
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRealization of high-power-efficiency white electroluminescence from a single polymer by energy-level engineering†
Shiyang
Shao
a,
Shumeng
Wang
a,
Xiushang
Xu
ab,
Yun
Yang
ab,
Jianhong
Lv
a,
Junqiao
Ding
 *a,
Lixiang
Wang
*a,
Xiabin
Jing
a and
Fosong
Wang
a
*a,
Lixiang
Wang
*a,
Xiabin
Jing
a and
Fosong
Wang
a
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: junqiaod@ciac.ac.cn; lixiang@ciac.ac.cn
bUniversity of the Chinese Academy of Sciences, Beijing 100039, P. R. China
First published on 19th September 2018
Abstract
Single white light-emitting polymers (SWPs) represent a high-fidelity system for generating white light emission from polymers without phase separation compared to polymer blend systems. However, their device performance so far has been limited because of the unwanted hole scattering caused by an energy-level mismatch between emitters and hosts, and the large injection barrier at the polymer/anode interface. Here, we report novel poly(arylene phosphine oxide)-based all-phosphorescent SWPs by using the combination of a high-HOMO-level blue phosphor and high-HOMO-level poly(arylene phosphine oxide) host to achieve a low turn-on voltage of 2.8 V, high external quantum efficiency of 18.0% and remarkable power efficiency of 52.1 lm W−1, which makes them the most efficient SWPs so far. This record power efficiency is realized by using the high-HOMO-level blue phosphor to eliminate the hole scattering effect and by using the high-HOMO-level polymer host to reduce the hole injection barrier. This result represents an important progress in SWPs to achieve efficiency surpassing that of the polymer blends currently used for solution-processed white organic light-emitting diodes (WOLEDs) and even comparable with that of the small molecules used for vacuum-deposited WOLEDs.
Introduction
White organic light-emitting diodes (WOLEDs) have continued to attract the attention of researchers owing to their application in light sources and full-color displays by coupling with color filters.1–3 In the past few years, lots of efforts have been devoted to small-molecule WOLEDs relying on vacuum-deposition technologies, with their power efficiency (PE) now comparable with those of fluorescent tubes (60–100 lm W−1).4–6 Nevertheless, WOLEDs based on polymers, which can be fabricated by low-cost solution-processing methods such as ink-jet printing and roll-to-roll printing,3,7–14 are less developed. In particular, their PE still lags far behind those of their small-molecule counterparts because of the relatively high driving voltages and low external quantum efficiencies.The conventional approach for white electroluminescent polymers uses blend systems, e.g. polymer/polymer15 or polymer/small molecules16,17 with complementary colors (blue and yellow, or blue, green and red), to cover the whole visible region. These multi-component systems inherently suffer from phase separation of different emitters which leads to deterioration of device efficiency and an undesired voltage- and/or time-induced color shift.18,19 An alternative approach is to develop single white light-emitting polymers (SWPs) by covalently incorporating complementary emissive species into one macromolecule, which can produce broad-band emissions from all the components via partial energy transfer from high-energy (blue) species to low-energy (yellow, or green and red) ones.20 Because all the emissive species are chemically dispersed in the polymer, the risk of phase separation can be avoided. Therefore, SWPs are envisioned as a high-fidelity system compared to polymer blends.
Since the first report of SWPs with individual blue and yellow fluorescence emission in 2004,21 a number of SWPs containing two22–27 or three colors28–32 have been reported. Particularly, incorporating phosphorescent emitters to harvest triplet excitons has been an important advance in boosting the device efficiency. For example, Ir(III),23,24,26,28 Pt(II)33 and Os(II) complexes31 have been introduced into polyfluorenes to produce fluorescent/phosphorescent hybrid SWPs exhibiting the best external quantum efficiency (EQE) of 5.4%.31 Additionally, all-phosphorescent SWPs reported have been synthesized by incorporating both blue and yellow phosphors into one polymer host, revealing EQEs up to 7.1%.23,24 Despite these progresses, the device performance of SWPs still lags behind that of polymer blends and cannot compete with that of small molecules—including the driving voltage, EQE and power efficiency (PE).
From the materials design aspect, to develop high-efficiency all-phosphorescent SWPs, there are at least two major challenges to be addressed. First, there lacks a polymer host that possesses both a high triplet energy (ET, >2.70 eV) and high HOMO level close to the Fermi level of the anode. For efficient white light emission, a high-ET host is required to prevent back energy transfer from the blue phosphor to the host.34 However, the high ET tends to result in a wide energy gap, leading to a low HOMO level and thus a large hole-injection barrier. For instance, the commonly used high-ET host poly(9-vinylcarbazole) (PVK) shows a HOMO level of −5.90 eV,35 giving an energy barrier of ∼0.7 eV for hole injection from the indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene):polystyrene sulfonate (PEDOT:PSS) anode (−5.20 eV) to the polymer. Second, there is a mismatch of energy levels between the blue phosphor and the polymer host, which brings about unwanted charge trapping or charge scattering effects for SWPs depending on the relative position of the HOMO/LUMO levels of the dopant to those of the host. Generally, if the HOMO/LUMO levels of the phosphor fall within those of the host, charge trapping will take place. In contrast, if the phosphor has a lower-lying HOMO or higher-lying LUMO, hole scattering or electron scattering occurs. Both these processes will lead to a decrease of current density and an increase of driving voltage for devices36 and therefore are undesired for SWPs. These effects are critical for blue phosphors because their content in SWPs is at a relatively high level (>5 mol%).23,24,26 However, it is known that the benchmark blue phosphor bis(2-(4,6-difluorophenyl)pyridine)(picolinate)iridium(III) (FIrpic) shows a low HOMO level of −5.64 eV, so hole scattering can occur if a high-HOMO-level polymer host is used. Therefore, regulating the energy level of the blue phosphor to match that of the host is of particular significance for the development of SWPs with low driving voltage and high power efficiency.
Here, we demonstrate novel poly(arylene phosphine oxide)-based all-phosphorescent SWPs to realize high-power-efficiency white electroluminescence from a single polymer, which is achieved by using a combination of a high-HOMO-level blue phosphor and high-HOMO-level nonconjugated poly(arylene phosphine oxide) host. The high-HOMO-level blue phosphor is used to eliminate the hole scattering between the blue phosphors and polymer hosts, while the high-HOMO-level nonconjugated polymer host is used to reduce the hole injection barrier from the anode to SWPs and meanwhile to prevent back energy transfer from the blue phosphor to the host. With this novel strategy, a low turn-on voltage (Von) of 2.8 V, a high external quantum efficiency of 18.0% and a record forward PE of 52.1 lm W−1 are achieved for SWPs. Such remarkable progress makes SWPs outperform the polymer blends currently used for solution-processed WOLEDs and comparable with the small molecules used for vacuum-deposited WOLEDs.6,37,38 We believe that this study will definitely push forward the development of SWPs for low-cost and large-scale illumination applications in the future.
