS studies as well as a photograph of the tip. It is custom-made from borosilicate glass and is similar to that used by Antonakis et al.113 The body consists of two concentric glass tubes for introduction of a solvent through the center tube and sheath gas flow through the outer tube. The mechanical action of the sheath gas flow approaching sonic velocities generates excess charges on droplets and transfers them to the gas phase,71,96,98,114 a phenomenon that generally occurs in devices used to generate particles by spraying solutions through small orifices, such as atomizers.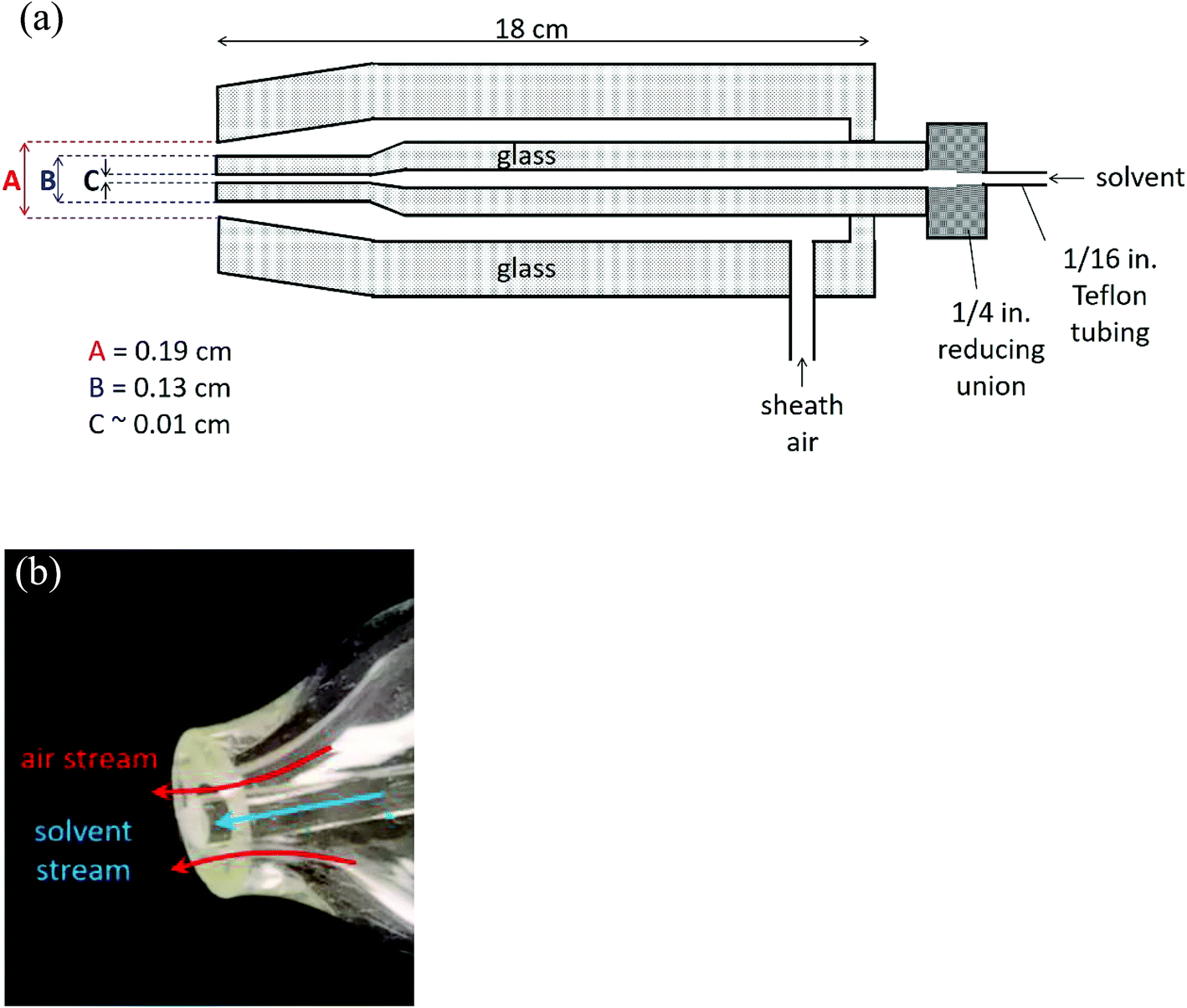 | ||
| Fig. 1 Easy ambient sonic-spray ionization nebulizer (a) schematic (not to scale) and (b) photograph of the nebulizer tip. | ||
The dimensions at the tip of the nebulizer combined with the sheath air volumetric flow rate of 8.0 L min−1, with a pressure of 17 psi on the nebulizer, result in a linear velocity of approximately 100 m s−1. In initial studies, the nebulizer was manually positioned 1–10 cm from the inlet of the mass spectrometer. While different flow rates and pressures have been used with similar nebulizers,113,115 the flow rate was chosen here to generate sufficient signal from solvent ions at these initial positions while using convenient flows of the available sheath gas. Ultimately, two nebulizer positions were chosen for further studies, as described below.
Solvent was introduced into the nebulizer through Teflon fittings adapted from the 1/4 in. end of the nebulizer to 1/16 in. Teflon tubing. The Teflon tubing was coupled to a syringe needle with a PEEK union and a tubing sleeve. Solvent was continuously added to the nebulizer using a syringe pump (New Era Pump Systems, Inc., model NE-1000). The solvent was a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol:water mixture with 0.2% (v/v) formic acid. Methanol (EMD Millipore Corp., LC-MS grade, 99.9%) and formic acid (Fisher, 88%) were used as received. A water purifier provided Nanopure water (Barnstead, Nanopure, 18.2 MΩ cm). Sheath air was obtained from a purge gas generator (Parker-Balston, model 75-62).
:1 methanol:water mixture with 0.2% (v/v) formic acid. Methanol (EMD Millipore Corp., LC-MS grade, 99.9%) and formic acid (Fisher, 88%) were used as received. A water purifier provided Nanopure water (Barnstead, Nanopure, 18.2 MΩ cm). Sheath air was obtained from a purge gas generator (Parker-Balston, model 75-62).
Ion signals were detected with a triple quadrupole mass spectrometer (Waters, Xevo TQ-S) with a 1/4 in. ceramic tube inlet that is typically used with a direct analysis in real time-MS ionization source (DART SVP, IonSense), which includes a short metal elbow (Vapur® Interface) between the ceramic inlet to the MS cone (Fig. 2). To use the nebulizer for ion generation, the DART probe was removed but the ceramic inlet and metal elbow remained in place. No voltage or heat was applied to the nebulizer, solvent, or sample in any experiments.
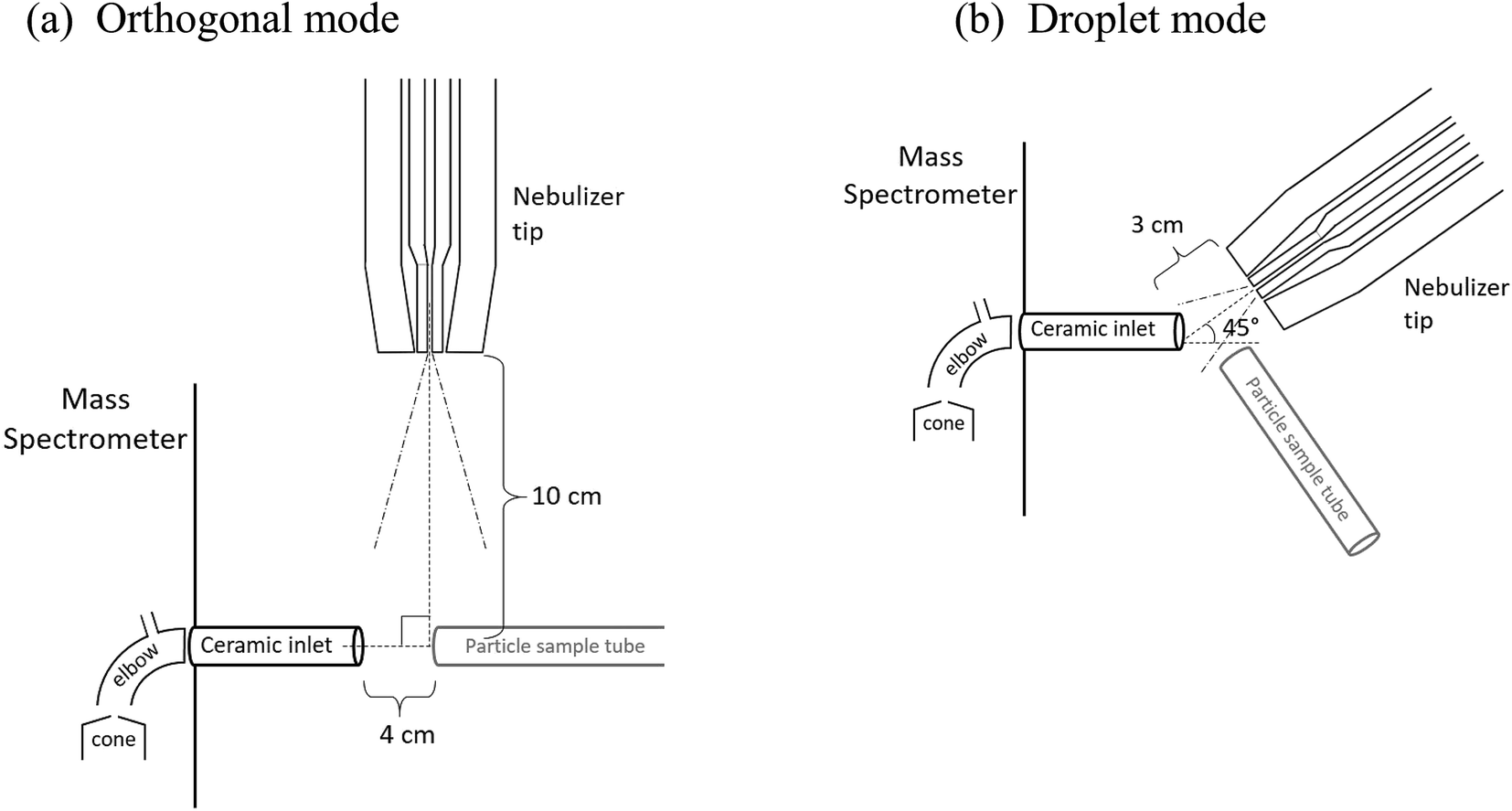 | ||
| Fig. 2 Schematic of the nebulizer and particle sample tube position with respect to the mass spectrometer inlet for (a) orthogonal mode and (b) droplet mode. In the droplet mode, the particle sample tube is directed into the plane of the page. | ||
Two configurations between the nebulizer and MS inlet were chosen for these studies, as shown in Fig. 2. In the first, the “orthogonal mode,” the nebulizer was placed at a 90° angle to the inlet at a distance of 10 cm with a solvent flow of 1.0 mL h−1. This arrangement allowed solvent droplets time to evaporate before reaching the inlet. In the second configuration, the “droplet mode,” the nebulizer was 3 cm from the MS inlet at an angle of 45° with a solvent flow of 6.0 mL h−1. In both cases, a sheath air flow of 8.0 L min−1 was used. For some experiments in the orthogonal mode, a stainless-steel Swagelok® tee was connected to the ceramic MS inlet to loosely hold the sample tubing in place.
