
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct synthesis of aryl-annulated [c]carbazoles by gold(I)-catalysed cascade reaction of azide-diynes and arenes†
Yuiki
Kawada‡
a,
Shunsuke
Ohmura‡
a,
Misaki
Kobayashi
a,
Wataru
Nojo
b,
Masaki
Kondo
cd,
Yuka
Matsuda
a,
Junpei
Matsuoka
a,
Shinsuke
Inuki
 a,
Shinya
Oishi
a,
Chao
Wang
cd,
Tatsuo
Saito
c,
Masanobu
Uchiyama
*cd,
Takanori
Suzuki
*b and
Hiroaki
Ohno
*a
a,
Shinya
Oishi
a,
Chao
Wang
cd,
Tatsuo
Saito
c,
Masanobu
Uchiyama
*cd,
Takanori
Suzuki
*b and
Hiroaki
Ohno
*a
aGraduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan. E-mail: hohno@pharm.kyoto-u.ac.jp
bDepartment of Chemistry, Faculty of Science, Hokkaido University, Sapporo 060-0810, Japan
cGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
dCluster of Pioneering Research (CPR), Advanced Elements Chemistry Laboratory, RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
First published on 10th September 2018
Abstract
The gold-catalysed annulation of conjugated alkynes bearing an azido group with arenes gave annulated [c]carbazoles. Using benzene, pyrrole, and indole derivatives as the nucleophiles, benzo[c]-, pyrrolo[2,3-c]-, and indolo[2,3-c]carbazoles were produced, respectively. The reaction proceeded through pyrrole and benzene ring construction accompanied by the formation of two carbon–carbon and one carbon–nitrogen bond and the cleavage of two aromatic C–H bonds. The mechanism of the reaction with pyrrole was investigated by density functional theory calculations. N,N′-dimethylated indolo[2,3-c]carbazole showed dual ultraviolet-visible-near-infrared and fluorescence spectral changes upon electrolysis.
Introduction
Carbazoles are important structural motifs that are found in a variety of organic molecules of current interest (Fig. 1).1 Benzo[c]carbazoles are commonly used in organic light-emitting diodes (OLEDs) owing to their charge-transport properties and thermal stability.2 Heteroaryl-annulated [c]carbazoles are the core structures of various bioactive natural products, such as eustifoline-D (furo[2,3-c]carbazole), arcyriaflavin A and dictyodendrins (pyrrolo[c]carbazole), and asteropusazole A (indolo[3,2-c]carbazole).1a Thus, the development of efficient synthetic methods for preparing benzo[c]carbazoles and their heteroaromatic congeners from readily accessible starting materials is an active pursuit in organic chemistry.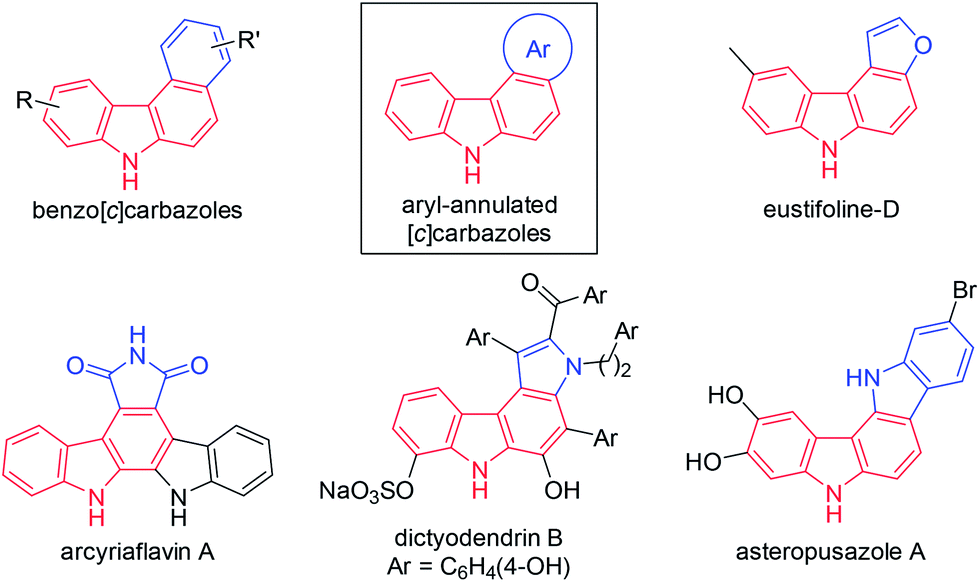 | ||
| Fig. 1 Aryl-annulated [c]carbazoles. | ||
The general synthetic approaches to carbazoles including aryl-annulated [c]carbazoles are shown in Scheme 1A and B.1a,3 Pyrrole ring formation based on a combination of carbon–nitrogen and carbon–carbon bond formation provides an efficient route to carbazole synthesis (Scheme 1A).4,5 Reliable coupling reactions such as the Suzuki–Miyaura, Buchwald, and oxidative coupling reactions can be employed for this purpose. Benzene ring formation using vinyl- or aryl-substituted indoles including Diels–Alder-type reactions,6 hydroarylation,7 and related reactions8 leads to various carbazoles including aryl-annulated carbazoles (Scheme 1B). However, the double cyclisation approach for synthesising aryl-annulated [c]carbazoles has not been investigated until recently.9–11 We expected that the gold carbenoid-based cascade cyclisation of conjugated diynes would directly provide aryl-annulated [c]carbazoles in a single operation via the sequential cleavage of two aromatic C–H bonds (Scheme 1C).