Results and discussion
Molecular design and synthesis
To design all-phosphorescent SWPs, the selection of the polymer host and phosphors is the key factor in determining the device efficiency. For the polymer host, a high ET is required to confine the triplet excitons on all the phosphors; meanwhile, appropriate HOMO/LUMO energy levels are needed to allow smooth carrier injection in the emissive layer (EML). Based on these requirements, our selection of the polymer host is focused on the following considerations. First, triphenylphosphine oxide (TPO) units are chosen as building blocks for the polymer backbone because of their high ET and appropriate LUMO level for efficient electron injection. Second, non-conjugated 3,3′-dimethyl diphenyl ether is incorporated into the poly(arylene phosphine oxide) backbone to isolate the TPO units to maintain a high ET (Fig. 1). The diphenyl ether moiety is expected to break the π-conjugation between the adjacent phenyl groups of the spacer, while the methyl groups are expected to increase the twisting angle between the TPO unit and the spacer which further decreases the conjugation along the polymer backbone. This molecular design endows the nonconjugated poly(arylene phosphine oxide) host with a high ET up to 2.85 eV (Fig. S1 and S2, ESI†), making it sufficient to act as the host for blue phosphors. Third, three kinds of carbazole derivatives—including tert-butyl-capped carbazole dendrons with first (Cz) or second (DtBu) generation, and n-butoxy-capped carbazole dendron with second generation (DOBu)—are selected as side chains to tune the HOMO level of the resulting polymers (PH1, PH2 and PH3). According to the density functional theory (DFT) calculations (Fig. 2), the HOMO level shifts to a higher energy by 0.29 eV through changing the capping groups from tert-butyl (DtBu) to n-butoxy (DOBu) due to the extended electron cloud across the electron-donating oxygen atoms. However, from DtBu to Cz, the HOMO level shifts to lower energy owing to the more localized electron population. With this design, PH2 containing the DOBu sidechain shows the highest experimental HOMO level of −5.20 eV, matching well with the Fermi level of the ITO/PEDOT:PSS anode. In comparison, PH1 and PH3 give experimental values of −5.37 and −5.64 eV, respectively, which are much lower than that of the anode (Fig. S3†).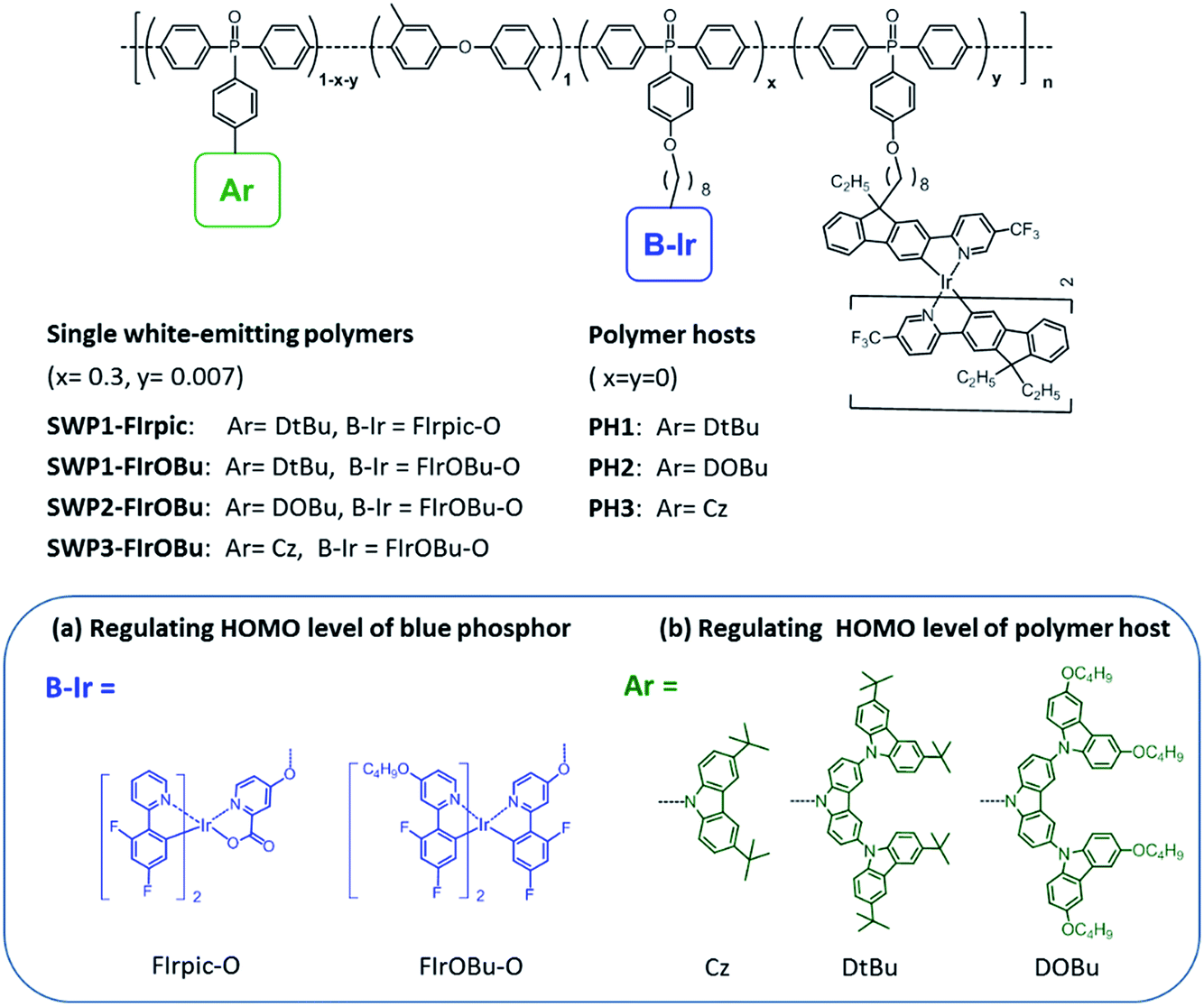 | ||
| Fig. 1 Molecular design and chemical structures of the single white light-emitting polymers (SWPs). | ||
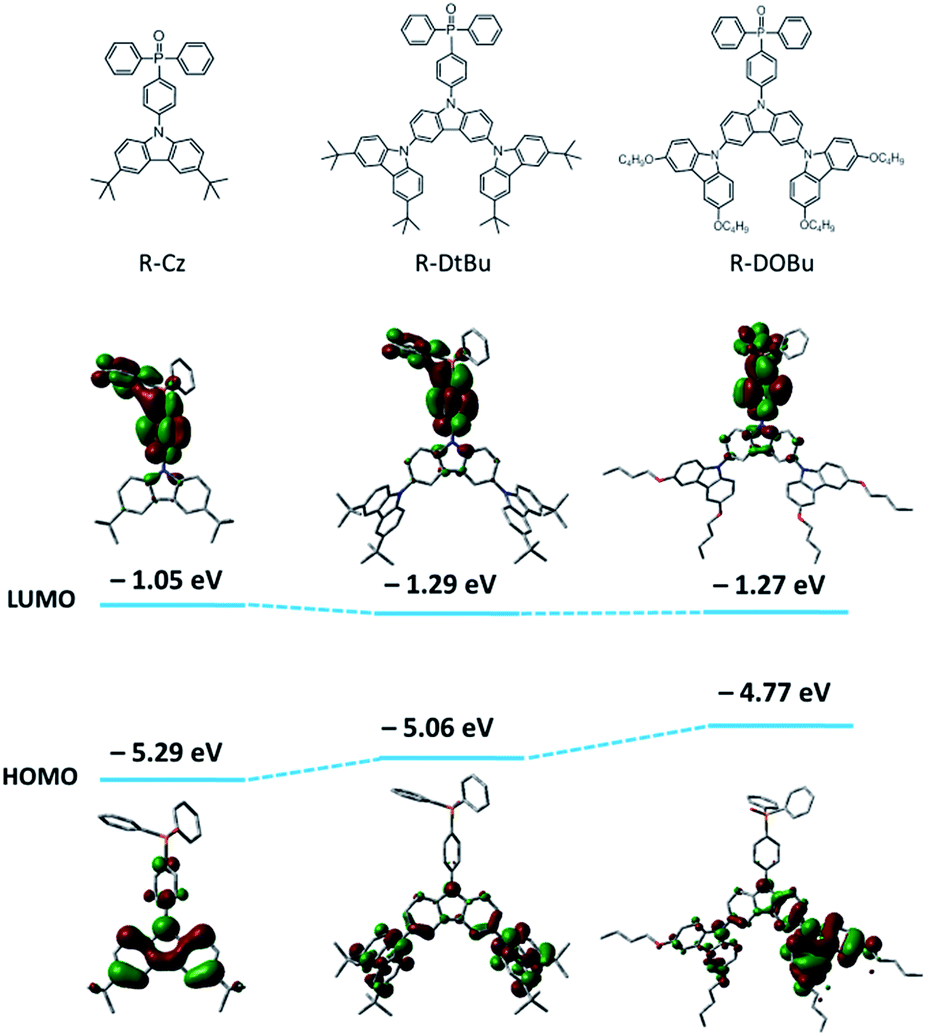 | ||