EASI-MS mass spectrometry of particles
Particles containing a dicarboxylic acid core and a different diacid coating of variable thicknesses were generated to probe the depth from the surface which could be detected by EASI-MS. EASI mass spectra of bare and coated particles, in both droplet and orthogonal modes, were collected by positioning the end of the aerosol sample tubing approximately 1–2 cm from the MS inlet for 30 s to 2 min. Spectra were collected in the range m/z 25–400 in negative ion mode. Vapors were removed from the sample by flowing the aerosol stream through a charcoal denuder before intersecting the nebulizer stream. The denuder was regenerated before each experiment by flowing N2 through while heating for 1–2 h and cooling under N2 flow. Spectral subtractions were not carried out for orthogonal mode spectra since little to no solvent ion signal is generated in this position. Raw peak intensities from orthogonal mode EASI-MS spectra were used to obtain ratios of surface to core.In the droplet mode, particles could dissolve in the charged droplets before being ionized, similar to electrospray ionization. Because of the close proximity of the solvent spray to the MS inlet, the tip of the sample tubing often collected solvent droplets and thus there was a significant contribution of the solvent to background spectra. The spectrum from the solvent spray, collected just before each particle sample and averaged over the same duration as the particle sample (30 s to 2 min), was subtracted from the sample signal for droplet mode spectra. Solvent-subtracted peak heights from [M − H]− ions associated with the core and coating materials were plotted as a function of coating temperature. In the orthogonal mode, the sample tubing remained dry.
EASI-MS signals generated from monodisperse particles were much smaller due to lower particle number and mass concentrations. Multiple reaction monitoring (MRM, both quadrupoles at fixed m/z) methods were utilized in monodisperse particle experiments to measure the diacids. Fragmentation of [M − H]− parent ions to [M–H–CO2]− daughter ions with a collision energy of 10-11 V were measured using the following transitions: m/z 89 > 45 for oxalic acid, m/z 103 > 59 for malonic acid, m/z 117 > 73 for succinic acid, and m/z 131 > 87 for glutaric acid. For pimelic acid, two transitions, m/z 159 > 115 (loss of CO2) and m/z 159 > 97 (loss of both CO2 and H2O), were summed to increase MS sensitivity since for diacids ≥C6, loss of CO2 becomes weak and loss of H2O becomes dominant.116 Raw MRM signals from the core and coating material, and their ratios, were plotted as a function of coating temperature.
Generation of coated particles
Coated particles were generated using the flow systems shown in Fig. 3. Aqueous solutions of 10–50 mM of the dicarboxylic acid selected as the core were atomized with a constant output atomizer (TSI, model 3076) at 34 psi. The flow exiting the atomizer at 1.9 L min−1 passed through two successive diffusion dryers filled with desiccant (that was dried before each experiment). Size distributions of uncoated, polydisperse particles were measured after flowing through the top of the coating assembly with the coating reservoir closed. Core particle size distributions were measured with a scanning mobility particle sizer (SMPS) consisting of an electrostatic classifier (TSI, model 3080) with a long differential mobility analyzer (DMA-1, TSI, model 3081) and a condensation particle counter (CPC, TSI, model 3776). Measurements were performed at stable particle concentrations, typically (1–3) × 106 cm−3 of core particles. Core particles were generated from succinic acid (SA, the C4 diacid), glutaric acid (GA, the C5 diacid) and pimelic acid (PA, the C7 diacid). Malonic acid (MA, the C3 diacid) and oxalic acid (OA, the C2 diacid) were used as coatings. All diacids were used as received from Sigma-Aldrich (OA, 98%; MA, ReagentPlus 99%; SA, BioXtra ≥99.0%; GA, 99%; PA, 98%). Adipic acid (Sigma-Aldrich, BioXtra ≥99.5%) was used for SMPS calibrations.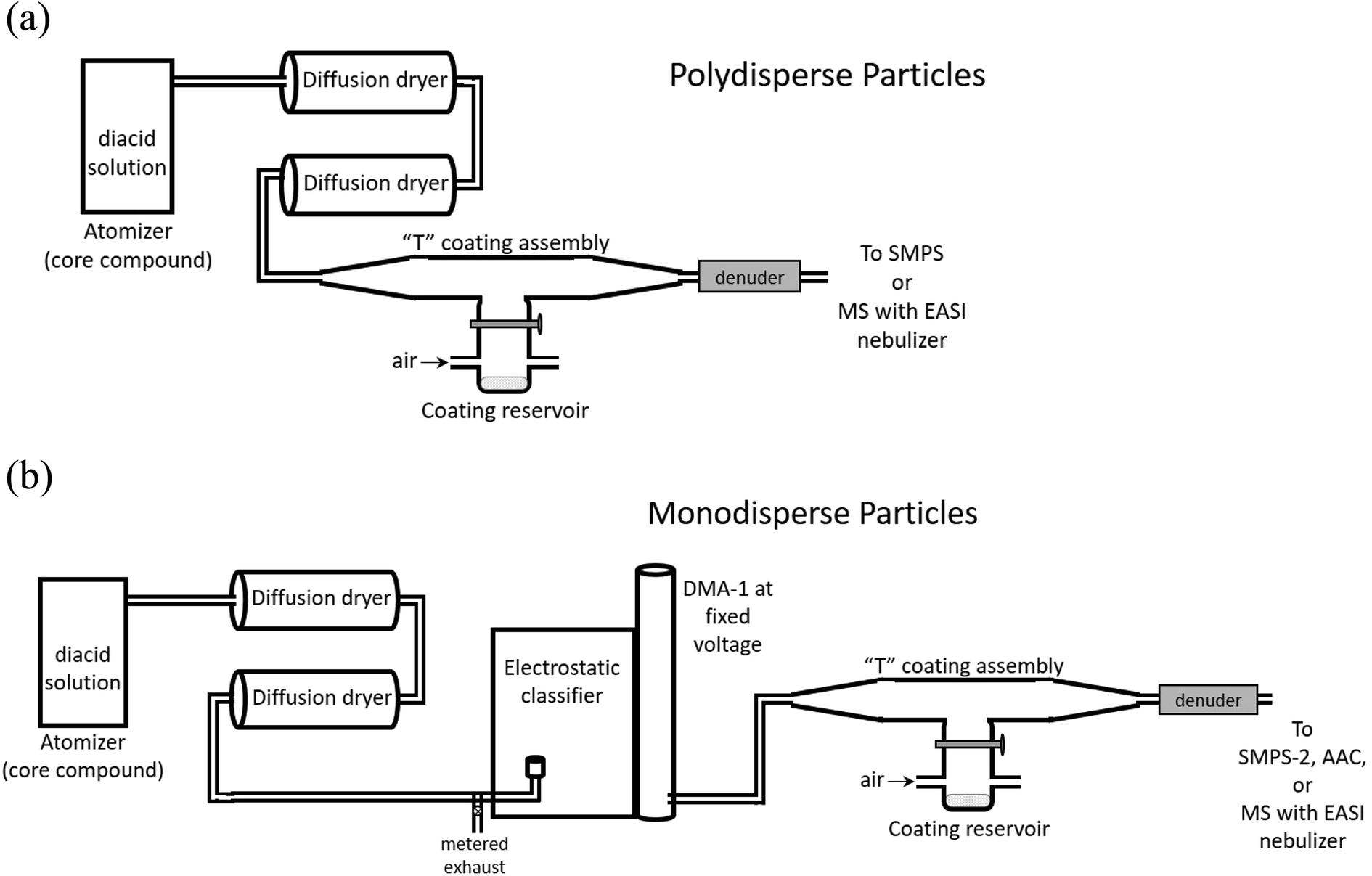 | ||
| Fig. 3 Schematic of the flow systems for generating (a) polydisperse, coated particles, or (b) monodisperse, coated particles. | ||
While sampling bare particles, the coating reservoir was kept closed as 0.6–0.8 L min−1 air flowed through the reservoir to the exhaust to maintain a steady-state concentration of coating vapor. Coating was initiated by opening the coating reservoir containing the second diacid to redirect this air flow up into the top of the “T” assembly, giving a total flow of 2.5–2.7 L min−1 of core particles plus coating vapor in air exiting the coater. The temperature of the reservoir was controlled with heating tape and a variable transformer. Reservoir temperature was measured with a thermocouple (Omega, type K) mounted under the heating tape on the outside of the glass reservoir. Note that the temperature of aerosol exiting the coater was significantly lower than the heated reservoir, ≤6 °C above room temperature. Diameter increases were not detectable during polydisperse particle coating, likely because of the breadth of the size distributions (typical geometric standard deviations of 2.1; see ESI, Fig. S1†), which was not sufficient to capture the change due to relatively thin coatings.
Monodisperse core particles were generated by setting DMA-1 to a constant voltage for selected particle diameters. SMPS-1 was calibrated using the method of Kidd et al. (2014)117 at 0.3 L min−1 sample flow and 3.0 L min−1 sheath flow. In the diameter range 180 nm > d > 300 nm, measurements were within ±1% of the reported diameters for carboxylate modified latex spheres (CMLs, Life Technologies, Grand Island, NY). For 110 nm CMLs, this SMPS measured diameters that were systematically ∼10% larger than selected, therefore most monodisperse particle experiments were carried out with larger selected diameters.
Upon exiting DMA-1, monodisperse core particles were coated by flowing through the T coating assembly (Fig. 3b). Flow through DMA-1 was restricted to 0.8–1.3 L min−1 to keep a flow rate ratio of sheath air to aerosol of ∼(3–4):1. Higher ratios resulted in narrower distributions but significant particle dilution. The flow of bare or coated monodisperse particles was directed to a second SMPS (SMPS-2, TSI, electrostatic classifier model 3080, DMA model 3081, CPC model 3775) which sampled at 0.3 L min−1 (sheath flow rate = 3.0 L min−1), while the remaining flow was exhausted. SMPS-2 was also calibrated117 and exhibited uncertainties of ±<5% in the diameter range 110 nm > d > 300 nm.
An aerodynamic aerosol classifier (AAC, Cambustion, Ltd., Cambridge, UK) was also used for a limited number of diameter measurements for bare and coated particles. As discussed in the ESI, AAC measurements provided evidence that glutaric acid particles underwent some evaporation during measurements by SMPS, which was partly alleviated during measurement by AAC due to its shorter measurement residence times.
The possibility of self-nucleation of the vapors of the coating compound to form new particles during heating was investigated by directing a portion of the flow from the heated reservoir, which was closed off from the core particles, to the SMPS. For MA, reservoir temperatures >122 °C induced self-nucleation to give particles with diameters of ∼50 nm (e.g. 115 particles per cm3 at a reservoir temperature of 125 °C). The small diameters of these self-nucleated particles resulted in number (and mass) concentrations much less than that of size-selected coated particles (100 nm or 250 nm, (1–10) × 104 cm−3) or polydisperse coated particles (∼100 nm geometric mean diameters, (1–3) × 106 cm−3). Nonetheless, to avoid self-nucleation, all MA coating experiments were carried out at MA reservoir temperatures <119 °C. For OA, self-nucleation began at 93 °C and generated ∼30 nm particles, thus coating experiments were limited to reservoir temperatures <89 °C.
Bulk composition analysis of coated particles with EASI-MS
Impaction of uncoated and coated particle samples was performed by flowing the particles through a charcoal denuder and onto Teflon filters (Fluoropore™ Membrane Filters, PTFE, 0.45 μm) with the aid of a pump (SKC, Inc., Leland Legacy) to ensure constant flow through the filter. Particle collection time varied from 15 min (for polydisperse samples) to 6 hours (for 100 nm monodisperse samples). Each filter was sonicated for 5 min with 10–20 mL of 1:1 methanol:water in an amber bottle, and formic acid was added to 0.2% after sonication. The solution was then placed in the nebulizer with the syringe pump and analyzed at a 3 cm distance (as in droplet mode) from the inlet. EASI-MS mass spectra collected for impacted particles reflect the bulk composition of the particles since the entire particle core and coating is dissolved in the solvent. The ratio of signal intensities from this bulk analysis was compared to ratios found for the separate particle stream of coated particles in droplet and orthogonal modes to determine the probe depth of each method.