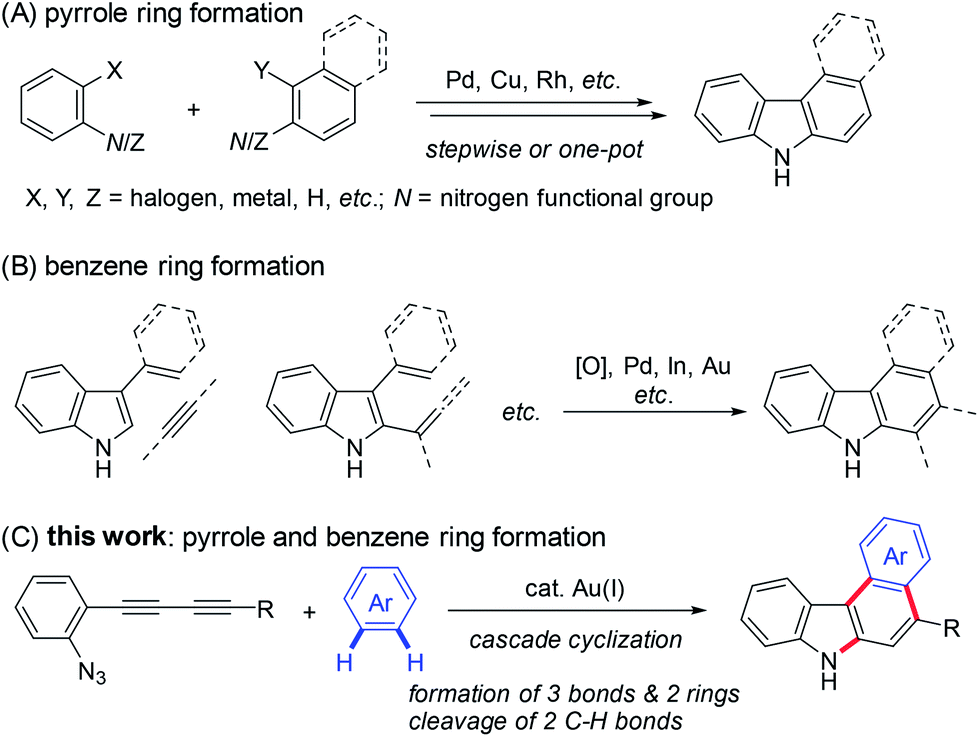 | ||
| Scheme 1 General synthetic approaches to carbazoles including aryl-annulated carbazoles and this work. | ||
Homogenous gold catalysis has emerged as a powerful tool for atom-economical transformations.12 The π-acidity of gold catalysts enables the activation of C–C multiple bonds to promote various transformations. Recent investigations using diynes in gold-catalysed reactions have revealed that both conjugated and unconjugated diynes are useful precursors of complex molecules.13 For example, we recently reported a gold-catalysed formal [4 + 2] reaction between 1,3-diynes and pyrroles for synthesising 4,7-disubstituted indoles (Scheme 2A, n = 0).14a This reaction proceeded through a double hydroarylation cascade involving the initial intermolecular hydroarylation of 1,3-diyne at the 2-position of pyrrole, followed by intramolecular hydroarylation. When using skipped diynes as substrates, the formal [5 + 2] reaction efficiently proceeded to produce 1,6-dihydrocyclohepta[b]pyrrole derivatives,14b which can be considered as homologs of 4,7-disubstituted indoles (Scheme 2A, n = 1).
 | ||
| Scheme 2 Related studies. | ||
We then turned our attention to synthesising aryl-annulated [c]carbazoles based on gold-carbenoid formation15 using conjugated diynes. In 2011, Gagosz16a and Zhang16b independently reported the gold(I)-catalysed synthesis of indoles bearing an electron-donating group at the 3-position (Scheme 2B).17 The reaction can be rationalised by the formation of a gold carbenoid intermediate followed by a nucleophilic reaction at the carbenoid moiety. As the coupling partners, alcohols and arenes can be used for the reaction to produce 3-substituted indoles. We envisaged that incorporating gold carbenoid chemistry into diyne cyclisation would provide direct access to aryl-annulated [c]carbazoles (Scheme 3). Thus, the gold(I)-mediated nucleophilic attack of the azido group of diyne 1 on the proximal alkyne followed by the elimination of nitrogen would produce gold carbenoid species A. The electrophilic aromatic substitution of benzene-type arenes with A would produce intermediate 2. Finally, intramolecular hydroarylation toward alkynes18 would occur to produce benzo[c]carbazole 3. The challenge of this strategy is controlling the regioselectivity when using pyrrole-type heteroarenes as the coupling partner: whereas the first nucleophilic attack at the pyrrole 3-position would produce pyrrolo[2,3-c]carbazole 6, the first nucleophilic attack at the pyrrole 2-position would give pyrrolo[3,2-c]carbazole 7, through 3-pyrrolylindole intermediates 4 and 5, respectively. Attention should also be given to the regioselectivity in the second arylation in the reaction with the benzene-type nucleophile (2 to 3).
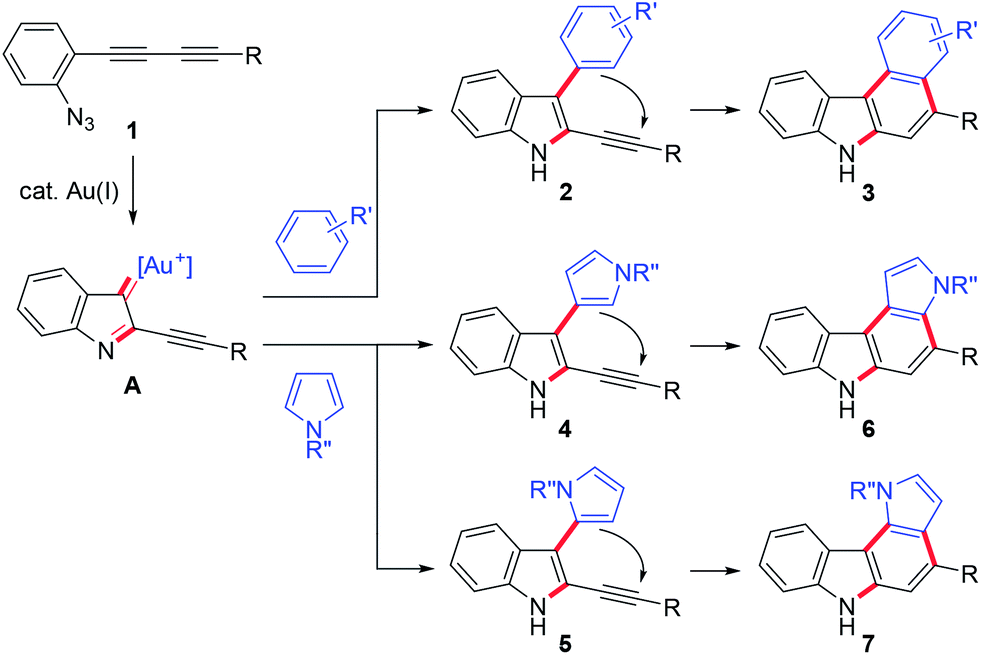 | ||
| Scheme 3 Possible reaction pathways. | ||
Herein, we report a full account of our study on the direct synthesis of aryl-annulated [c]carbazoles by the regioselective gold-catalysed annulation of conjugated diynes and arenes such as benzene, pyrrole, and indole derivatives.19 Computational investigations for elucidating the mechanism as well as redox and fluorescence properties of the pyrrolo[2,3-c]carbazoles are also presented.