| Fig. 2 Calculated HOMO/LUMO levels for the building blocks of the polymer hosts (B3LYP/6-31G(d)). The calculated HOMO level shifts from −5.29 eV to −5.06 eV and −4.77 eV in the order of R-Cz, R-DtBu and R-DOBu, consistent with the experimental results (−5.64, −5.37 eV and −5.20 eV for PH3, PH1 and PH2, respectively). | ||
Another consideration for our design of SWPs involves the selection of blue and yellow phosphors. For blue phosphors, two iridium complexes, e.g. FIrpic and the homemade tri(4-butoxy-2-(2,4-difluorophenyl)pyridine)iridium(III) (FIrOBu),39 with different energy levels are chosen. Compared with FIrpic (HOMO: −5.64 eV), the HOMO level of FIrOBu (−5.28 eV) is up-shifted by 0.36 eV through introducing electron-donating n-butoxy substituents into the pyridine units (Fig. S4†). This high HOMO level is expected to suppress the hole scattering effect between the phosphor and host. As for the yellow phosphor, the Ir complex tri(5-trifluoromethyl-2-(9,9-diethylfluoren-2-yl)pyridine)iridium(III) (Ir(FlPy-CF3)3) featuring a short triplet-state lifetime (τ = 0.1 μs) is selected for the SWPs. As demonstrated previously,40 Ir(FlPy-CF3)3 showed much higher device efficiency and smaller efficiency roll-off than its counterpart (fbi)2Ir(acac) (bis(2-(9,9-diethyl-fluoren-2-yl)-1-phenyl-benzoimidazol-N, C3) iridium (acetylacetonate)) which had a longer τ of 1.5 μs, because the collision-induced quenching effect—including triplet–triplet annihilation and triplet–polaron quenching—was inhibited for Ir(FlPy-CF3)3.
With the hosts and dopants in hand, four kinds of all-phosphorescent SWPs (SWP1-FIrpic, SWP1-FIrOBu, SWP2-FIrOBu and SWP3-FIrOBu) are designed by covalently tethering the blue and yellow phosphors to the side chain of poly(arylene phosphine oxide) hosts through alkyl chains. The saturated alkyl chain keeps the electronic properties of the host and the phosphor independent from each other. To get balanced blue and yellow light emission for white light emission, the content of the blue and yellow phosphors are tuned according to the feed ratio, which is 30 mol% and 0.7 mol%, respectively, for the optimized SWPs.
Schemes 1 and 2 show the synthetic routes for the poly(arylene phosphine oxide)s. The dibrominated monomers Br-Cz, Br-DtBu and Br-DOBu were produced from bis-(4-bromophenyl)phosphine oxide (1) and the corresponding aryl iodides (2, 3, and 4) through palladium-catalyzed coupling in high yields (>75%). The polymers were then synthesized by Suzuki–Miyaura polymerization from the corresponding bromide monomers and borated monomer (5), with typical number-average