Results and discussion
EASI-MS ion intensities were first measured in the absence of samples to examine the signal associated with the solvent as a function of distance between the nebulizer tip and the MS inlet. Typical solvent peaks were observed in positive ion mode at m/z 33 and 65 from [M + H]+ and [2M + H]+ ions of methanol and in negative ion mode at m/z 45 and 91 from [M − H]− and [2M − H]− ions of formic acid. Fig. 4 shows the decrease in solvent peak intensity as a function of distance in both negative (Fig. 4a and c) and positive ion mode (Fig. 4b and d). No solvent peaks were observed when the nebulizer was placed at a 90° angle to the inlet and a distance of 10 cm with the solvent flow decreased to 1.0 mL h−1 (orthogonal mode). Fig. 4e shows that introduction of a sample flow of dry polydisperse glutaric acid (GA) particles resulted in [GA–H]− peak detection in both the droplet mode and the orthogonal mode, with smaller intensities in the latter case. This suggests that increasing the distance of the nebulizer from the inlet allows solvent droplets enough time to evaporate, but still generates sufficient ions to interact with the solid GA particles.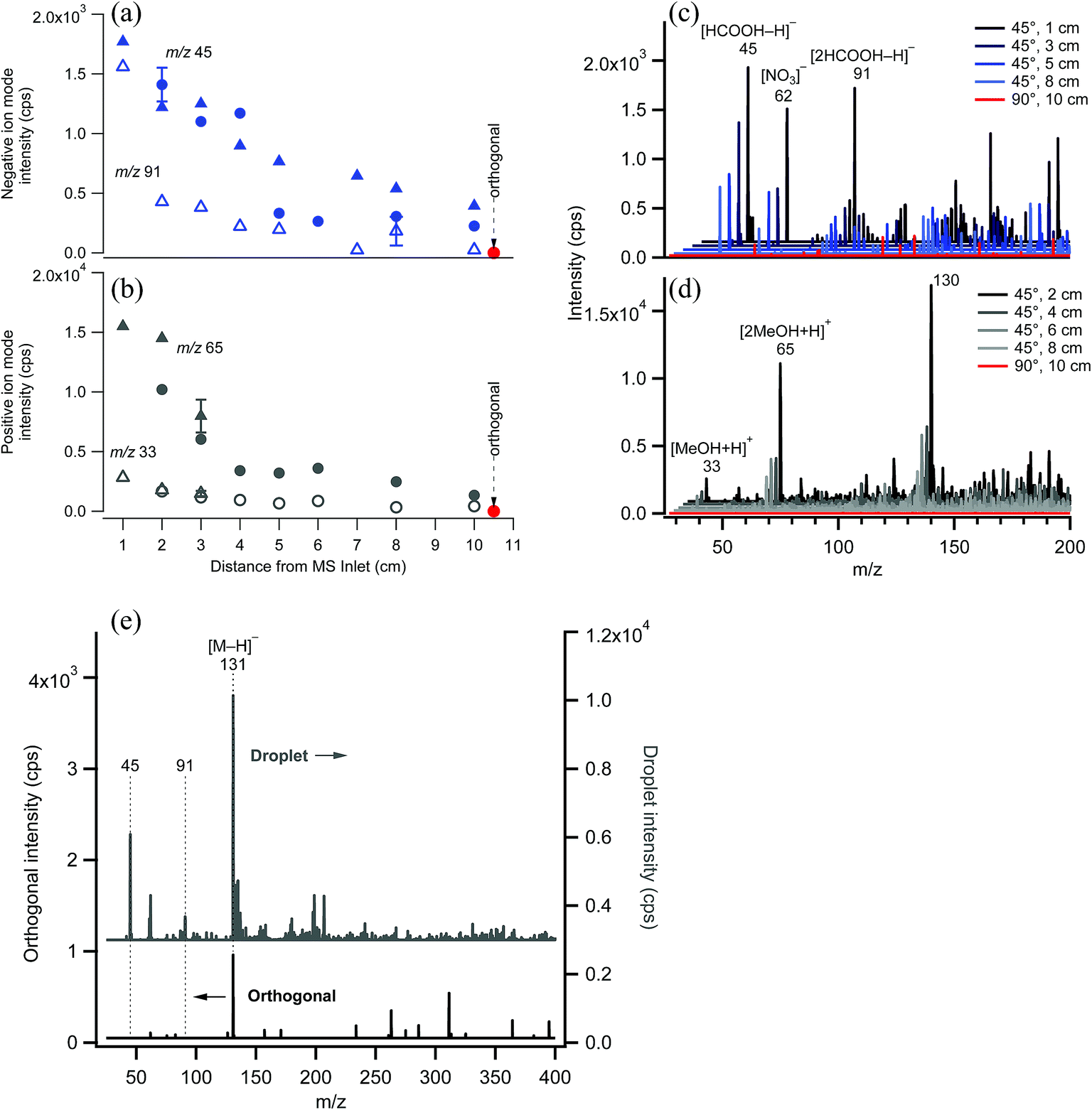 | ||
| Fig. 4 MS intensity as a function of increasing distance of the nebulizer from the MS inlet for (a) negative ion mode [HCOOH–H]− solvent peak and (b) positive ion mode solvent peaks [MeOH + H]+ and [2MeOH + H]+ and their corresponding EASI mass spectra (c, d). Different symbols represent repeated measurements with slightly different nebulizer position and error bars are 1s standard deviations from replicate measurements. The peak at m/z 130 in (d) is due to an unknown solvent contaminant. (e) Typical EASI-MS mass spectra of solid, bare glutaric acid particles in droplet (grey) and orthogonal (black) modes showing the presence and absence, respectively, of typical solvent peaks. | ||
Polydisperse glutaric acid (C5 diacid) particles coated with malonic acid (C3 diacid)
The surface probe depth capability of this technique was investigated by coating polydisperse GA particles with MA. Fig. 5a shows EASI-MS spectra collected in orthogonal mode as a function of increasing MA temperature in the coating reservoir. As the coating temperature increased, increased intensities for MA were observed and the GA signal decreased. This trend is shown in Fig. 5b, and suggests that EASI ions interact with and eject species preferentially from the particle surface.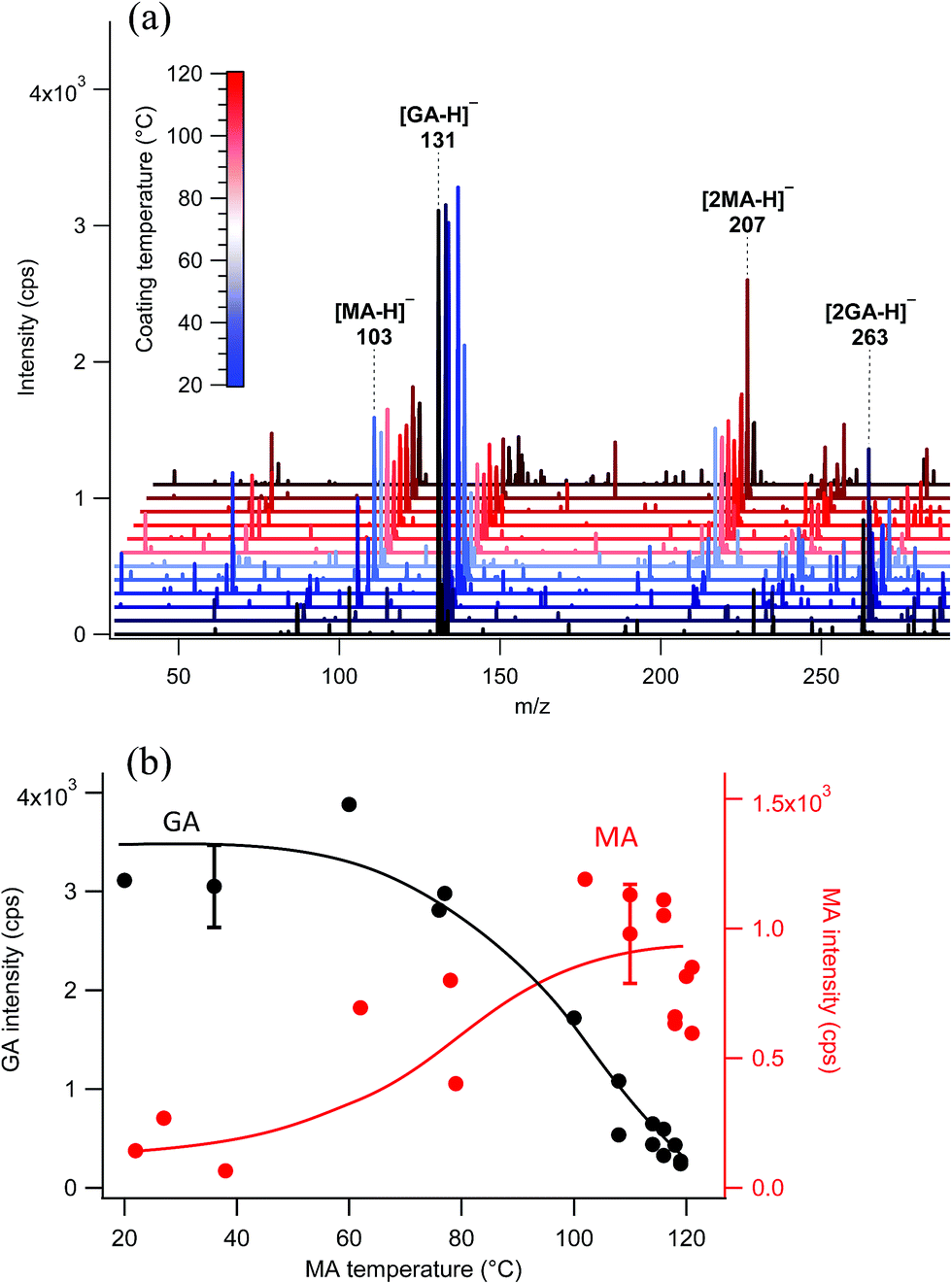 | ||
| Fig. 5 EASI-MS orthogonal mode analysis of polydisperse glutaric acid (GA) particles coated with malonic acid (MA) at increasing coating reservoir temperatures. (a) EASI-MS spectra collected in orthogonal mode. Spectra were averaged over approximately one minute. (b) Signal intensities of GA (black) and MA (red) as a function of MA coating temperature. Error bars represent 1s standard deviations and the solid lines are guidelines only. | ||
The peak intensity ratio of MA coating to GA core was calculated as an indication of the surface sensitivity of the technique and is shown in Fig. 6a as a function of MA coating temperature. Since particles flowed through a charcoal denuder to remove vapor phase contributions, these ratios represent the signal detected from the particle surface relative to its bulk. There is some variability in the signal intensities at each temperature (Fig. 5b) due to changes in sample tubing placement between measurements as well as variability in the particle concentrations that are sampled by the mass spectrometer. The ratio of coating to core eliminates much of this variability and shows an increasing trend in the amount of MA coating detected relative to the GA core as the MA temperature and particle coating thickness increase. For comparison, the ratios of MA to GA from bulk analysis of particles collected on a filter and extracted into solution are also shown in Fig. 6a at three coating temperatures (blue triangles). The higher MA/GA ratios observed from orthogonal nebulizer particle sampling relative to those from the bulk analysis (extracted and fully dissolved particles) indicate that orthogonal EASI-MS probes less of the GA in the particle core. Additionally, once a coating temperature of 116 °C was reached during this experiment, the MA coater was closed and an orthogonal EASI-MS spectrum was taken of the bare GA particles at room temperature without the charcoal denuder (Fig. 6a, open circle). The low MA/GA ratio of this data point shows that there was very little MA detected either in (a) the particle phase, since these particles were intended to be bare GA, or in (b) the vapor phase that could be present from MA taken up on the sample tubing and subsequently desorbing as the flow of uncoated GA particles passed through it. These results suggest that EASI-MS in orthogonal mode provides a surface sensitive method for particle analysis.
 | ||
| Fig. 6 (a) Ratios of MA coating on polydisperse GA particles calculated from EASI-MS peak intensities collected in orthogonal mode. The open circle represents a spectrum taken when the coating reservoir was closed off during the experiment to form bare GA particles and with no denuder. Blue triangles (a and b) are from bulk analysis of collected and extracted polydisperse particles. (b) Ratios of MA coating on polydisperse GA particles collected in droplet mode. Peak intensities from droplet mode were corrected by subtracting spectra of solvent averaged over 1 min. Typical 1s standard deviations are included from replicate measurements and are smaller than the markers for the droplet mode analysis. The solid lines are guidelines only. | ||
Similar experiments were carried out on these MA-coated polydisperse GA particles using the nebulizer in droplet mode, in which visibly larger solvent droplets interact directly with the particle stream and the nebulizer is much closer to the MS inlet. Fig. 6b shows the droplet mode ratios of MA coating to GA core observed as a function of MA temperature. EASI-MS spectra are provided in the ESI (Fig. S2†). Ratios for the same bulk analysis from Fig. 6a are also included in Fig. 6b for comparison. The similarity of the ratios obtained in droplet mode to those from the bulk analysis suggests that the droplet mode allows complete dissolution of the particles. This is not surprising since, as noted in the Experimental section, the tip of the particle sample tubing often collected solvent droplets because of its proximity to the nebulizer in this mode.