Results and discussion
Reaction with benzene derivatives
Azido-substituted diynes 1 were easily prepared through Cadiot–Chodkiewicz coupling20 between 2-ethynylaniline and bromoalkynes (Scheme 4). The resulting anilines bearing a conjugated diyne moiety were converted to 1a–gvia the Sandmeyer reaction with sodium azide (see ESI†).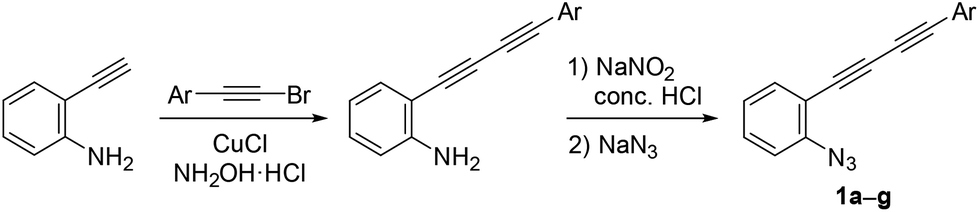 | ||
| Scheme 4 Preparation of azide-diynes. | ||
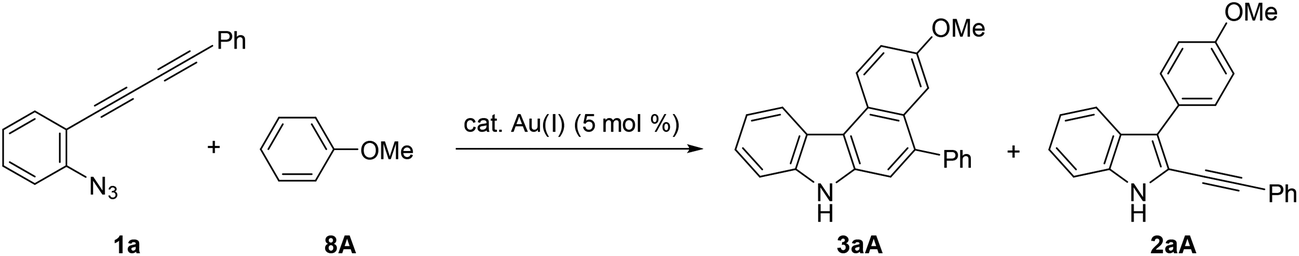
We initially screened a variety of different gold catalysts (5 mol%) for the synthesis of benzo[c]carbazole using diyne 1a and anisole 8A (10 equiv.) (Table 1, entries 1–5). Ph3PAuSbF6 in 1,2-dichloroethane (DCE) did not promote even the first arylation of the desired transformation (entry 1). IPr, XPhos, and BrettPhos (Fig. 2) were ineffective ligands for bis-cyclisation; however, 3-phenylindole intermediate 2aA was formed in 36–65% yields (entries 2–4). Fortunately, the annulation reaction was promoted by JohnPhosAu(MeCN)SbF6 (entry 5) to provide the desired fused carbazole 3aA in 44% yield. Next, we examined the choice of reaction solvent using JohnPhosAu(MeCN)SbF6 as the catalyst. The reaction using benzene, propan-2-ol, and 1,4-dioxane gave the monocyclisation product 2aA (63–87% yields) without forming carbazole 3aA (entries 6–8). Carrying out the reaction in 1,1,2,2-tetrachloroethane (TCE) at 140 °C increased the yield of 3aA to 55% (entry 9). In this case, the decomposition of JohnPhosAu(MeCN)SbF6 at high reaction temperature was anticipated. Thus, the first arylation was conducted at 80 °C and, after the disappearance of the starting material and formation of 2aA (monitored by TLC), the reaction temperature was raised to 140 °C for the second arylation, giving rise to a higher yield of fused carbazole 3aA (75% yield, entry 10). Finally, an examination of the stoichiometry revealed that the reaction using excess anisole (as solvent) and 5 mol% BrettPhosAu(MeCN)SbF6 at 140 °C improved the yield to 86% (entry 12), whereas the reaction at 80 °C did not reach completion (entry 11). Thus, we used the conditions shown in entry 10 (10 equiv. of arene, condition A) and entry 12 (arene as the solvent, condition B) for further investigations of benzo[c]carbazole synthesis.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalystb | Solventc | Temperature (time) | Yieldd (%) | |
| 3aA | 2aA | ||||
| a Reactions were carried out using 1a (1 equiv.), 8A (10 equiv.), and the gold catalyst (5 mol%). b The ligand structures are shown in Fig. 2. BrettPhosAu(MeCN)SbF6, JohnPhosAu(MeCN)SbF6, and IPrAuNTf2 were prepared in advance. The other catalysts were prepared in situ by mixing the AuCl ligand with AgNTf2 or AgSbF6. c DCE = 1,2-dichloroethane, TCE = 1,1,2,2-tetrachloroethane. d Isolated yields. | |||||
| 1 | Ph3PAuCl/AgSbF6 | DCE | 80 °C (44 h) | 0 | 0 |
| 2 | IPrAuNTf2 | DCE | 80 °C (24 h) | 0 | 36 |
| 3 | XPhosAuCl/AgNTf2 | DCE | 80 °C (21 h) | 0 | 57 |
| 4 | BrettPhosAu(MeCN)SbF6 | DCE | 80 °C (30 h) | 0 | 65 |
| 5 | JohnPhosAu(MeCN)SbF6 | DCE | 80 °C (26 h) | 44 | 26 |
| 6 | JohnPhosAu(MeCN)SbF6 | Benzene | 80 °C (10 h) | 0 | 78 |
| 7 | JohnPhosAu(MeCN)SbF6 | Propan-2-ol | 80 °C (10 h) | 0 | 63 |
| 8 | JohnPhosAu(MeCN)SbF6 | 1,4-Dioxane | 80 °C (10 h) | 0 | 87 |
| 9 | JohnPhosAu(MeCN)SbF6 | TCE | 140 °C (13 h) | 55 | 0 |
| 10 | JohnPhosAu(MeCN)SbF 6 | TCE (condition A) | 80 °C (1 h), 140 °C (16 h) | 75 | 0 |