molecular weights of 30–100 kDa as obtained by a gel permeation chromatography (GPC) method using polystyrene as the standard and tetrahydrofuran as the eluent. It is noteworthy that unlike the harsh conditions used for previous poly(arylene ether)-based hosts (strong base at 165 °C), the Suzuki–Miyaura polymerization conditions used here (weak base under 90 °C) are mild enough for the fluorinated Ir(III) complexes, avoiding the risk of decomposition of the blue phosphor.41 The actual content of the Ir complexes in the SWPs is quantified by comparing the integration of the 1H NMR signals of the Ir complexes with that of carbazole segments. It is found that the Ir loading is close to the feed ratio. The detailed procedure for the synthesis and characterization of the monomers and polymers can be found in the ESI.†
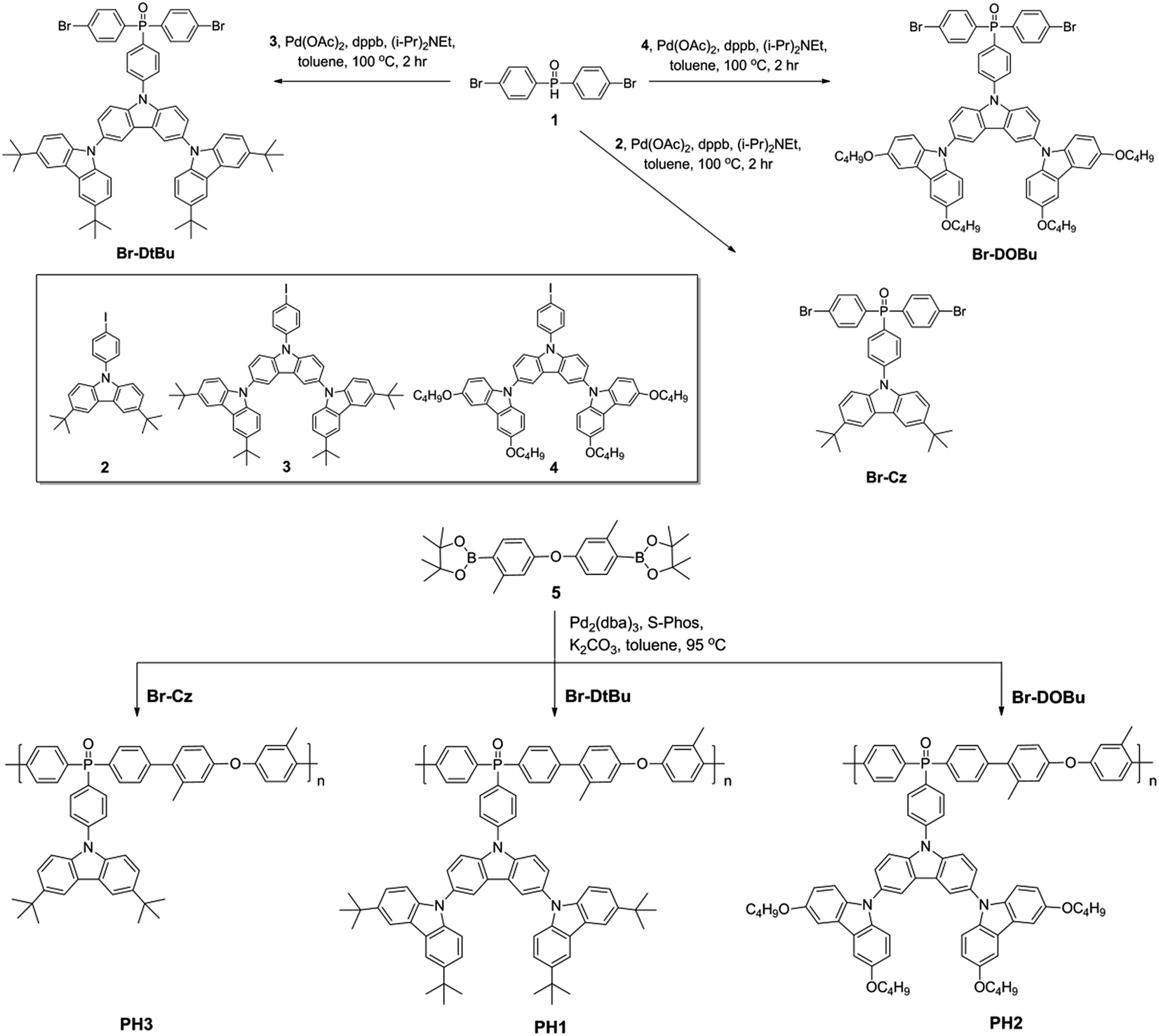 | ||
| Scheme 1 Synthetic procedure for the poly(arylene phosphine oxide) hosts. | ||
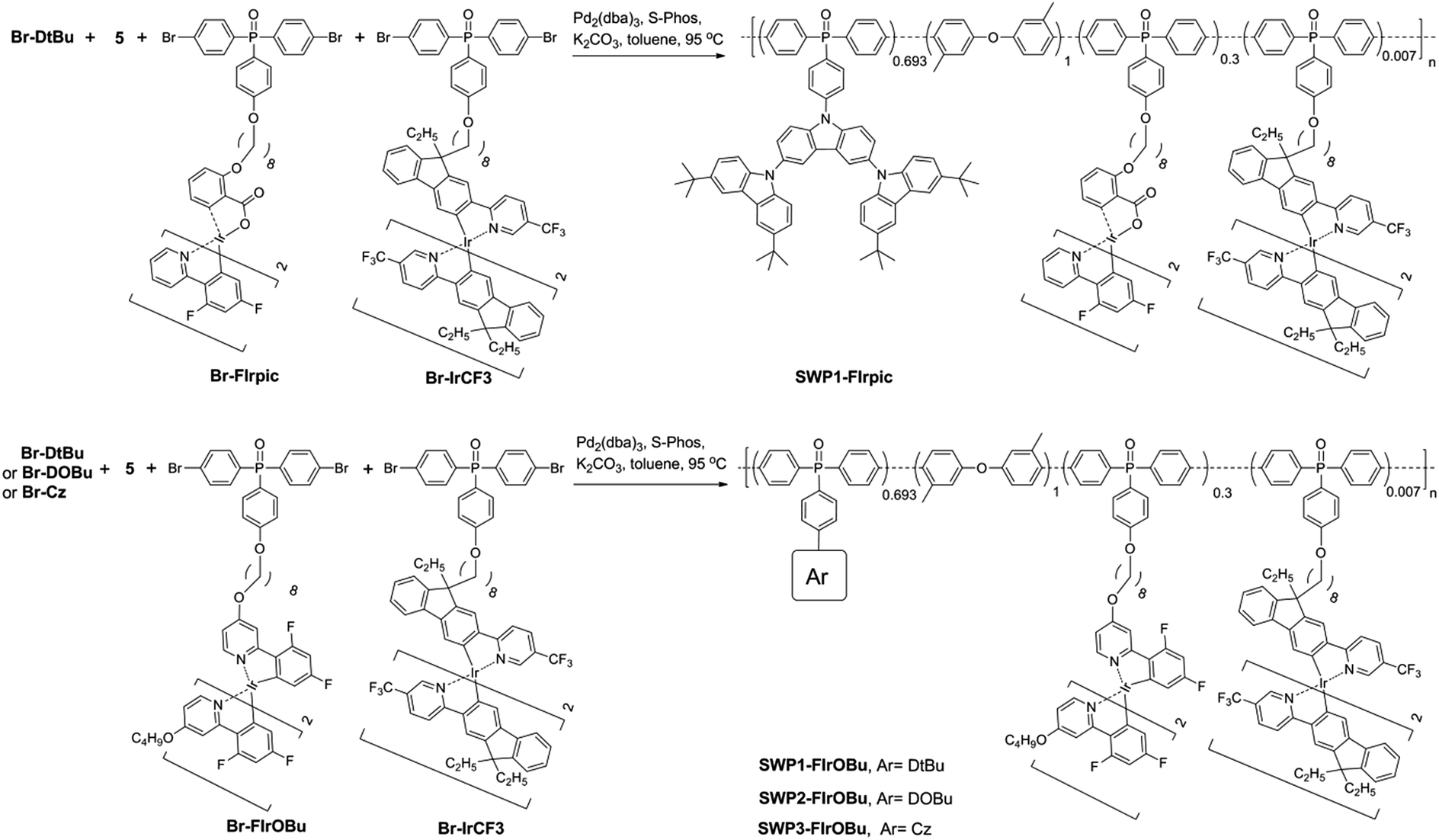 | ||
| Scheme 2 Synthetic procedure for the SWPs. | ||
Energy-level matching of the blue phosphor and polymer host
The energy level of the phosphor determines the charge transport behavior to a large extent. To avoid unwanted charge trapping/scattering effects, the energy level of the phosphor should match that of the host. This requirement is important for the blue phosphor because of its typically high doping concentration (30 mol% in this study) in SWPs.To investigate the effect of the energy level of the blue phosphor on the EL performance of the SWPs, two devices were fabricated using SWP1-FIrpic and SWP1-FIrOBu as the emissive layer. The device structure is ITO (130 nm)/PEDOT:PSS (30 nm)/SWPs (40 nm)/SPPO13 (55 nm)/LiF (1 nm)/Al (100 nm). Here SPPO13 represents 9,9′-spirobis(fluorine)-2,7-diylbis(diphenylphosphine oxide) and acts as the electron-transporting layer (ETL). As shown in Fig. 3, both SWP1-FIrpic and SWP1-FIrOBu show individual blue and yellow light emissions, which produce white light emission with Commission Internationale de L'Eclairage (CIE) coordinates of (0.41, 0.45) and (0.43, 0.44), respectively. However, SWP1-FIrpic exhibits a moderate device performance with a Von of 4.2 V, a maximum EQE of 8.3% and a maximum PE of 12.2 lm W−1 (Table 1). In comparison, SWP1-FIrOBu shows a much lower Von of 3.4 V, higher maximum EQE of 18.7% and higher maximum PE of 40.8 lm W−1. Given the comparable photoluminescence quantum yield (PLQY) of FIrpic (0.53) and FIrOBu (0.59), it is intriguing to note that the PE of SWP1-FIrOBu is more than 3 times that of SWP1-FIrpic.