Bulk analysis of mixtures of malonic acid and glutaric acid in solution indicated that MA could cause ion suppression of the GA, leading to increases in the MA/GA ratio of approximately a factor of two (Fig. S3–S5†). However, MA/GA ratios obtained from the bulk analysis are still ≤0.5 for all coating thicknesses and, despite this uncertainty, are lower than those obtained with orthogonal mode (MA/GA = 1–3.5) for coated particles. The aqueous solubilities of MA and GA in water are both very high (∼4–5 M)118 and similar solubilities in 1:1 methanol:water are expected. Thus, droplet mode EASI-MS can efficiently extract particles into the solvent to provide a bulk analysis, while orthogonal EASI-MS generates ion signals preferentially from particle surfaces with much less solvent interaction and a smaller probe depth.
Monodisperse glutaric acid particles coated with malonic acid
The probe depth of EASI-MS experiments with coated polydisperse particles is difficult to quantify because of the broad distributions of atomized GA for which diameter changes during coating were not detectable by SMPS. Therefore, experiments were carried out in which monodisperse GA particles were generated by size-selection with a DMA and then coated with MA using the coating apparatus in Fig. 3b. Selective MRM methods were used in these experiments due to the lower particle number concentrations and hence smaller signals in these experiments. Raw signals of MA and GA are shown as a function of MA temperature for monodisperse 220 nm GA particles coated with MA in Fig. 7a. Similar to the coated polydisperse particles, the GA core signal decreased and the MA signal increased as the reservoir temperature increased, indicating that an increasing amount of surface MA material was detected as the particle coating became thicker. Bulk analysis was carried out in which the coated monodisperse particles were collected on a filter and extracted into solution. Fig. 7b shows both the MA/GA ratios calculated from the raw signals in Fig. 7a as well as MA/GA ratios from their bulk analysis. Again, the bulk ratios are lower than orthogonal mode analysis of the particle stream, indicating that orthogonal EASI-MS probes less GA in the particle core than is present in the whole particle, i.e., it preferentially detects the surface.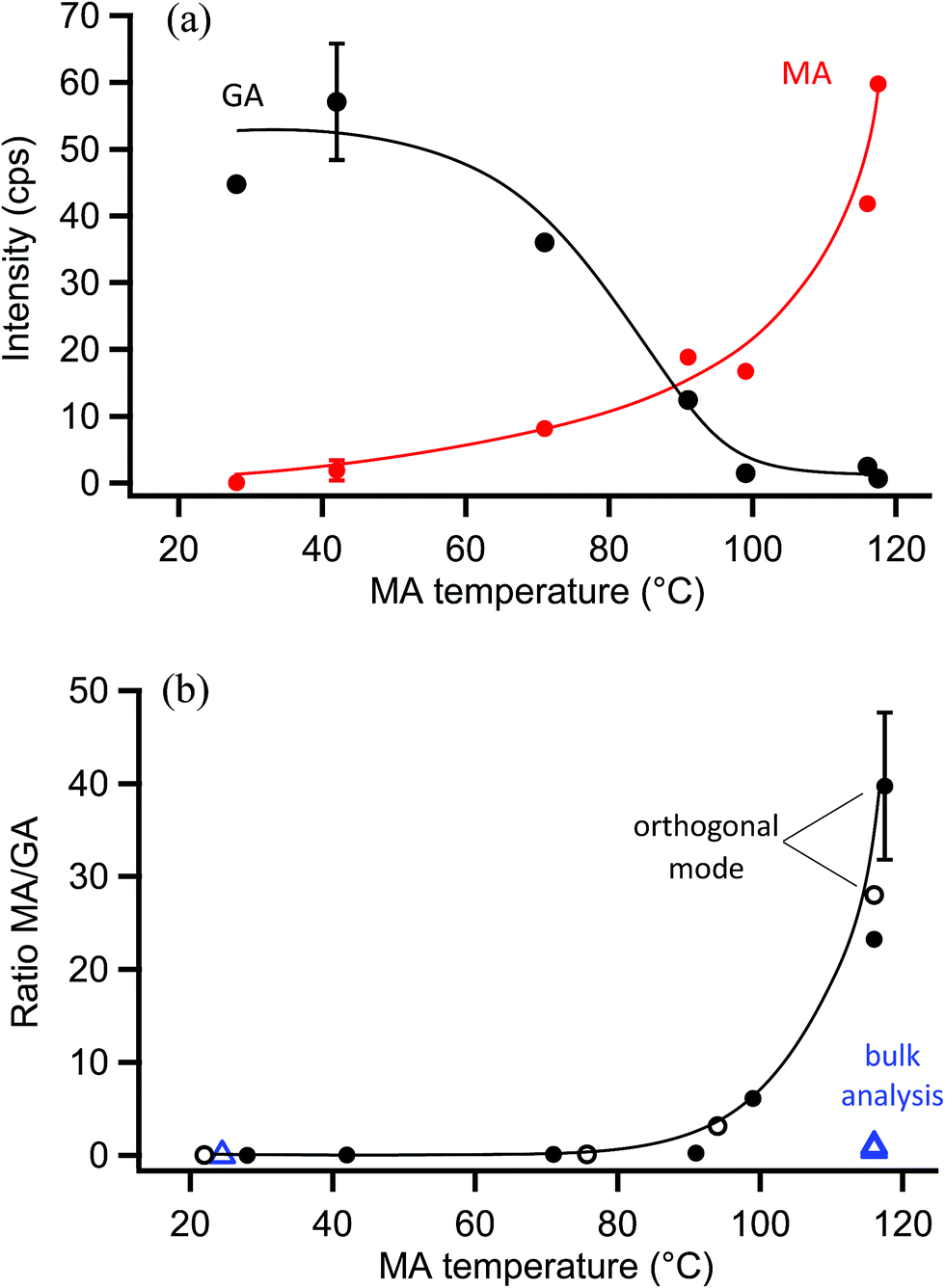 | ||
| Fig. 7 Measurements from orthogonal EASI-MS experiments on monodisperse (250 nm size-selected) glutaric acid (GA) particles coated with malonic acid (MA). (a) Signal intensities from orthogonal mode EASI-MS spectra of 220 nm GA particles (black) coated with MA (red) as a function of MA coating reservoir temperature. MRM intensities were averaged over approximately one minute. (b) Solid circles are the MA/GA ratio calculated from orthogonal EASI-MS data in (a); open circles are MA/GA ratios from another experiment on MA-coated 220 nm GA particles. Blue triangles are ratios from the bulk analysis of collected and extracted MA-coated 220 nm GA particles (250 nm size-selected). Typical 1s standard deviations are included from replicate measurements and are smaller than the markers for the bulk analysis. Solid lines are guidelines only. | ||
Fig. 8 shows measured size distributions of monodisperse GA particles coated with MA, in which the mode diameter increased as a function of MA reservoir temperature. As discussed in the ESI (Fig. S6 and S7†), some particle evaporation resulted in decreases in the expected diameter of bare GA particles of 10–13% relative to the DMA-selected diameter, with larger deviations for smaller selected diameters. Despite this evaporation, bare GA particle diameters were stable and exhibited reproducible increases in diameter as a function of MA coating temperature (Fig. S8†). The largest increase in diameter measured was 54 nm, observed at a reservoir temperature of 117 °C and giving an MA coating thickness of 27 nm. Measurement of MA-coated GA particle diameters using an AAC were in good agreement, with the thickest coatings of ∼33–35 nm measured at 117 °C (Fig. S9†). Because the AAC measures particle aerodynamic diameters, which depend on particle densities, this coating thickness estimate is based on the particle density increasing from that of pure GA (dGA = 1.42 g cm−3) to a density of 1.5 g cm−3. This density is calculated using a volume-weighted ratio of 1:1 MA:GA (dMA = 1.6 g cm−3) as indicated by SMPS measurements of coating thickness and is described in further detail in the ESI.† At these largest MA coating thicknesses estimated to be 27–35 nm, orthogonal EASI-MS spectra show very little spectral signal for the GA core (Fig. 7a).
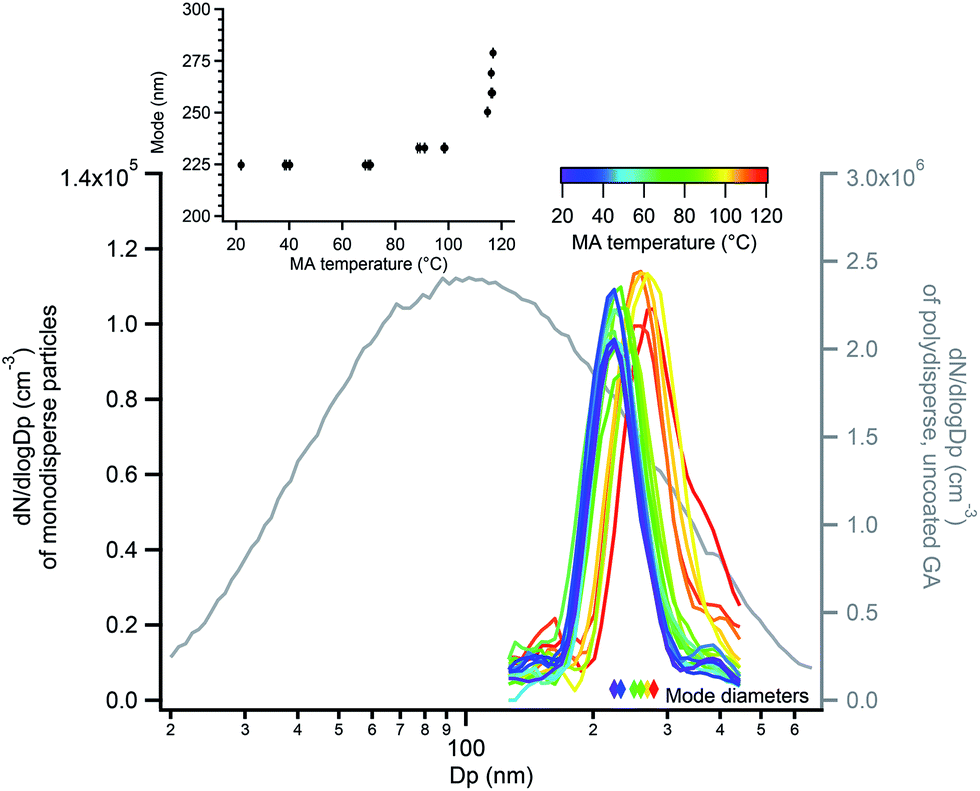 | ||
| Fig. 8 Size distributions of monodisperse 220 nm GA particles coated with MA (left axis), in which the mode diameter increased as a function of MA coating reservoir temperature (inset and colored diamonds). The grey line (right axis) shows the polydisperse GA size distribution measured before size selection. | ||
The thinnest MA coating detectable by SMPS, approximately 4 nm in this diameter range, was detected at 90–100 °C reservoir temperature (Fig. 8 inset). As measured with orthogonal EASI-MS, the GA intensity for monodisperse 220 nm GA particles became undetectable at approximately 90-95 °C when the coating thickness was 4 nm. These measurements suggest that the probe depth of orthogonal EASI-MS is ∼4 nm for the MA/GA system and is quantified further below.