| 11 | BrettPhosAu(MeCN)SbF6 | Anisole | 80 °C (15 h) | 13 | 28 |
| 12 | BrettPhosAu(MeCN)SbF 6 | Anisole (condition B) | 140 °C (19.5 h) | 86 | 0 |
 | ||
| Fig. 2 Structures of screened ligands. | ||
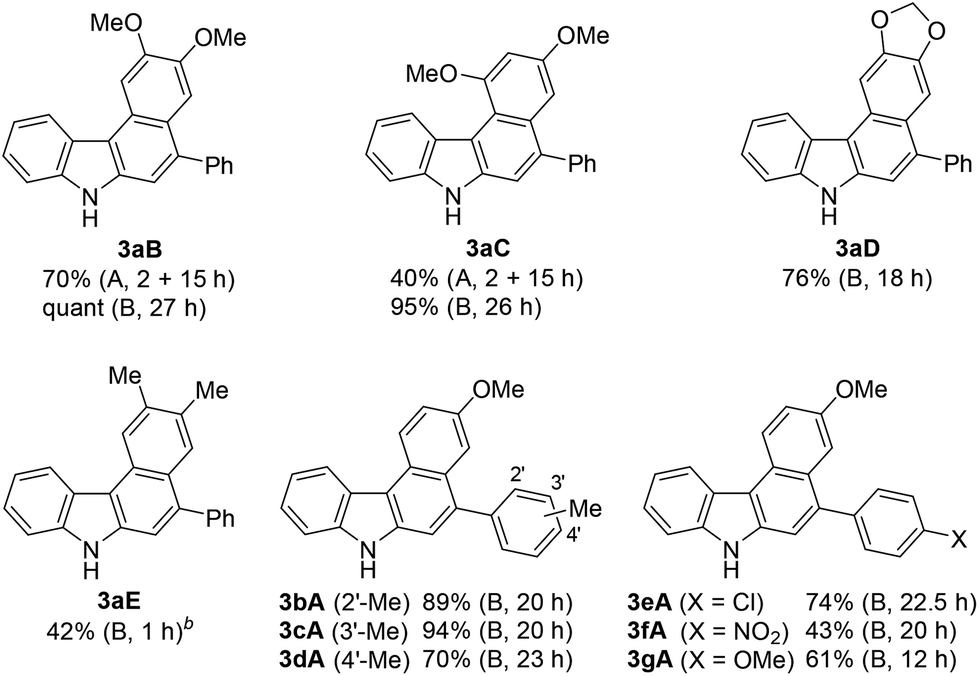
Using these optimised reaction conditions, we then explored the scope of the reaction. Variation of the substitution on the aryl moiety of nucleophile 8 was initially investigated (Table 2). 1,2-Dimethoxybenzene (8B) and 1,3-dimethoxybenzene (8C) served as suitable nucleophiles for gold-catalysed annulation when used as the solvent (condition B) to give benzo[c]carbazoles 3aB (quant) and 3aC (95%) in excellent yields. In these cases, the reaction using 10 equiv. of nucleophile (condition A) also permitted 70% and 40% yields of 3aB and 3aC, respectively. The reaction with benzodioxole (8D) afforded pentacyclic benzo[c]carbazole (3aD) in 76% yield. Less nucleophilic o-xylene (8E) also gave the corresponding benzo[c]carbazole (3aE) in moderate yield (42%), although an increased loading of the gold catalyst (20 mol%) was required. Benzene and toluene did not provide fused carbazoles owing to their lower reactivities. In all cases using arenes 8A–E, the cascade reaction proceeded in a regioselective manner: the first arylation occurred at the para-position of the electron-donating substituent of 8, and the second hydroarylation occurred at the less-sterically-hindered carbon of the introduced aryl group. We next investigated the reaction using various diynes 1b–g under condition B.21 A methyl substituent at the ortho-, meta-, or para-position of the terminal phenyl group was tolerated, producing the corresponding benzo[c]carbazoles (3bA–3dA) in good to excellent yields (70–94%). Similarly, the reaction of 1e–g bearing an electron-donating or -withdrawing group (Cl, NO2, or OMe) at the para-position gave the desired products 3eA–3gA (43–74% yield). The lower yield of the nitro derivative 3fA can be attributed to the less efficient coordination ability of the electron-deficient alkyne(s) to the gold catalyst, which would decrease the probability of the catalyst being activated.
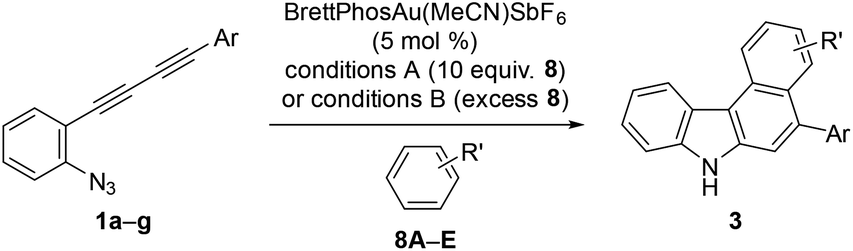
Reaction with pyrrole and indole derivatives
Next, we investigated the synthesis of pyrrolocarbazoles by the reaction with pyrroles 9 (Table 3). The gold-catalysed reaction of conjugated diyne 1a with NH-pyrrole 9A produced an isomeric mixture of two annulation products 6aA and 7aA in ca. 62% yield, along with several unidentified minor products (entry 1). In this case, pyrrolo[3,2-c]carbazole 7aA was obtained as the major isomer (6aA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7aA = 25:75). This result can be readily understood by the more nucleophilic nature of the C2-position of NH-pyrrole than that of the C3-position.22 Expecting that the regioselectivity of nucleophilic attack could be controlled by the steric and electronic factors of pyrrole, we subsequently evaluated the impact of substitution at the pyrrole nitrogen (entries 2–6). As expected, regioselectivity was significantly affected by the N-substituent: N-Boc pyrrole 9F showed the highest regioselectivity to produce pyrrolo[2,3-c]carbazole 6aF (6:7 = 92:8, entry 6), whereas N-benzylpyrrole 9B preferentially produced the corresponding [3,2-c]-isomer 7aB (6:7 = 18:82, entry 2).