 | ||
| Fig. 3 (a) Energy levels of the polymer host and the blue dopants for the SWP1-FIrpic and SWP1-FIrOBu devices, with the arrows showing the charge injection, transport and trapping process as well as the energy transfer (ET) pathway; (b and c) EL spectra and PE–L curves of SWP1-FIrpic and SWP1-FIrOBu devices (ITO (130 nm)/PEDOT:PSS (30 nm)/SWPs (40 nm)/SPPO13 (55 nm)/LiF (1 nm)/Al (100 nm)); (d) J–V characteristics of the hole-only devices (ITO (130 nm)/PEDOT:PSS (30 nm)/PH1: 0–30 wt% FIrpic or FIrOBu (90 nm)/Au (60 nm)); (e) transient EL decay curves of the blue devices (ITO (130 nm)/PEDOT:PSS (30 nm)/PH1: 15 wt% FIrpic or FIrOBu (40 nm)/SPPO13 (55 nm)/LiF (1 nm)/Al (100 nm)). | ||
| Polymer | V on (V) | Maximum value/at 100 cd m−2/at 1000 cd m−2 | L max (cd m−2) | CIE (x,y)e@1000 cd m−2 | |
|---|---|---|---|---|---|
| PEb (lm W−1) | EQEc (%) | ||||
| a Voltage at 1 cd m−2. b Power efficiency. c External quantum efficiency. d Maximum luminance. e CIE coordinates. | |||||
| SWP1-FIrpic | 4.2 | 12.2/12.1/8.6 | 8.3/7.8/7.0 | 4100 | 0.41, 0.45 |
| SWP1-FIrOBu | 3.4 | 40.8/38.0/25.4 | 18.7/18.6/16.0 | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 100 |
0.43, 0.44 |
| SWP2-FIrOBu | 2.8 | 52.1/41.0/27.3 | 18.0/17.2/14.4 | 16700 |
0.42, 0.45 |
| SWP3-FIrOBu | 3.6 | 32.9/30.6/23.2 | 15.8/15.8/13.4 | 11900 |
0.42, 0.44 |
Another finding is that SWP1-FIrpic shows much lower current density (J) than SWP1-FIrOBu at the same driving voltage (V) (Fig. S5†). This behavior is similar to that of the physically blended blue devices containing PH1 doped with FIrpic or FIrOBu as the emissive layer (Fig. S6†). Given the same device structure, the difference in device performance between SWP1-FIrpic and SWP1-FIrOBu can be ascribed to the different energy alignment of the phosphors relative to the polymer host. As shown in Fig. 3a, owing to the low HOMO level, FIrpic acts as a hole scattering center in SWP1-FIrpic, leading to an increase of transit path for holes and thus a decrease of hole current density. This effect further causes the electrons on FIrpic (captured from either SPPO13 or the polymer host) to not recombine readily with the holes and so accumulate near the EML/ETL interface. Both the reduced hole current density and the electron accumulation will lead to an inferior device performance such as high driving voltage and low EQE. In contrast, hole scattering is avoided in SWP1-FIrOBu because the HOMO level of FIrOBu is slightly higher than that of the polymer host, so holes can transport smoothly to the EML/ETL interface to recombine immediately with the electrons, thus giving a much better device performance.
To confirm the hole scattering effect, hole-only devices based on polymer blends of PH1: 0–30 wt% FIrpic and PH1: 0–30 wt% FIrOBu were prepared. As shown in Fig. 3d, for the FIrpic-doped devices, the current density at the same voltage decreases as the doping concentration increases from 0 to 30 wt%. This result indicates that the hole transport can be impeded by FIrpic, which is consistent with the hole scattering effect. By contrast, for the FIrOBu-doped devices, the current density is slightly increased as the doping concentration increases, probably because FIrOBu can play the role of a hole transporter in the active layer.
Further evidence for the hole scattering effect comes from the time-resolved EL intensity of the blue light-emitting devices. As shown in Fig. 3e, a transient overshoot after voltage turn-off is observed for the device fabricated with an emissive layer of PH1: 15 wt% FIrpic. Upon a negative bias of −3 V, the overshoot time is distinctly shortened, accompanied by enhanced overshoot intensity. In contrast, no overshoot of EL intensity is detected for the device based on the layer of PH1: 15 wt% FIrOBu at a bias of either 0 V or −3 V. The observed transient overshoot for the FIrpic device indicates that there are accumulated electrons on FIrpic, which can recombine with the holes in the emissive layer to give delayed emission after voltage turn-off.42 Comparatively, in the FIrOBu device with no scattering effect, the holes transport smoothly and recombine rapidly with the electrons, and therefore no residual holes and accumulated electrons are available to produce delayed emission.