A blank experiment was carried out in which the coating assembly was empty during heating and orthogonal EASI-MS measurements of the particles were made as above. Particles were again size-selected at 250 nm with DMA-1 and measured with SMPS at each temperature before EASI-MS spectra were collected. Fig. S10† demonstrates that there was no evidence of GA decomposition, increased GA evaporation, or particle aggregation as the air stream was heated for coating. Measurements of the temperature of the aerosol stream exiting the coating assembly when the reservoir temperature was 115 °C showed that the particle stream warmed only slightly to 28 °C relative to the room temperature of 22 °C.
An additional experiment was carried out in which smaller, size-selected monodisperse GA particles with 100 nm diameters were coated with MA and measured with EASI-MS in orthogonal mode. Although SMPS measurement of the bare GA particles yielded diameters of 62 nm due to evaporative loss, these diameters were stable and increases in diameter were observed as MA coating temperature was increased (Fig. S11a†). After each diameter measurement, EASI-MS spectra were collected in which GA signals decreased and MA signals increased with MA reservoir temperature and hence thickness (Fig. S11b†). Coating thicknesses up to 22 nm and MA/GA ratios up to 35 (Fig. S11c†) were observed at 105 °C. A bulk measurement of 62 nm GA particles (also size-selected at 100 nm) coated with MA at 105 °C was made from particles collected on a filter and extracted as above. The MA/GA ratio from the bulk measurement was <1, despite possible GA suppression that could increase the MA/GA ratio, again much lower than EASI-MS orthogonal mode ratios (Fig. S11c†).
For orthogonal mode experiments, the MA/GA ratios (∼40, Fig. 7b and S11c†) at the highest reservoir temperature are much higher for both monodisperse 62 nm and 220 nm particles than for coated polydisperse particles where this ratio was ∼3 (Fig. 6). One major difference is the higher particle concentrations in the polydisperse case relative to the monodisperse case. Polydisperse particles had typical surface area concentrations of 2 × 10−3 cm2 cm−3. Note that multiply charged particles with the same electrical mobility are present in both mono- and polydisperse particle samples119 and this total surface area includes these multiply charged particles. The available vapor phase concentration of MA is ∼1.3 × 1011 molecules per cm3 at 30 °C,120 which is the approximate temperature of the aerosol stream at the exit of the coating apparatus at the highest MA coating temperatures. The number of gas phase MA (MW 104 g mol−1) collisions with the particle surfaces available is 8.2 × 1014 collisions per cm2 per s. At a polydisperse residence time of 0.8 s in the coater and a surface area concentration of 2 × 10−3 cm2 cm−3, the number of gas-surface collisions is estimated to be 1 × 1012 per cm3 of air. This is an order of magnitude greater than the concentration of MA (∼1.3 × 1011 molecules per cm3) so that MA may have become depleted, leaving more GA core exposed and hence lower ratios of MA/GA, as observed for polydisperse particles. For the monodisperse particles, the number of gas–particle collisions per cm3 air was at least an order of magnitude smaller, so that significant depletion of MA was less likely.
Monodisperse succinic acid (C4 diacid) particles coated with malonic acid (C3 diacid)
Similar experiments were carried out with 250 nm monodisperse succinic acid (SA) particles coated with MA. The measured diameter of bare SA particles was 241 nm, within the uncertainty of the 250 nm selection by SMPS-2 and consistent with the lower volatility of SA.120,121Fig. 9a shows size distributions of uncoated and coated monodisperse SA particles, along with the polydisperse SA distribution from which they were generated. During coating, the particle mode increased from 241 nm to 259 nm as MA condensed on the monodisperse SA particles, with a maximum particle growth of 18 nm (Fig. 9a inset), equivalent to a coating thickness of 9 nm. EASI-MS MRM spectra were collected for bare and MA-coated SA particles after particle size measurements at each temperature, again using a denuder to remove coating vapors. Fig. 9b shows that the SA intensity decreased and MA intensity increased as a function of MA reservoir temperature. Ratios of MA/SA from these raw signals are shown in Fig. 9c to increase to 20–25 at 9 nm coating, again supporting preferential surface detection for these diacid core–shell particles.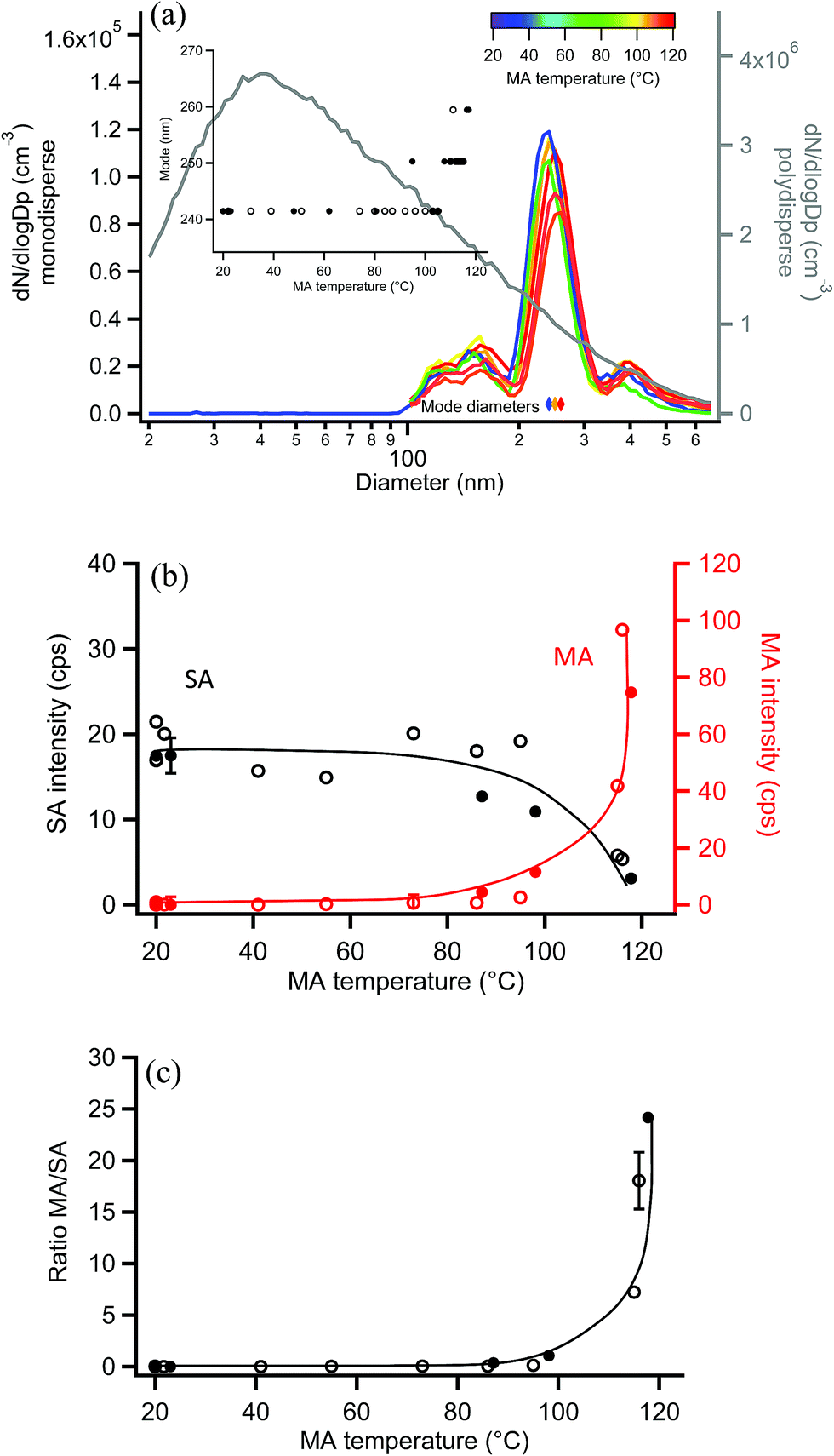 | ||
| Fig. 9 Orthogonal EASI-MS experiment with MA-coated monodisperse 250 nm succinic acid (SA) particles showing (a) selected size distributions in which the mode diameter (inset and colored diamonds) increased as a function of MA coating reservoir temperature. The grey line (right axis) shows the polydisperse SA size distribution measured before size selection. (b) Signal intensities from orthogonal mode EASI-MS spectra of SA particles (black) coated with MA (red) as a function of MA coating temperature. MRM intensities were averaged over approximately one minute. (c) MA/SA ratios calculated from orthogonal EASI-MS data in (b). Solid and open circles represent two repeated experiments in (a)–(c). Error bars represent typical 1s uncertainties. Solid lines are guidelines only. | ||
The measured MA coating is thinner for this odd diacid vapor on the even SA diacid particle than coatings observed for MA on the odd diacid, GA. Alternating odd-even effects in the diacids have been observed in other systems, for example, in the uptake of amine vapors onto diacids,122,123 and in their physical properties such as melting points,123 sublimation temperatures,123 and vapor pressures.120 This may be related to the packing arrangement for even diacids, which has a more stable configuration than for the odd diacids. Odd diacid packing leads to larger spacing between the chains and increased torsional strain123 that may provide opportunities for increased uptake of other odd diacids. As in the coating of MA on GA particles, the minimum increase in coating that was detected on SA particles was 4–5 nm at a reservoir temperature of ∼100 °C, which resulted in a ratio of MA/SA of ∼1.