:7aA = 25:75). This result can be readily understood by the more nucleophilic nature of the C2-position of NH-pyrrole than that of the C3-position.22 Expecting that the regioselectivity of nucleophilic attack could be controlled by the steric and electronic factors of pyrrole, we subsequently evaluated the impact of substitution at the pyrrole nitrogen (entries 2–6). As expected, regioselectivity was significantly affected by the N-substituent: N-Boc pyrrole 9F showed the highest regioselectivity to produce pyrrolo[2,3-c]carbazole 6aF (6:7 = 92:8, entry 6), whereas N-benzylpyrrole 9B preferentially produced the corresponding [3,2-c]-isomer 7aB (6:7 = 18:82, entry 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Pyrrole | R | Time (h) | Yieldb (%) | Ratioc (6:7) |
| a Reaction conditions: 9 (5 equiv.), BrettPhosAu(MeCN)SbF6 (5 mol%), DCE, and 80 °C. b Combined isolated yields. c Determined by 1H NMR spectroscopy. d Contained small amounts of impurities. e Reaction carried out in TCE at 140 °C using 10 mol% of the catalyst. f Separation of the minor isomer from other by-products was difficult. | |||||
| 1 | 9A | H | 8 | <62%d | 25:75 |
| 2 | 9B | Bn | 10 | 62% | 18:82 |
| 3e | 9C | Ts | 0.5 | 34% | 58:42 |
| 4 | 9D | CO2Me | 1.5 | 62% | 81:19 |
| 5 | 9E | Piv | 1.5 | 60% | 82:18f |
| 6 | 9F | Boc | 1.5 | 60% | 92:8 |
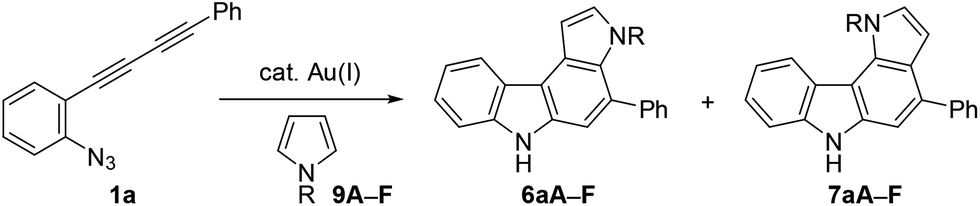
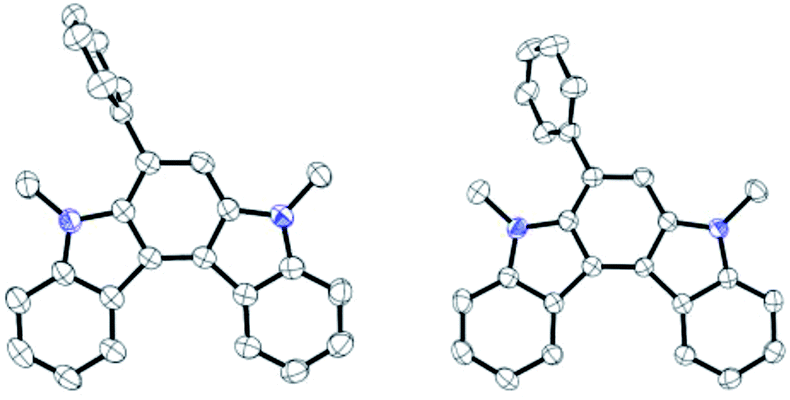
The structural elucidation of 6aF and 7aF was unambiguously made by X-ray crystallographic analyses of the methylation products 6aF-Me2 and 7aF-Me2 (Fig. 3). The pyrrolocarbazole moiety adopted a planar geometry as expected, and the twist angle of the phenyl group was 71.1° (for 6aF-Me2) and 26.2–44.0° (for 7aF-Me2).23 The larger twist angle of the phenyl group in 6aF-Me2 was attributed to the presence of an N-methyl group in close proximity to the phenyl group.
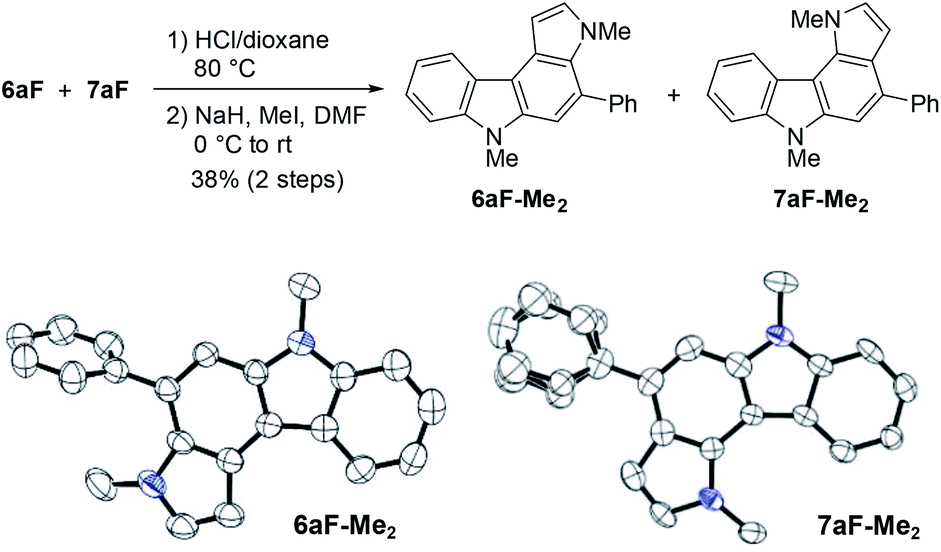 | ||
| Fig. 3 Synthesis and X-ray structures of dimethylated pyrrolocarbazoles. The phenyl group in the latter adopted two orientations in the crystal structure. | ||
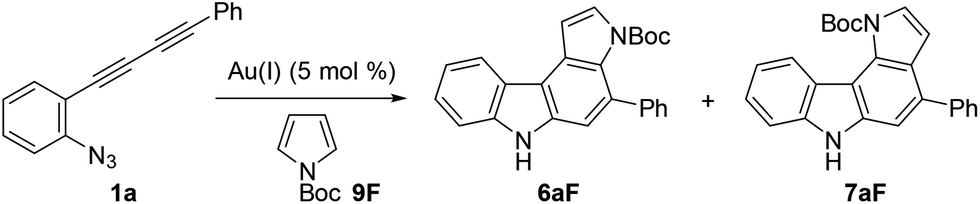
We then optimised the reaction conditions for pyrrolocarbazole formation using diyne 1a, N-Boc-pyrrole 9F (5 equiv.), and various gold catalysts (5 mol%) (Table 4). Whereas Ph3PAuCl/AgNTf2 showed low reactivity (<5% yield, entry 1), other gold complexes bearing IPr, JohnPhos, XPhos, or BrettPhos as the ligand resulted in the formation of pyrrolo[2,3-c]carbazole 6aF in sufficient regioselectivities (>91:9) and moderate yields (55–62%, entries 2–5). Using the most efficient ligand BrettPhos in terms of regioselectivity (6:7 = 94:6, entry 5), two other silver salts were tested (AgSbF6 and AgOTf, entries 6 and 7, respectively); however, the regioselectivity was not improved. The use of a gold complex prepared in advance slightly improved the reactivity (reaction completed within 0.5 h) and regioselectivity (6:7 = 95:5, entries 8 and 9). Solvent screening and investigations of reaction temperature did not improve the yields and product ratios (see ESI†), whereas the reaction at 80 °C was found to be acceptable (entry 10). From these results, we used the conditions shown in entry 8 (condition C) and entry 10 (condition D) for further studies.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Yieldb (%) | Ratioc (6:7) |