Regulating the HOMO levels of the polymer hosts
Another key issue for the design of SWPs involves the energy-level matching of the polymer host and the electrode. In particular, to minimize the hole injection barrier, a polymer host with a HOMO level close to the Fermi level of the anode is required. For this purpose, regulation of the HOMO level of the poly(arylene phosphine oxide) host is carried out for the SWPs. As shown in Fig. 4a, PH1 containing the tert-butyl-capped second-generation carbazole dendron shows a HOMO level of −5.37 eV, leading to an energy barrier of 0.17 eV for hole injection from the ITO/PEDOT:PSS anode (−5.20 eV) to SWP1-FIrOBu, while for PH2 containing the n-butoxy-capped second-generation carbazole dendron, the HOMO level (−5.20 eV) is close to the anode. Therefore, the hole injection barrier at the polymer/anode interface is negligible for SWP2-FIrOBu. In comparison, for SWP3-FIrOBu, the polymer host PH3 having the tert-butyl-capped first-generation carbazole sidechain shows a low HOMO level of −5.64 eV, giving a large barrier of 0.44 eV for hole injection. To show the importance of the HOMO-level regulation, hole-only devices based on PH1, PH2 and PH3 were fabricated. As shown in Fig. 4b, the driving voltage for the same current density (e.g. 1 mA cm−2) is substantially decreased from PH3 (9.5 V) to PH1 (6.5 V) and PH2 (5.3 V), suggesting that the high HOMO level of the polymer host is favorable for hole injection. | ||
| Fig. 4 (a) Energy levels of the polymer hosts with different HOMO levels. The red arrows denote the hole-injection barriers from the anode to the polymers. (b) J–V curves of the hole-only devices (ITO (130 nm)/PEDOT:PSS (30 nm)/PH1–PH3 (90 nm)/Au (60 nm)). (c) EL spectra of SWPs with different HOMO levels. (d–f) J–V, L–V and PE–V characteristics of the SWPs (ITO (130 nm)/PEDOT:PSS (30 nm)/SWPs (40 nm)/SPPO13 (55 nm)/LiF (1 nm)/Al (100 nm)). | ||
To explore the influence of the HOMO levels of the host on the EL performance of SWPs, devices based on SWP1-FIrOBu, SWP2-FIrOBu and SWP3-FIrOBu were fabricated with the configuration of ITO (130 nm)/PEDOT:PSS (30 nm)/SWPs (40 nm)/SPPO13 (55 nm)/LiF (1 nm)/Al (100 nm). All the SWPs give simultaneous blue and yellow emissions with CIE coordinates located in the white light zone. In particular, SWP2-FIrOBu exhibits CIE coordinates of (0.41 ± 0.01, 0.44 ± 0.01) in the luminance range from 100 to 5000 cd m−2 (Fig. 4c). The small variation of CIE coordinates at practical luminance implies that they are potential candidates for illumination sources with a stable spectrum. In addition, due to the high ETs of the polymer hosts, all the SWPs give impressive EQEs of 15.8–18.6% at 100 cd m−2 and 13.4–16.0% at 1000 cd m−2 (Table 1).
Remarkably, SWP2-FIrOBu exhibits very low driving voltages of 2.8/3.8/4.8 V at 1/100/1000 cd m−2, respectively, and is superior to SWP1-FIrOBu showing driving voltages of 3.4/4.4/5.4 V at the same luminances. In contrast, these values for SWP3-FIrOBu increased to 3.8/4.6/5.6 V, respectively. These results indicate that a HOMO level close to the Fermi level of the anode is favorable for reducing the driving voltage of the SWPs. Thanks to the low driving voltage, SWP2-FIrOBu shows a maximum PE of 52.1 lm W−1 without the use of any out-coupling enhancement technology. At 100 cd m−2 and 1000 cd m−2, the PE decays to 41.0 lm W−1 and 27.3 lm W−1, respectively. The considerable efficiency roll-off indicates that there is still triplet–triplet or triplet–polaron annihilation in the emissive layer. Further improving the charge balance of the polymers (e.g. by covalently incorporating charge-transporting units in the mainchain or physically blending with charge-transporting materials) to broaden the charge combination zone may be prospective to address this problem. Despite this, these power efficiencies represent an important progress for SWPs considering the PE of SWPs at a practically relevant brightness (for example, 100 cd m−2) has long been kept below 15 lm W−1 in the past decade (Table S1†).
The remarkable power efficiencies of the SWPs surpass those of the polymer blends ever reported and are even comparable with those of the small molecules relying on the vacuum-deposition method (see Fig. 5 and Table S1†). From the aspects of the design of materials and devices, we propose that the key to the high-power-efficiency SWPs is to satisfy the following requirements:
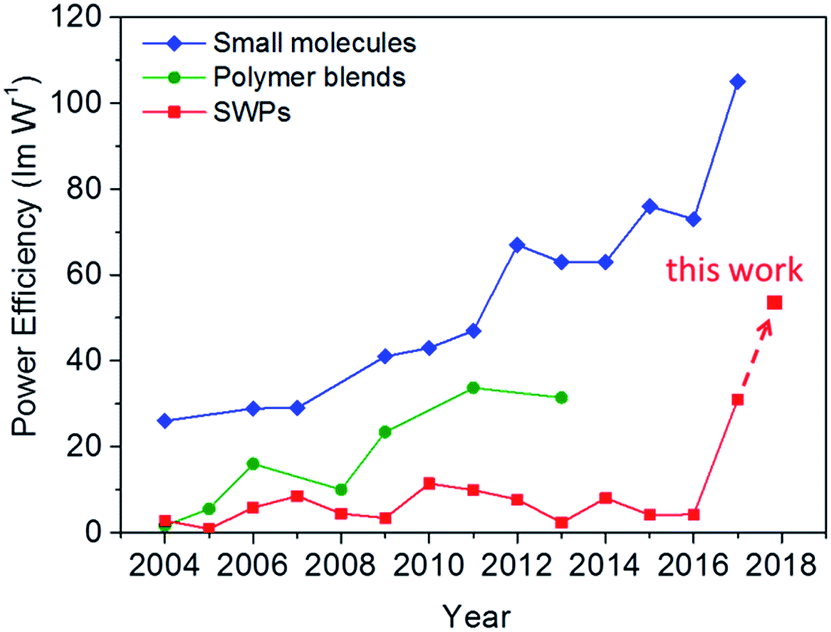 | ||
| Fig. 5 Recent progress in power efficiency for organic white light-emitting materials. The values are collected from the most efficient materials reported in the literature in the past decade (see Table S1†). All the values are given without the use of out-coupling enhancement technology for better comparison. | ||
(1) appropriate energy levels of both the polymer host and phosphor to realize barrier-free charge injection and to avoid unwanted charge trapping and charge scattering processes: from a survey of the state-of-the-art small molecule WOLEDs,6,37,38 a Von of less than 3.0 V is basically needed to achieve a PE higher than 50 lm W−1, thus requiring smooth charge injection and transport from the anode to the recombination zone. To meet this requirement, multi-layer device structures that contain extra charge injection and transporting layers are always adopted for small molecule WOLEDs. However, applying such multi-layer device structures to SWPs through solution-based processing methods is challenging. In view of this, fine regulation of the energy levels of both the polymer host and phosphor is critical for SWPs to overcome all the barriers in charge injection and transport. For the polymer host, a HOMO level close to the Fermi level of the anode is needed to eliminate the hole injection barrier at the polymer/anode host interface. For phosphors, their energy levels should match those of the host to avoid unwanted charge trapping and charge scattering processes. In this study, the combination of the high-HOMO-level blue phosphor and high-HOMO-level poly(arylene phosphine oxide) host in SWP2-FIrOBu plays the key role in satisfying these requirements to realize a low Von of 2.8 V. Taking into account the band gap of 2.7 eV for FIrOBu, it is reasonable to deduce that the charge injection and transport in SWP2-FIrOBu is almost barrier-free.