Monodisperse pimelic acid (C7 diacid) particles coated with malonic acid (C3 diacid)
EASI-MS experiments were also carried out on monodisperse pimelic acid (PA) particles coated with MA, and similar results were obtained. Coating of monodisperse PA particles with MA resulted in the mode diameter increasing from 241 nm to a maximum thickness of 300 nm, as shown in Fig. 10a, giving a maximum coating thickness of 30 nm. Fig. 10b shows the results of EASI-MS orthogonal experiments in which decreases in PA intensity and increases in MA intensity were observed as the MA reservoir temperature and hence coating increased. The resulting MA/PA ratio increased with coating thickness (Fig. 10c) as orthogonal mode EASI-MS probed the outer surface of the particles.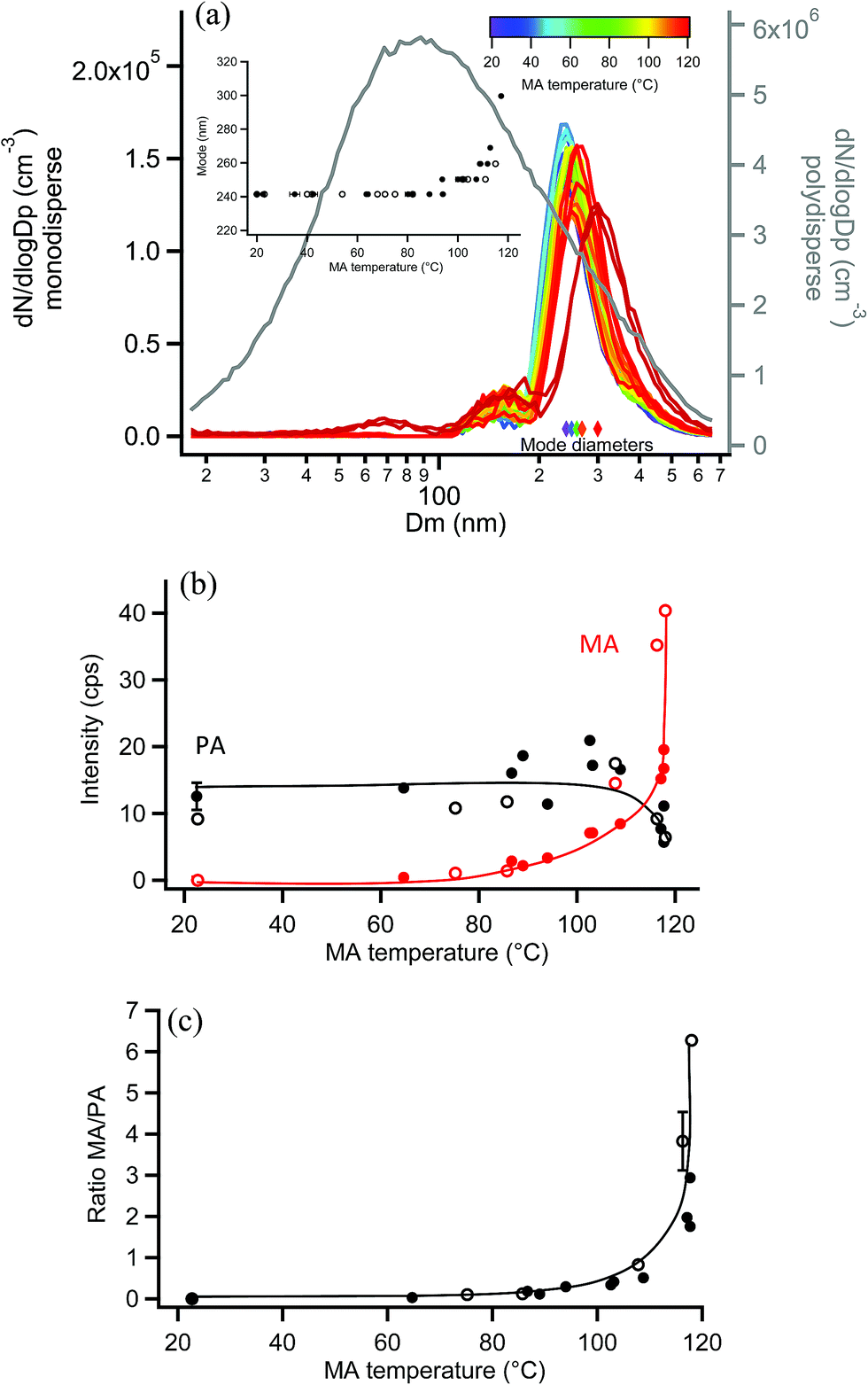 | ||
| Fig. 10 Orthogonal EASI-MS experiment with MA-coated monodisperse 250 nm pimelic acid (PA) particles showing (a) size distributions in which the mode diameter (inset and colored diamonds) increased as a function of MA coating reservoir temperature. The grey line (right axis) shows the polydisperse PA size distribution measured before size selection. (b) Signal intensities from orthogonal mode EASI-MS spectra of PA particles (black) coated with MA (red) as a function of MA coating temperature. MRM intensities were averaged over approximately one minute. (c) MA/PA ratios calculated from orthogonal EASI-MS data in (b). Solid and open circles represent two repeated experiments in (a)–(c). Error bars represent typical 1s uncertainties. Solid lines are guidelines only. | ||
In these experiments on MA-coated PA particles, MA intensities reached comparable levels (i.e., ∼40 cps) relative to MA-coated GA particles, indicating detection of a thick MA coating. The thick odd diacid coatings formed on both odd-carbon PA and GA diacid particles are further evidence that strained packing of the odd diacids allows for increased uptake of MA. However, Fig. 10b shows that PA intensities did not exhibit the same relative decrease as other coated particles following coating. A decrease of only ∼50% in the PA core signal occurred despite the thick, 30 nm MA coating detected. Furthermore, maximum MA/PA ratios of only ∼3–6 were observed, compared to the coating/core ratios of 20–40 for the other diacid systems. The observed low MA/PA ratios may be the result of uneven particle coating, in which the PA core is still partly exposed and probed. Monodisperse PA particle size distributions were broader than other diacid particles and further broadened during coating as shown in Fig. S12.† Such broadening indicates that a large range of coating thicknesses was generated at the highest MA coating temperatures. Additionally, MA islands on the surface may have resulted from the mismatched chain lengths of the C3 and C7 diacids, leaving regions of bare PA. Thus, a non-uniform coating may be responsible for the smaller coating/core ratios for MA-coated PA particles.
Monodisperse succinic acid (C4 diacid) particles coated with oxalic acid (C2 diacid)
As a final system, oxalic acid (OA) was chosen to coat monodisperse SA particles. Measurements by SMPS, shown in Fig. 11a, indicated that OA did not appreciably coat the 250 nm monodisperse SA particles. This is not surprising given the high vapor pressure of OA.120 If a uniform 2 nm coating was formed, resulting in a diameter increase of 4 nm, the coated particle diameter would be detected in the same SMPS diameter bin (the DMA bin width is 9 nm in this 250 nm measurement region so no change in size would be detected). Orthogonal EASI-MS measurements of the SA particles with an undetectable coating (≤2 nm) showed that the SA intensity did not decrease with increasing reservoir temperature and very little OA was detected (after passage through the denuder to remove vapors) until the OA reservoir temperature reached 86–88 °C. Fig. 11b shows the small increases in the OA signal detected at these highest coating temperatures. Fig. 11c shows the corresponding OA/SA ratios, which are clearly much smaller than for any other systems examined here. A thin or partial coating of OA may have formed on SA particles as detected by orthogonal EASI-MS, but since it was not detected with SMPS, the orthogonal method must have a lower limit of ≥2 nm probe depth.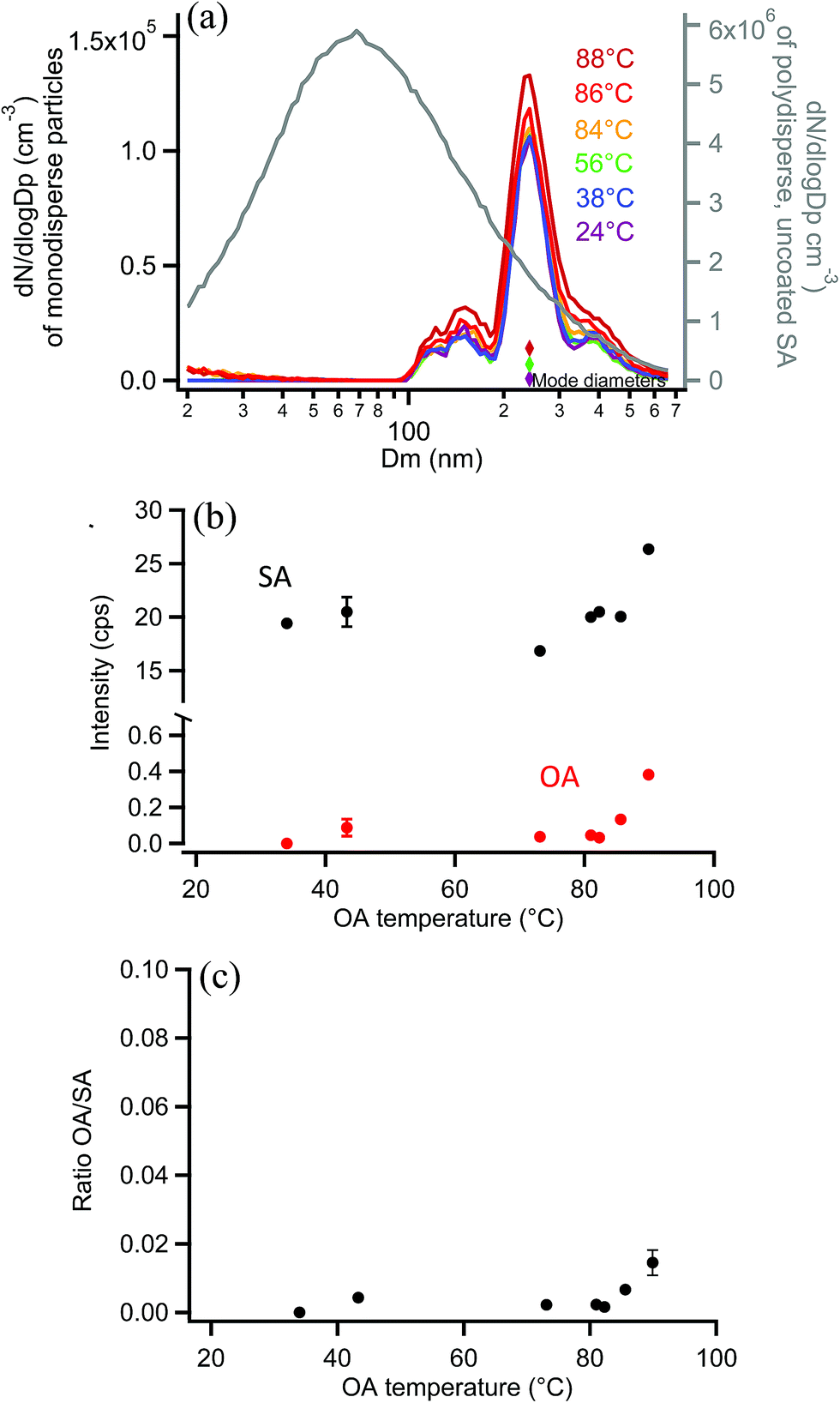 | ||
| Fig. 11 Orthogonal EASI-MS experiment with OA-coated monodisperse 250 nm succinic acid (SA) particles showing (a) size distributions in which the mode diameter (colored diamonds) remained constant as a function of OA coating reservoir temperature. The grey line (right axis) shows the polydisperse SA size distribution measured before size selection. (b) Signal intensities from orthogonal mode EASI-MS spectra of SA particles (black) and OA (red) as a function of OA temperature. MRM intensities were averaged over approximately one minute. (c) OA/SA ratios calculated from orthogonal EASI-MS data in (b). Error bars represent typical 1s uncertainties. | ||
Comparison with on-line surface sensitive methods
A few analytical methods have been demonstrated to allow surface sensitive analysis of particles in real time. For example, one useful ambient ionization technique that has allowed surface sensitive detection is DART-MS, which also requires little sample preparation.71,124 DART has been shown to have a probe depth of 1–30 nm on a variety of organic particles, depending on their phase, viscosity, and vapor pressures of the components.29,30,78,79 The use of heated gas streams in DART studies (typically 350 °C) to increase sensitivity by increasing desorption into the gas phase, can cause thermal decomposition, increased fragmentation, or chemical reactions that change the identity of the sample.71,124 While lower temperature DART measurements can be made to avoid introducing artifacts, this decreases sensitivity. In addition, OH radicals can be generated in this source and react with the analyte.In a previous study in this laboratory, EESI-MS measurements of MA-coated polydisperse GA particles resulted in a MA/GA ratio of 1.7 at the thickest coatings.91 Ratios of surface/core observed with EASI-MS obtained for GA particles with the thickest MA coatings here were similar, MA/GA ∼1–3.5. There are additional complexities associated with EESI-MS.91 For example, the voltage applied to the EESI spray capillary affects the stability of the solvent spray cone, and hence signal intensity, and may need to be varied with distance from the MS inlet. Additionally, the proximity of the EESI capillary with the sample particle flow may cause the capillary to become contaminated with sample and block or break the fragile tip. EASI-MS has a significant advantage in that no voltage is needed to generate charged droplets with a range of sizes.
A major advantage of EASI-MS is its ability to probe either the surface or the bulk of particles in real time with little to no sample preparation. This is carried out through relatively easy changes in nebulizer orientation and solvent flow rate. Probe depth is likely to depend to some degree on the ionization efficiency and instrumental sensitivity to specific compounds, and possibly the surface free energy of the particles.91 With the probe depths of 2–4 nm demonstrated here, the orthogonal EASI-MS method is remarkably surface sensitive for these core–shell diacid systems.
Conclusions
Easy ambient sonic-spray ionization mass spectrometry (EASI-MS), applied to a separate stream of solid, laboratory-generated particles, was demonstrated to be useful in two modes, providing either bulk or preferentially surface composition depending on the experimental configuration. Droplet mode EASI-MS efficiently extracts particles into the solvent to provide bulk analysis, as confirmed by comparison to analysis of the same particles collected, extracted, and analyzed by including the extract in the nebulizer solution. On the other hand, orthogonal mode EASI-MS generates ion signals preferentially from particle surfaces. Orthogonal mode EASI-MS analysis of monodisperse core–shell particles with known coating thicknesses enabled quantification of the probe depth, which is shown to be 2–4 nm. This is in contrast to the full dissolution that occurs for droplet mode and demonstrates that EASI-MS is a useful surface-sensitive technique for analysis of inhomogeneous airborne particles.Probe depths of a few nm are desirable for studies of laboratory-generated and ambient organic particles, since there is increasing evidence of surface composition that differs from the composition that nucleates the particles.33–35,52,125 Knowledge of the difference between surface and bulk composition of ambient and laboratory-generated particles is central in understanding their growth, CCN activity, heterogeneous chemistry, and interactions with biological surfaces leading to health effects.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Army Research Office (Grant #W911NF1710105), NSF (Grant #1647386) and the NSF Major Research Instrumentation (MRI) program (Grant #1337080). The authors thank J. C. Meyer and the UC Irvine glassblowing shop, C. Nickolaus of Cambustion, Ltd. (Cambridge, UK) and M. Irwin for facilitating the loan of the aerodynamic aerosol classifier, and the Nizkorodov group for use of their SMPS. We also thank Professor J. P. D. Abbatt for helpful discussions and Victor Griganavicius (Waters) for mass spectrometer support.References
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prevot, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Science, 2009, 326, 1525–1529 CrossRef CAS.