| a Reaction conditions: 9F (5 equiv.), gold catalyst (5 mol%), TCE, and 110 °C. b Combined isolated yields. c Determined by 1H NMR spectroscopy. d Contained small amounts of impurities. e The reaction was conducted in DCE at 80 °C. | ||||
| 1 | Ph3PAuCl/AgNTf2 | 24 | <5d | 87:13 |
| 2 | IPrAuCl/AgNTf2 | 1 | 60 | 91:9 |
| 3 | JohnPhosAuCl/AgNTf2 | 1 | 56 | 92:8 |
| 4 | XPhosAuCl/AgNTf2 | 1 | 62 | 93:7 |
| 5 | BrettPhosAuCl/AgNTf2 | 1 | 55 | 94:6 |
| 6 | BrettPhosAuCl/AgSbF6 | 3 | 51 | 89:11 |
| 7 | BrettPhosAuCl/AgOTf | 20 | <12d | 75:25 |
| 8 | BrettPhosAu(MeCN)SbF6, (TCE, 110 °C: condition C) | 0.5 | 58 | 95:5 |
| 9 | BrettPhosAuNTf2 | 0.5 | 58 | 95:5 |
| 10 | BrettPhosAu(MeCN)SbF6 (DCE, 80 °C: condition D)e | 1.5 | 60 | 92:8 |
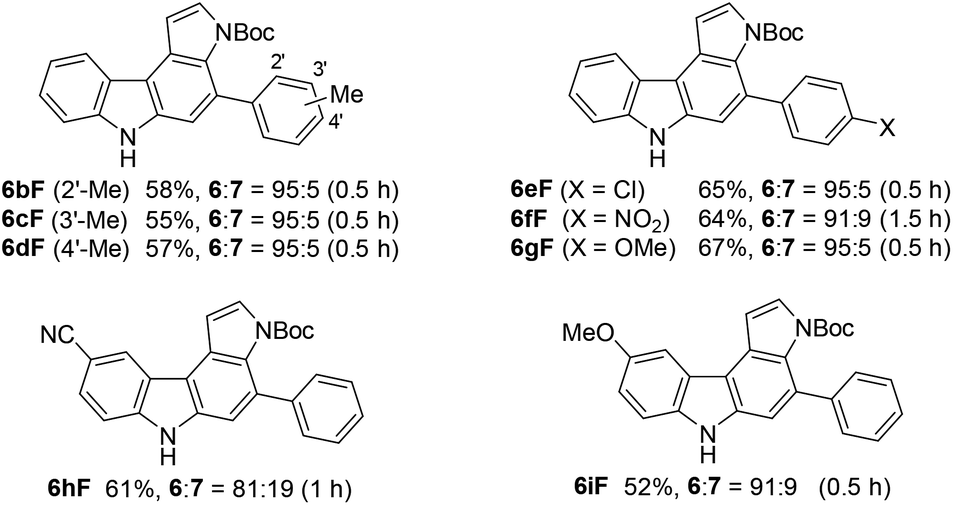
We subsequently investigated the scope of pyrrolo[2,3-c]carbazole formation (Table 5). The conjugated diynes 1b–i bearing electron-donating or -withdrawing substituents on both the aryl groups reacted smoothly with pyrrole 9F to afford the corresponding carbazoles 6bF–6iF under condition C. The position of the methyl group or introduction of chloro or methoxy substituents at the terminal aryl group did not significantly affect the reaction, and the desired annulation products were efficiently produced (6:7 = 95:5). The regioselectivity was slightly decreased when using electron-deficient diyne 1f substituted by a nitro group. Diynes 1h and 1i substituted by a cyano or methoxy group at the para-position to the azido group also showed relatively low selectivities (6:7 = 81:19–91:9).
|
|
|---|
| a Reaction conditions: 9F (5 equiv.), BrettPhosAu(MeCN)SbF6 (5 mol%), TCE, and 110 °C (condition C). |
|

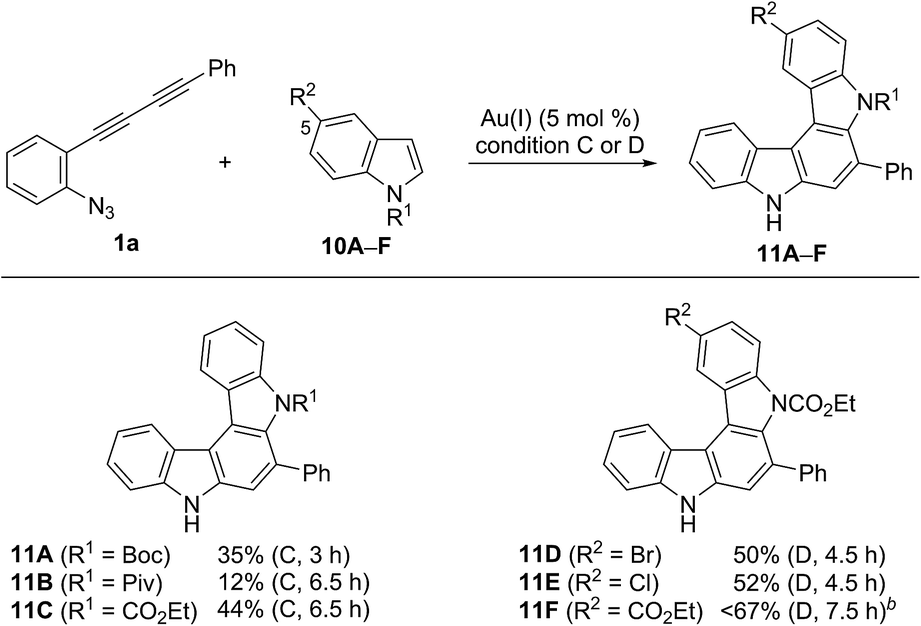
We then applied indole derivatives as the nucleophile for the annulation reaction (Table 6). The reactions of azide-diyne 1a with N-protected indoles 10A–C (R1 = Boc, Piv, or CO2Et) under condition C regioselectively gave the indolo[2,3-c]carbazoles 11A–C as well as several unidentified by-products. The structure of 11A was confirmed by X-ray analysis after cleavage of the N-Boc group and dimethylation,24 similar to the cases of 6aF and 7aF (Fig. 3). Indoles possess reactive sites other than the desired 2- and 3-positions, which may cause undesired side reactions.16b Thus, the introduction of an electron-withdrawing group at the 5-position of indole was examined. As expected, indoles 10D–F bearing a bromo, chloro, or ethoxycarbonyl group at the 5-position reacted more efficiently to afford indolo[2,3-c]carbazoles 11D–F in better yields (50–67%) under condition D.