(2) A high-ET polymer host to confine excitons on phosphors: as an indispensable component for white light emission, the blue phosphor requests a polymer host with an ET > 2.70 eV to avoid quenching of high-energy triplet excitons. However, in previous reports, most polymer hosts are based on conjugated backbones (e.g. polyfluorenes and polycarbazoles35) which show ETs lower than 2.70 eV, rendering them not suitable for all-phosphorescent SWPs. In this study, the incorporation of the non-conjugated 3,3′-dimethyl diphenyl ether spacer endows the poly(arylene phosphine oxide) host with an ET up to 2.85 eV, which is critical for avoiding quenching of the blue excitons. Indeed, if this spacer is replaced by more conjugated counterparts, the resulting polymers show much lower ETs (e.g. PH4 (ET = 2.70 eV) and PH5 (ET = 2.45 eV), Fig. S1 and S2†). As a result, the blue excitons can be substantially quenched, which is verified by the fact that the phosphorescence lifetime of the polymer host: 15 wt% FIrpic film becomes much shorter, and the EQE of the corresponding blue phosphorescent device is decreased from 11.5% to 0.5% (Fig. S8†).
(3) Balanced charge transport to enhance the carrier recombination ratio: we find that the EQEs of the SWPs are strongly dependent on the ETL. When 1,3,5-tri(m-pyrid-4-yl-phenyl)benzene (TmPyPB)43 is used as the ETL instead of SPPO13, the maximum EQE of the SWP2-FIrOBu device is decreased from 18.0% to 12.3%, which is accompanied by an increase of current density at the same voltage (Fig. S10†). However, inserting a thin layer (8 nm) of 3,6-bis(diphenylphosphoryl)-9-[4-(diphenylphosphoryl)phenyl]-carbazole (TPCz) between the SWP2-FIrOBu and TmPyPB layers leads to a decrease of current density and an increase of EQE from 12.3% to 16.7%. This observation suggests that there are excess electrons in the recombination zone if Tm4PyPB is used as the ETL considering its high electron mobility (1.0 × 10−3 cm2 V−1 s−1), which leads to a low recombination ratio of holes and electrons.
It should be pointed out that the design of high-power-efficiency SWPs needs systematic molecular engineering. Besides the above-mentioned issues, other factors, such as the carrier mobility of the polymer host, the photoluminescence quantum yield and excited state lifetimes of the phosphors and the topological structure used to combine the polymer host and phosphors, are also important considerations. High-power-efficiency SWPs can be achieved only when these issues are taken into account in a comprehensive way.
Conclusions
In summary, we have reported poly(arylene phosphine oxide)-based all-phosphorescent SWPs by using a combination of a high-HOMO-level blue phosphor and high-HOMO-level polymer host to realize a power efficiency of over 50 lm W−1. The use of the high-HOMO-level blue phosphor is to eliminate the hole-scattering effect caused by the energy-level mismatch between the blue phosphor and the polymer host, and the high-HOMO-level polymer host is to reduce the hole injection barrier at the polymer/anode interface, which synergetically leads to a very low turn-on voltage of 2.8 V, a high external quantum efficiency of 18.0% and a high forward PE of 52.1 lm W−1. This record efficiency makes these poly(arylene phosphine oxide)-based SWPs outperform the multi-component polymer blends currently in use for solution-processed WOLEDs and even comparable with the vacuum-deposited molecules used for vacuum-deposited WOLEDs. Such a progress in SWPs provides an incentive for researchers to make future efforts on this low-cost lighting technology. Our further research on boosting the power efficiency (e.g. by using high-HOMO-level polymer hosts with the thermally activated delayed fluorescence effect) and enhancing the device stability of the SWPs (e.g. by using potentially stable non-fluorinated blue dopants to replace the current fluorinated ones (FIrpic and FIrOBu)) is currently underway.Experimental
Materials
Synthesis and characterization of the materials used in this study can be found in the ESI.†OLED fabrication
The white light-emitting OLEDs were fabricated on ultraviolet-ozone treated ITO-glass substrates (15 Ω per square). A 30 nm-thick PEDOT:PSS (Clevious P AI4083) film was deposited on the substrates and then baked at 120 °C for 45 min, which was then transferred into a N2-filled glove box. Solutions of SWPs in chlorobenzene were spin-coated on the PEDOT:PSS layer as the emissive layer with a thickness of ∼30 nm. After annealing at 100 °C for 30 min, the devices were transferred into a vacuum chamber. Successively, SPPO13 (55 nm) (or TmPyPB (55 nm) and TPCz (8 nm)/TmPyPB (47 nm)) was thermally evaporated on top of the emissive layer at a pressure less than 4 × 10−4 Pa to act as the electron-transporting layer. Finally, a 1 nm-thick film of LiF and 100 nm-thick film of Al were deposited as the cathode through a shadow mask to give an active area of 3.5 × 4.0 mm2. The hole-only devices were fabricated according to a similar procedure, except that a 60 nm-thick Au layer was evaporated on top of the active layer as the cathode.OLED characterization
The J–V–L characteristics of the devices were measured at room temperature under ambient atmosphere using a Keithley 2400/2000 source meter coupled with a calibrated silicon photodiode. The EL spectra and CIE coordinates were recorded with a PR650 SpectraScan colorimeter. The EQE of the devices was calculated based on the current density, luminance and EL spectrum assuming a Lambertian emission distribution.Transient EL measurements
The decay curves of the transient EL intensity were measured by electrically exciting the devices with a pulse generator (Agilent 8114A, 100V/2A). After going through a monochromator, the transient EL signals are collected with a photomultiplier tube. The obtained results were presented using an oscilloscope (Agilent Model 54825A, 500 MHz/2 GSa/s).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Z. Zhang from the Fujian Institute of Research on the Structure, Chinese Academy of Sciences for helpful discussion on density functional theory calculation results and Dr Q. Guo from the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences for help on transient EL measurements. We also acknowledge financial support from the National Natural Science Foundation of China (No. 51573182, 51203149 and 91333205) and the 973 Project (No. 2015CB655000).References
- J. Kido, M. Kimura and K. Nagai, Science, 1995, 267, 1332 CrossRef CAS PubMed.
- K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater., 2010, 22, 572 CrossRef CAS PubMed.
- L. Ying, C. L. Ho, H. Wu, Y. Cao and W. Y. Wong, Adv. Mater., 2014, 26, 2459 CrossRef CAS PubMed.
- Y.-M. Xie, L.-S. Cui, Y. Liu, F.-S. Zu, Q. Li, Z.-Q. Jiang and L.-S. Liao, J. Mater. Chem. C, 2015, 3, 5347 RSC.
- J. Wang, J. Chen, X. Qjao, S. M. Alshehri, T. Aharnad and D. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 10093 CrossRef CAS PubMed.
- S.-F. Wu, S.-H. Li, Y.-K. Wang, C.-C. Huang, Q. Sun, J.-J. Liang, L.-S. Liao and M.-K. Fung, Adv. Funct. Mater., 2017, 27, 1701314 CrossRef.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Bredas, M. Logdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS.