- D. K. Farmer, C. D. Cappa and S. M. Kreidenweis, Chem. Rev., 2015, 115, 4199–4217 CrossRef CAS.
- C. George, M. Ammann, B. D'Anna, D. J. Donaldson and S. A. Nizkorodov, Chem. Rev., 2015, 115, 4218–4258 CrossRef CAS.
- T. Moise, J. M. Flores and Y. Rudich, Chem. Rev., 2015, 115, 4400–4439 CrossRef CAS.
- B. Nozière, M. Kalberer, M. Claeys, J. Allan, B. D'Anna, S. Decesari, E. Finessi, M. Glasius, I. Grgić, J. F. Hamilton, T. Hoffmann, Y. Iinuma, M. Jaoui, A. Kahnt, C. J. Kampf, I. Kourtchev, W. Maenhaut, N. Marsden, S. Saarikoski, J. Schnelle-Kreis, J. D. Surratt, S. Szidat, R. Szmigielski and A. Wisthaler, Chem. Rev., 2015, 115, 3919–3983 CrossRef.
- U. Pöschl and M. Shiraiwa, Chem. Rev., 2015, 115, 4440–4475 CrossRef PubMed.
- R. Zhang, G. Wang, S. Guo, M. L. Zamora, Q. Ying, Y. Lin, W. Wang, M. Hu and Y. Wang, Chem. Rev., 2015, 115, 3803–3855 CrossRef CAS PubMed.
- B. J. Finlayson-Pitts and J. N. Pitts Jr, Chemistry of the Upper and Lower Atmosphere - Theory, Experiments, and Applications, Academic Press, San Diego, 2000 Search PubMed.
- J. C. Chow, P. Doraiswamy, J. G. Watson, L. W. A. Chen, S. S. H. Ho and D. A. Sodeman, J. Air Waste Manage. Assoc., 2008, 58, 141–163 CrossRef CAS.
- K. A. Pratt and K. A. Prather, Mass Spectrom. Rev., 2012, 31, 1–16 CrossRef CAS.
- A. Laskin, J. Laskin and S. A. Nizkorodov, Environ. Chem., 2012, 9, 163–189 CrossRef CAS.
- J. Laskin, A. Laskin and S. A. Nizkorodov, Anal. Chem., 2018, 90, 166–189 CrossRef CAS.
- D. G. Nash, T. Baer and M. V. Johnston, Int. J. Mass Spectrom., 2006, 258, 2–12 CrossRef CAS.
- K. A. Pratt and K. A. Prather, Mass Spectrom. Rev., 2012, 31, 17–48 CrossRef CAS.
- J. Laskin, A. Laskin and S. A. Nizkorodov, Int. Rev. Phys. Chem., 2013, 32, 128–170 Search PubMed.
- C. Zuth, A. L. Vogel, S. Ockenfeld, R. Huesmann and T. Hoffmann, Anal. Chem., 2018, 90, 8816–8823 CrossRef CAS.
- H. J. Tobias and P. J. Ziemann, Environ. Sci. Technol., 2000, 34, 2105–2115 CrossRef CAS.
- J. N. Smith, K. F. Moore, P. H. McMurry and F. L. Eisele, Aerosol Sci. Technol., 2004, 38, 100–110 CrossRef CAS.
- J. L. Jimenez, J. T. Jayne, Q. Shi, C. E. Kolb, D. R. Worsnop, I. Yourshaw, J. H. Seinfeld, R. C. Flagan, X. F. Zhang, K. A. Smith, J. W. Morris and P. Davidovits, J. Geophys. Res., 2003, 108, 8425 CrossRef.
- J. G. Slowik, K. Stainken, P. Davidovits, L. R. Williams, J. T. Jayne, C. E. Kolb, D. R. Worsnop, Y. Rudich, P. F. DeCarlo and J. L. Jimenez, Aerosol Sci. Technol., 2004, 38, 1206–1222 CrossRef CAS.
- E. Gard, J. E. Mayer, B. D. Morrical, T. Dienes, D. P. Fergenson and K. A. Prather, Anal. Chem., 1997, 69, 4083–4091 CrossRef CAS.
- A. Zelenyuk and D. Imre, Aerosol Sci. Technol., 2005, 39, 554–568 CrossRef CAS.
- S. Wang, C. A. Zordan and M. V. Johnston, Anal. Chem., 2006, 78, 1750–1754 CrossRef CAS.
- A. Zelenyuk and D. Imre, Int. Rev. Phys. Chem., 2009, 28, 309–358 Search PubMed.
- D. M. Murphy, D. J. Cziczo, K. D. Froyd, P. K. Hudson, B. M. Matthew, A. M. Middlebrook, R. E. Peltier, A. Sullivan, D. S. Thomson and R. J. Weber, J. Geophys. Res., 2006, 111, D23S32 Search PubMed.
- K. A. Prather, C. D. Hatch and V. H. Grassian, Annu. Rev. Anal. Chem., 2008, 1, 485–514 CrossRef CAS.
- A. Zelenyuk, J. Yang, C. Song, R. A. Zaveri and D. Imre, J. Phys. Chem. A, 2008, 112, 669–677 CrossRef CAS.
- M. N. Chan, T. Nah and K. R. Wilson, Analyst, 2013, 138, 3749–3757 RSC.
- T. Nah, M. Chan, S. R. Leone and K. R. Wilson, Anal. Chem., 2013, 85, 2087–2095 CrossRef CAS.
- E. Woods, G. D. Smith, R. E. Miller and T. Baer, Anal. Chem., 2002, 74, 1642–1649 CrossRef CAS.
- K. Jorabchi and L. M. Smith, Anal. Chem., 2009, 81, 9682–9688 CrossRef CAS.
- M. Ehn, E. Kleist, H. Junninen, T. Petaja, G. Lonn, S. Schobesberger, M. Dal Maso, A. Trimborn, M. Kulmala, D. R. Worsnop, A. Wahner, J. Wildt and T. F. Mentel, Atmos. Chem. Phys., 2012, 12, 5113–5127 CrossRef CAS.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipila, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann, B. Lee, F. Lopez-Hilfiker, S. Andres, I. H. Acir, M. Rissanen, T. Jokinen, S. Schobesberger, J. Kangasluoma, J. Kontkanen, T. Nieminen, T. Kurten, L. B. Nielsen, S. Jorgensen, H. G. Kjaergaard, M. Canagaratna, M. Dal Maso, T. Berndt, T. Petaja, A. Wahner, V. M. Kerminen, M. Kulmala, D. R. Worsnop, J. Wildt and T. F. Mentel, Nature, 2014, 506, 476–479 CrossRef CAS PubMed.
- T. Jokinen, T. Berndt, R. Makkonen, V. M. Kerminen, H. Junninen, P. Paasonen, F. Stratmann, H. Herrmann, A. B. Guenther, D. R. Worsnop, M. Kulmala, M. Ehn and M. Sipila, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7123–7128 CrossRef CAS.
- P. M. Winkler, J. Ortega, T. Karl, L. Cappellin, H. R. Friedli, K. Barsanti, P. H. McMurry and J. N. Smith, Geophys. Res. Lett., 2012, 39, 6 CrossRef.
- J. Zhao, J. Ortega, M. Chen, P. H. McMurry and J. N. Smith, Atmos. Chem. Phys., 2013, 13, 7631–7644 CrossRef.
- C. Kidd, V. Perraud and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2014, 16, 22706–22716 RSC.
- Y. Zhao, L. M. Wingen, V. Perraud and B. J. Finlayson-Pitts, Atmos. Chem. Phys., 2016, 16, 3245–3264 CrossRef CAS.
- E. Abramson, D. Imre, J. Beranek, J. Wilson and A. Zelenyuk, Phys. Chem. Chem. Phys., 2013, 15, 2983–2991 RSC.
- M. Kuwata and S. T. Martin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17354–17359 CrossRef CAS.
- L. Renbaum-Wolff, J. W. Grayson, A. P. Bateman, M. Kuwata, M. Sellier, B. J. Murray, J. E. Shilling, S. T. Martin and A. K. Bertram, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8014–8019 CrossRef CAS PubMed.
- E. Saukko, H. Kuuluvainen and A. Virtanen, Atmos. Meas. Tech., 2012, 5, 259–265 CrossRef.
- E. Saukko, A. T. Lambe, P. Massoli, T. Koop, J. P. Wright, D. R. Croasdale, D. A. Pedernera, T. B. Onasch, A. Laaksonen, P. Davidovits, D. R. Worsnop and A. Virtanen, Atmos. Chem. Phys., 2012, 12, 7517–7529 CrossRef CAS.
- T. D. Vaden, D. Imre, J. Beranek, M. Shrivastava and A. Zelenyuk, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2190–2195 CrossRef CAS.
- A. Virtanen, J. Joutsensaari, T. Koop, J. Kannosto, P. Yli-Pirila, J. Leskinen, J. M. Makela, J. K. Holopainen, U. Poschl, M. Kulmala, D. R. Worsnop and A. Laaksonen, Nature, 2010, 467, 824–827 CrossRef CAS.
- A. Virtanen, J. Kannosto, H. Kuuluvainen, A. Arffman, J. Joutsensaari, E. Saukko, L. Hao, P. Yli-Pirila, P. Tiitta, J. K. Holopainen, J. Keskinen, D. R. Worsnop, J. N. Smith and A. Laaksonen, Atmos. Chem. Phys., 2011, 11, 8759–8766 CrossRef CAS.
- C. Kidd, V. Perraud, L. M. Wingen and B. J. Finlayson-Pitts, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7552–7557 CrossRef CAS.
- C. Pfrang, M. Shiraiwa and U. Poschl, Atmos. Chem. Phys., 2011, 11, 7343–7354 CrossRef CAS.
- M. Shiraiwa, M. Ammann, T. Koop and U. Poschl, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11003–11008 CrossRef CAS PubMed.
- M. Shiraiwa and J. H. Seinfeld, Geophys. Res. Lett., 2012, 39, L24801 CrossRef.
- C. Denjean, P. Formenti, B. Picquet-Varrault, E. Pangui, P. Zapf, Y. Katrib, C. Giorio, A. Tapparo, A. Monod, B. Temime-Roussel, P. Decorse, C. Mangeney and J. F. Doussin, Atmos. Chem. Phys., 2015, 15, 3339–3358 CrossRef CAS.
- D. F. Zhao, A. Buchholz, B. Kortner, P. Schlag, F. Rubach, A. Kiendler-Scharr, R. Tillmann, A. Wahner, J. M. Flores, Y. Rudich, A. K. Watne, M. Hallquist, J. Wildt and T. F. Mentel, Geophys. Res. Lett., 2015, 42, 10920–10928 CrossRef CAS.
- A. Muller, Y. Miyazaki, S. G. Aggarwal, Y. Kitamori, S. K. R. Boreddy and K. Kawamura, J. Geophys. Res.: Atmos., 2017, 122, 9301–9318 Search PubMed.
- E. O. Fors, E. Swietlicki, B. Svenningsson, A. Kristensson, G. P. Frank and M. Sporre, Atmos. Chem. Phys., 2011, 11, 8343–8361 CrossRef CAS.
- A. Gray Bé, M. A. Upshur, P. Liu, S. T. Martin, F. M. Geiger and R. J. Thomson, ACS Cent. Sci., 2017, 3, 715–725 CrossRef.