Reaction mechanism
Although the nucleophilicity of arenes including heteroarenes was well investigated previously, their relative reactivity with N-Boc-pyrrole is not well understood.25 To better understand the reaction mechanism as well as the relative reactivities of arenes employed in this study, several further experiments were performed. First, the exposure of 2aA (obtained during the reaction optimisations shown in Table 1) to the gold-catalysed reaction conditions led to its complete conversion to the corresponding benzo[c]carbazole 3aA in 90% yield (Scheme 5). Second, the reaction of 1a with pyrrole 9F was intentionally stopped before it reached completion, which afforded alkyne-substituted indoles 4aF and 5aF in 25% yield (4:5 = 69:31) along with the recovered starting material. These alkynylindoles were consumed completely under gold-catalysed reaction conditions to produce pyrrolocarbazoles 6aF and 7aF in 67% and 82% yield, respectively. These results strongly indicated that the reactions proceeded through a stepwise nucleophilic attack of the arenes on the gold carbenoid followed by intramolecular hydroarylation, as per our intended reaction pathway.
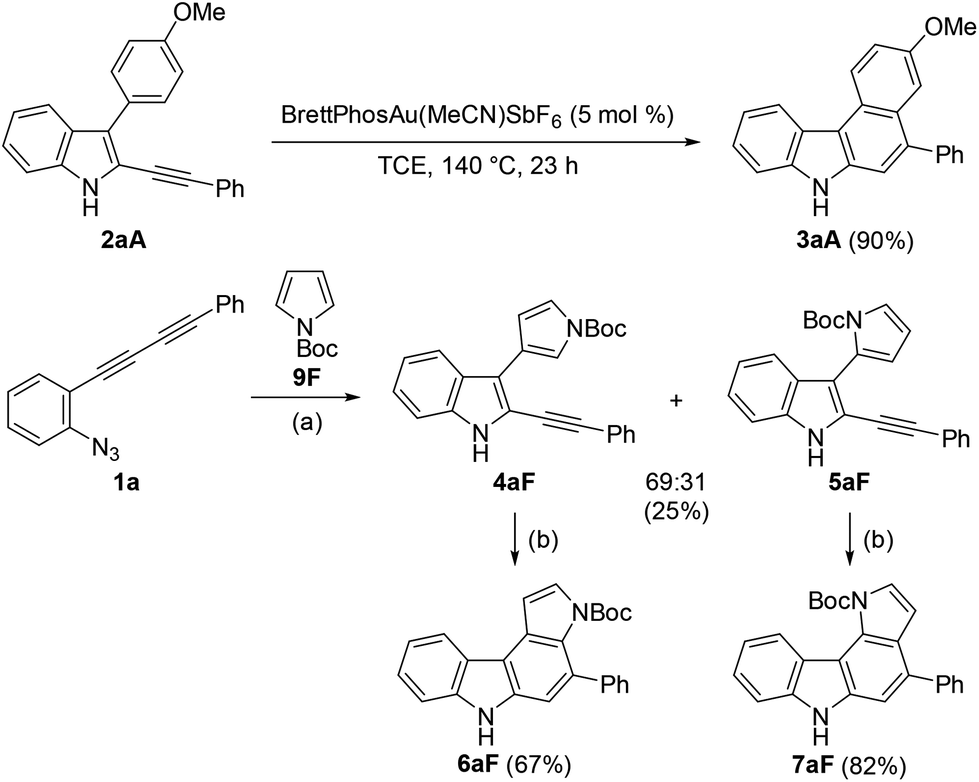 | ||
| Scheme 5 Hydroarylation of the monocyclisation intermediates. Reaction conditions: (a) 9F (5 equiv.), BrettPhosAu(MeCN)SbF6 (5 mol%), DCE, 60 °C, and 1.5 h. (b) BrettPhosAu(MeCN)SbF6 (5 mol%), TCE, 110 °C, and 0.5 h. | ||
Competition experiments using two different arenes were then carried out (Scheme 6). The gold-catalysed reaction of 1a with anisole 8A (10 equiv.) and toluene 8F (10 equiv.) gave the anisole-derived products 2aA (30%) and 3aA (29%) along with a small amount of toluene derivative 2aF (2%) (eqn (1) in Scheme 6). Thus, the first arylation was highly dependent on the nucleophilicity of the arene.25 The competition between anisole 8A (5 equiv.) and N-Boc-pyrrole 9F (5 equiv.) led to the formation of the anisole-derived monocyclised product 2aA (27%) and pyrrole-derived biscyclised product 6aF (41%), the latter being the preferred product (eqn (2) in Scheme 6). This result suggested that N-Boc-pyrrole 9F was a slightly more efficient partner in the first arylation than anisole 8A, and that anisole-derived intermediate 2aA was significantly less reactive for the second arylation than the pyrrole-derived intermediate. The competition reaction using dimethoxybenzene 8B and N-Boc-pyrrole 9F gave the biscyclisation products 3aB (37%) and 6aF (34%) in comparable yields (eqn (3) in Scheme 6). This result suggested that the second arylation was accelerated by the additional methoxy group located at the para-position to the reacting