- B. W. D'Andrade and S. R. Forrest, Adv. Mater., 2004, 16, 1585 CrossRef.
- M. Berggren, D. Nilsson and N. D. Robinson, Nat. Mater., 2007, 6, 3 CrossRef CAS PubMed.
- H. Sasabe and J. Kido, J. Mater. Chem. C, 2013, 1, 1699 RSC.
- M. S. White, M. Kaltenbrunner, E. D. Glowacki, K. Gutnichenko, G. Kettlgruber, I. Graz, S. Aazou, C. Ulbricht, D. A. M. Egbe, M. C. Miron, Z. Major, M. C. Scharber, T. Sekitani, T. Someya, S. Bauer and N. S. Sariciftci, Nat. Photonics, 2013, 7, 811 CrossRef CAS.
- Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed.
- C. Li, R. S. Nobuyasu, Y. Wang, F. B. Dias, Z. Ren, M. R. Bryce and S. Yan, Adv. Opt. Mater., 2017, 5, 1700435 CrossRef.
- S. Wang, L. Zhao, B. Zhang, J. Ding, Z. Xie, L. Wang and W.-Y. Wong, iScience, 2018, 6, 128 CrossRef PubMed.
- L. Yu, J. Liu, S. Hu, R. He, W. Yang, H. Wu, J. Peng, R. Xia and D. D. C. Bradley, Adv. Funct. Mater., 2013, 23, 4366 CrossRef CAS.
- H. Wu, G. Zhou, J. Zou, C. L. Ho, W. Y. Wong, W. Yang, J. Peng and Y. Cao, Adv. Mater., 2009, 21, 4181 CrossRef CAS.
- J. Zou, H. Wu, C. S. Lam, C. Wang, J. Zhu, C. Zhong, S. Hu, C. L. Ho, G. J. Zhou, H. Wu, W. C. H. Choy, J. Peng, Y. Cao and W. Y. Wong, Adv. Mater., 2011, 23, 2976 CrossRef CAS PubMed.
- F. C. Chen, G. F. He and Y. Yang, Appl. Phys. Lett., 2003, 82, 1006 CrossRef CAS.
- Y. Y. Noh, C. L. Lee, J. J. Kim and K. Yase, J. Chem. Phys., 2003, 118, 2853 CrossRef CAS.
- L. Bao and M. D. Heagy, Curr. Org. Chem., 2014, 18, 740 CrossRef CAS.
- G. L. Tu, Q. G. Zhou, Y. X. Cheng, L. X. Wang, D. G. Ma, X. B. Jing and F. S. Wang, Appl. Phys. Lett., 2004, 85, 2172 CrossRef CAS.
- J. Liu, S. Shao, L. Chen, Z. Xie, Y. Cheng, Y. Geng, L. Wang, X. Jing and F. Wang, Adv. Mater., 2007, 19, 1859 CrossRef CAS.
- D. A. Poulsen, B. J. Kim, B. Ma, C. S. Zonte and J. M. J. Frechet, Adv. Mater., 2010, 22, 77 CrossRef CAS PubMed.
- S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2012, 134, 20290 CrossRef CAS PubMed.
- S. J. Ananthakrishnan, E. Varathan, V. Subramanian, N. Somanathan and A. B. Mandal, J. Phys. Chem. C, 2014, 118, 28084 CrossRef CAS.
- F. Xu, J. H. Kim, H. U. Kim, J. H. Jang, K. S. Yook, J. Y. Lee and D. H. Hwang, Macromolecules, 2014, 47, 7397 CrossRef CAS.
- B. Liu, F. Dang, Z. Tian, Z. Feng, D. Jin, W. Dang, X. Yang, G. Zhou and Z. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 16360 CrossRef CAS PubMed.
- J. X. Jiang, Y. H. Xu, W. Yang, R. Guan, Z. Q. Liu, H. Y. Zhen and Y. Cao, Adv. Mater., 2006, 18, 1769 CrossRef CAS.
- J. Liu, L. Chen, S. Shao, Z. Xie, Y. Cheng, Y. Geng, L. Wang, X. Jing and F. Wang, Adv. Mater., 2007, 19, 4224 CrossRef CAS.
- F.-I. Wu, X.-H. Yang, D. Neher, R. Dodda, Y.-H. Tseng and C.-F. Shu, Adv. Funct. Mater., 2007, 17, 1085 CrossRef CAS.
- C. H. Chien, S. F. Liao, C. H. Wu, C. F. Shu, S. Y. Chang, Y. Chi, P. T. Chou and C. H. Lai, Adv. Funct. Mater., 2008, 18, 1430 CrossRef CAS.
- R. Abbel, C. Grenier, M. J. Pouderoijen, J. W. Stouwdam, P. E. L. G. Leclere, R. P. Sijbesma, E. W. Meijer and A. P. H. J. Schenning, J. Am. Chem. Soc., 2009, 131, 833 CrossRef CAS PubMed.
- P. T. Furuta, L. Deng, S. Garon, M. E. Thompson and J. M. J. Frechet, J. Am. Chem. Soc., 2004, 126, 15388 CrossRef CAS PubMed.
- M. Sudhakar, P. I. Djurovich, T. E. Hogen-Esch and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7796 CrossRef CAS PubMed.
- A. van Dijken, J. Bastiaansen, N. M. M. Kiggen, B. M. W. Langeveld, C. Rothe, A. Monkman, I. Bach, P. Stossel and K. Brunner, J. Am. Chem. Soc., 2004, 126, 7718 CrossRef CAS PubMed.
- K. K. Tsung and S. K. So, Appl. Phys. Lett., 2008, 92, 103315 CrossRef.
- X. Ouyang, X.-L. Li, L. Ai, D. Mi, Z. Ge and S.-J. Su, ACS Appl. Mater. Interfaces, 2015, 7, 7869 CrossRef CAS PubMed.
- M. Du, Y. Feng, D. Zhu, T. Peng, Y. Liu, Y. Wang and M. R. Bryce, Adv. Mater., 2016, 28, 5963 CrossRef CAS PubMed.
- D. Xia, B. Wang, B. Chen, S. Wang, B. Zhang, J. Ding, L. Wang, X. Jing and F. Wang, Angew. Chem., Int. Ed., 2014, 53, 1048 CrossRef CAS PubMed.
- B. Zhang, G. Tan, C. S. Lam, B. Yao, C. L. Ho, L. Liu, Z. Xie, W. Y. Wong, J. Ding and L. Wang, Adv. Mater., 2012, 24, 1873 CrossRef CAS PubMed.
- S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2012, 134, 15189 CrossRef CAS PubMed.
- C. Weichsel, L. Burtone, S. Reineke, S. I. Hintschich, M. C. Gather, K. Leo and B. Luessem, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 075204 CrossRef.
- S.-J. Su, T. Chiba, T. Takeda and J. Kido, Adv. Mater., 2008, 20, 2125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization details, computational results, photophysical and electrochemical properties, and supplementary device performance of the polymers. See DOI: 10.1039/c8sc03753a |
| This journal is © The Royal Society of Chemistry 2018 |