- R. M. Harrison and J. Yin, Sci. Total Environ., 2000, 249, 85–101 CrossRef CAS.
- G. Oberdörster, E. Oberdörster and J. Oberdörster, Environ. Health Perspect., 2005, 113, 823–839 CrossRef PubMed.
- S. E. Mancebo and S. Q. Wang, J. Eur. Acad. Dermatol. Venereol., 2015, 29, 2326–2332 CrossRef CAS PubMed.
- R. G. Cooks, Z. Ouyang, Z. Takats and J. M. Wiseman, Science, 2006, 311, 1566 CrossRef CAS.
- Z. Takáts, J. M. Wiseman, B. Gologan and R. G. Cooks, Science, 2004, 306, 471–473 CrossRef PubMed.
- A. Venter, M. Nefliu and G. R. Cooks, Trends Anal. Chem., 2008, 27, 284–290 CrossRef CAS.
- H. Chen, G. Gamez and R. Zenobi, J. Am. Soc. Mass Spectrom., 2009, 20, 1947–1963 CrossRef CAS.
- C. N. McEwen and S. Trimpin, Int. J. Mass Spectrom., 2011, 300, 167–172 CrossRef CAS.
- M. Z. Huang, S. C. Cheng, Y. T. Cho and J. Shiea, Anal. Chim. Acta, 2011, 702, 1–15 CrossRef CAS PubMed.
- G. A. Harris, A. S. Galhena and F. M. Fernandez, Anal. Chem., 2011, 83, 4508–4538 CrossRef CAS PubMed.
- M. E. Monge, G. A. Harris, P. Dwivedi and F. M. Fernández, Chem. Rev., 2013, 113, 2269–2308 CrossRef CAS PubMed.
- P. M. Peacock, W.-J. Zhang and S. Trimpin, Anal. Chem., 2017, 89, 372–388 CrossRef CAS PubMed.
- R. M. Alberici, R. C. Simas, G. B. Sanvido, W. Romão, P. M. Lalli, M. Benassi, I. B. Cunha and M. N. Eberlin, Anal. Bioanal. Chem., 2010, 398, 265–294 CrossRef CAS.
- C. Wu, A. L. Dill, L. S. Eberlin, R. G. Cooks and D. R. Ifa, Mass Spectrom. Rev., 2013, 32, 218–243 CrossRef CAS PubMed.
- M. Domin and R. Cody, Ambient Ionization Mass Spectrometry, Royal Society of Chemistry, England, 2015 Search PubMed.
- E. T. Jansson, M. T. Dulay and R. N. Zare, Anal. Chem., 2016, 88, 6195–6198 CrossRef CAS PubMed.
- C. N. McEwen, R. G. McKay and B. S. Larsen, Anal. Chem., 2005, 77, 7826–7831 CrossRef CAS PubMed.
- C. McEwen and S. Gutteridge, J. Am. Soc. Mass Spectrom., 2007, 18, 1274–1278 CrossRef CAS PubMed.
- F. Zydel, S. Trimpin and C. N. McEwen, J. Am. Soc. Mass Spectrom., 2010, 21, 1889–1892 CAS.
- E. A. Bruns, V. Perraud, J. Greaves and B. J. Finlayson-Pitts, Anal. Chem., 2010, 82, 5922–5927 CrossRef CAS PubMed.
- R. B. Cody, J. A. Laramée, J. M. Nilles and H. D. Durst, JEOL News, 2005, 40, 8–12 Search PubMed.
- J. F. Davies and K. R. Wilson, Chem. Sci., 2015, 6, 7020–7027 RSC.
- Y. Zhao, M. C. Fairhurst, L. M. Wingen, V. Perraud, M. J. Ezell and B. J. Finlayson-Pitts, Atmos. Meas. Tech., 2017, 10, 1373–1386 CrossRef CAS.
- K. Z. Aregahegn, D. Shemesh, R. B. Gerber and B. J. Finlayson-Pitts, Environ. Sci. Technol., 2017, 51, 2660–2668 CrossRef CAS PubMed.
- A. K. Badu-Tawiah, L. S. Eberlin, Z. Ouyang and G. R. Cooks, Annu. Rev. Phys. Chem., 2013, 64, 481–505 CrossRef CAS PubMed.
- P. J. Roach, J. Laskin and A. Laskin, Analyst, 2010, 135, 2233–2236 RSC.
- C. M. Hong, F. C. Tsai and J. Shiea, Anal. Chem., 2000, 72, 1175–1178 CrossRef CAS PubMed.
- C. Y. Lee and J. Shiea, Anal. Chem., 1998, 70, 2757–2761 CrossRef CAS PubMed.
- H. Chen, A. Venter and R. G. Cooks, Chem. Commun., 2006, 19, 2042–2044 RSC.
- W. S. Law, R. Wang, B. Hu, C. Berchtold, L. Meier, H. Chen and R. Zenobi, Anal. Chem., 2010, 82, 4494–4500 CrossRef CAS PubMed.
- L. Meier, S. Schmid, C. Berchtold and R. Zenobi, Eur. J. Mass Spectrom., 2011, 17, 345 CrossRef CAS PubMed.
- L. A. Doezema, T. Longin, W. Cody, V. Perraud, M. L. Dawson, M. J. Ezell, J. Greaves, K. R. Johnson and B. J. Finlayson-Pitts, RSC Adv., 2012, 2, 2930–2938 RSC.
- A. J. Horan, Y. Gao, W. A. Hall IV and M. V. Johnston, Anal. Chem., 2012, 84, 9253–9258 CrossRef CAS PubMed.
- P. J. Gallimore and M. Kalberer, Environ. Sci. Technol., 2013, 41, 7324–7331 CrossRef PubMed.
- S. Kumbhani, T. Longin, L. M. Wingen, C. Kidd, V. Perraud and B. J. Finlayson-Pitts, Anal. Chem., 2018, 90, 2055–2062 CrossRef CAS PubMed.
- P. J. Gallimore, C. Giorio, B. M. Mahon and M. Kalberer, Atmos. Chem. Phys., 2017, 17, 14485–14500 CrossRef CAS.
- A. Hirabayashi, M. Sakairi and H. Koizumi, Anal. Chem., 1994, 66, 4557–4559 CrossRef CAS.
- A. Hirabayashi, M. Sakairi and H. Koizumi, Anal. Chem., 1995, 67, 2878–2882 CrossRef CAS PubMed.
- R. Haddad, R. Sparrapan and M. N. Eberlin, Rapid Commun. Mass Spectrom., 2006, 20, 2901–2905 CrossRef CAS PubMed.
- S. F. Teunissen, A. Fernandes, M. N. Eberlin and R. M. Alberici, Trends Anal. Chem., 2017, 90, 135–141 CrossRef CAS.
- A. J. Ingram, C. L. Boeser and R. N. Zare, Chem. Sci., 2016, 7, 39–55 RSC.
- S. F. Teunissen and M. N. Eberlin, J. Am. Soc. Mass Spectrom., 2017, 28, 2255–2261 CrossRef CAS PubMed.
- D. Schuetzle, D. Cronn, A. L. Crittenden and R. J. Charlson, Environ. Sci. Technol., 1975, 9, 838–845 CrossRef CAS.
- R. J. O'Brien, J. H. Crabtree, J. R. Holmes, M. C. Hoggan and A. H. Bockian, Environ. Sci. Technol., 1975, 9, 577–582 CrossRef.
- Y. Yokouchi and Y. Ambe, Atmos. Environ., 1986, 20, 1727–1734 CrossRef CAS.
- K. Kawamura and I. R. Kaplan, Environ. Sci. Technol., 1987, 21, 105–110 CrossRef CAS.
- K. Kawamura and K. Ikushima, Environ. Sci. Technol., 1993, 27, 2227–2235 CrossRef CAS.
- K. Kawamura and K. Usukura, J. Oceanogr., 1993, 49, 271–283 CrossRef CAS.
- R. Sempéré and K. Kawamura, Atmos. Environ., 1994, 28, 449–459 CrossRef.
- K. Kawamura, H. Kasukabe, O. Yasui and L. Barrie, Geophys. Res. Lett., 1995, 22, 1253–1256 CrossRef CAS.
- A. Chebbi and P. Carlier, Atmos. Environ., 1996, 30, 4233–4249 CrossRef CAS.
- K. Kawamura, R. Seméré, Y. Imai, Y. Fujii and M. Hayashi, J. Geophys. Res.: Atmos., 1996, 101, 18721–18728 CrossRef CAS.
- E. D. Baboukas, M. Kanakidou and N. Mihalopoulos, J. Geophys. Res.: Atmos., 2000, 105, 14459–14471 CrossRef CAS.
- A. Röhrl and G. Lammel, Chemosphere, 2002, 46, 1195–1199 CrossRef.
- M. Narukawa, K. Kawamura, K. Okada, Y. Zaizen and Y. Makino, Tellus B, 2011, 55, 777–786 CrossRef.
- K. Kawamura and S. Bikkina, Atmos. Res., 2016, 170, 140–160 CrossRef CAS.
- M. M. Antonakis, A. Tsirigotaki, K. Kanaki, C. J. Milios and S. A. Pergantis, J. Am. Soc. Mass Spectrom., 2013, 24, 1250–1259 CrossRef CAS PubMed.
- R. M. Alberici, P. H. Vendramini and M. N. Eberlin, Anal. Methods, 2017, 9, 5029–5036 RSC.
- A. Özdemir, J.-L. Lin, Y. S. Wang and C.-H. Chen, RSC Adv., 2014, 4, 61290–61297 RSC.
- J. S. Grossert, P. D. Fancy and R. L. White, Can. J. Chem., 2005, 83, 1878–1890 CrossRef CAS.
- C. Kidd, V. Perraud and B. J. Finlayson-Pitts, Atmos. Environ., 2014, 82, 56–59 CrossRef CAS.
- S. H. Yalkowsky, Y. He and P. Jain, Handbook of Aqueous Solubility Data, CRC Press, Boca Raton, 2nd edn, 2010 Search PubMed.
- W. C. Hinds, Aerosol Technology: Properties, Behavior, and Measurement of Airborne Particles, John Wiley & Sons, Inc., 1999 Search PubMed.
- M. Bilde, K. Barsanti, M. Booth, C. D. Cappa, N. M. Donahue, E. U. Emanuelsson, G. McFiggans, U. K. Krieger, C. Marcolli, D. Tropping, P. Ziemann, M. Barley, S. Clegg, B. Dennis-Smither, M. Hallquist, A. M. Hallquist, A. Khlystov, M. Kulmala, D. Mogensen, C. J. Percival, F. Pope, J. P. Reid, M. da Silva, T. Rosenoern, K. Salo, V. P. Soonsin, T. Yli-Juuti, N. L. Prisle, J. Pagels, J. Rarey, A. A. Zardini and I. Riipinen, Chem. Rev., 2015, 115, 4115–4156 CrossRef CAS PubMed.
- C. D. Cappa, E. R. Lovejoy and A. R. Ravishankara, J. Phys. Chem. A, 2007, 111, 3099–3109 CrossRef CAS PubMed.
- M. C. Fairhurst, M. J. Ezell, C. Kidd, P. S. J. Lakey, M. Shiraiwa and B. J. Finlayson-Pitts, Phys. Chem. Chem. Phys., 2017, 19, 4827–4839 RSC.
- V. R. Thalladi, M. Nüsse and R. Boese, J. Am. Chem. Soc., 2000, 122, 9227–9236 CrossRef CAS.
- Y. Dong, Direct Analysis in Real Time Mass Spectrometry: Principles and Practices of DART-MS, Wiley, 2017 Search PubMed.
- A. C. Vander Wall, P. S. J. Lakey, E. Rossich-Molina, V. Perraud, L. M. Wingen, J. Xu, D. Soulsby, R. B. Gerber, M. Shiraiwa and B. J. Finlayson-Pitts, Environ. Sci.: Processes Impacts, 2018 10.1039/c8em00348c.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03851a |
| This journal is © The Royal Society of Chemistry 2019 |