carbon.26 We then examined the kinetic isotope effect (eqn (4) in Scheme 6). The competition reaction using N-Boc-pyrroles 9F (2.5 equiv.) and 9F-d4 (2.5 equiv.) under condition C gave the corresponding pyrrolocarbazoles 6aF and 7aF, where the D/H ratios were 1:1 in both products. Thus, deprotonation was not the rate-determining step for the formation of these products. This result suggested that electrophilic aromatic substitution was more likely for the first arylation than C–H insertion.27
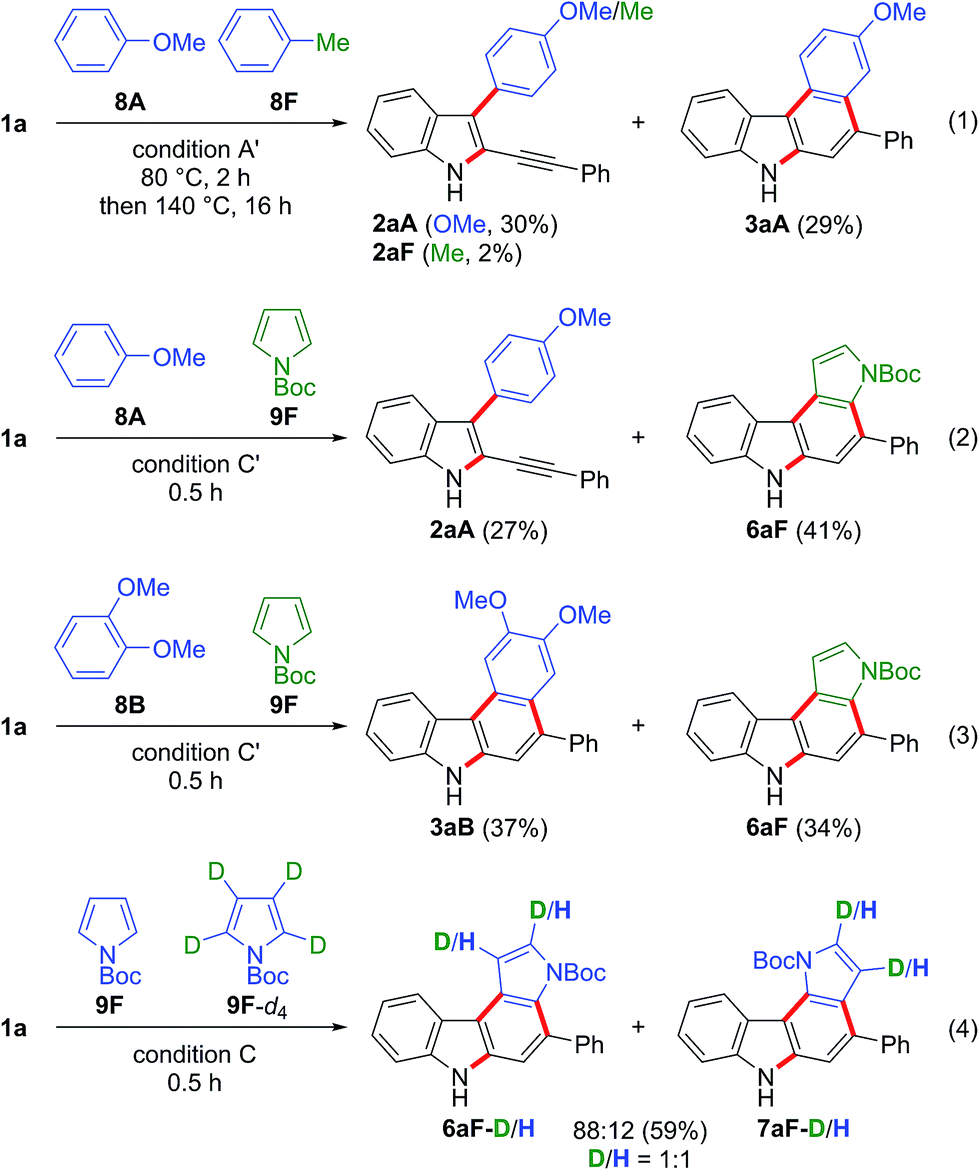 | ||
| Scheme 6 Competition experiments between different nucleophiles. Condition A′: nucleophiles (10 equiv. each), JohnPhosAu(MeCN)SbF6 (10 mol%), TCE, 80 °C and then 140 °C. Condition C/C′: nucleophiles (2.5 equiv. each for condition C; 5 equiv. each for condition C′), BrettPhosAu(MeCN)SbF6 (5 mol%), TCE, and 110 °C. | ||
To further elucidate the reaction mechanism, we undertook density functional theory (DFT) calculations. The calculations were conducted at the M06L/6-31G** (for H, C, N, and P) and SDD (for Au) levels using the formation of pyrrolo[2,3-c]carbazole from 1a and N-methylpyrrole as the model reaction (Fig. 4A). As previously proposed,16 the reaction is initiated by the intramolecular nucleophilic attack of the azide group on the activated alkyne through TS1/2 to form an indolyl-gold intermediate INT2 with a small barrier of 10.9 kcal mol−1 and a rather large endothermicity (10.5 kcal mol−1 higher than INT1). This unfavourable energy loss is compensated for by successive reaction(s). INT2 ejects nitrogen to form a gold carbenoid intermediate INT3-1 with a large stabilisation energy (45.6 kcal mol−1). Next, the key arylation step occurs by the intermolecular nucleophilic attack of N-methylpyrrole on the gold carbenoid INT3-2 through TS3/4, with a small barrier of 1.4 kcal mol−1, to produce INT4. The gold rearrangement from C to N, with a reasonable barrier of 16.6 kcal mol−1, gives an N-aurated indole intermediate INT5-1. This occurs with the simultaneous re-aromatisation, protodeauration, and re-complexation of the gold catalyst with the internal acetylene, and exothermically provides the pyrrole-substituted indole intermediate INT5-2. Finally, 6-endo-dig cyclisation of INT5-2 is promoted by the gold catalyst to produce pyrrolo[2,3-c]carbazole (PD), which regenerates the active gold catalyst. The entire reaction profile is illustrated in Fig. 4B. All the transition states have reasonable energy barriers (1.4–13.1 kcal mol−1). The overall exothermicity is very large because of the formation of one C–N bond, two C–C bonds, and two aromatic rings. This provides the driving force for the overall reaction.
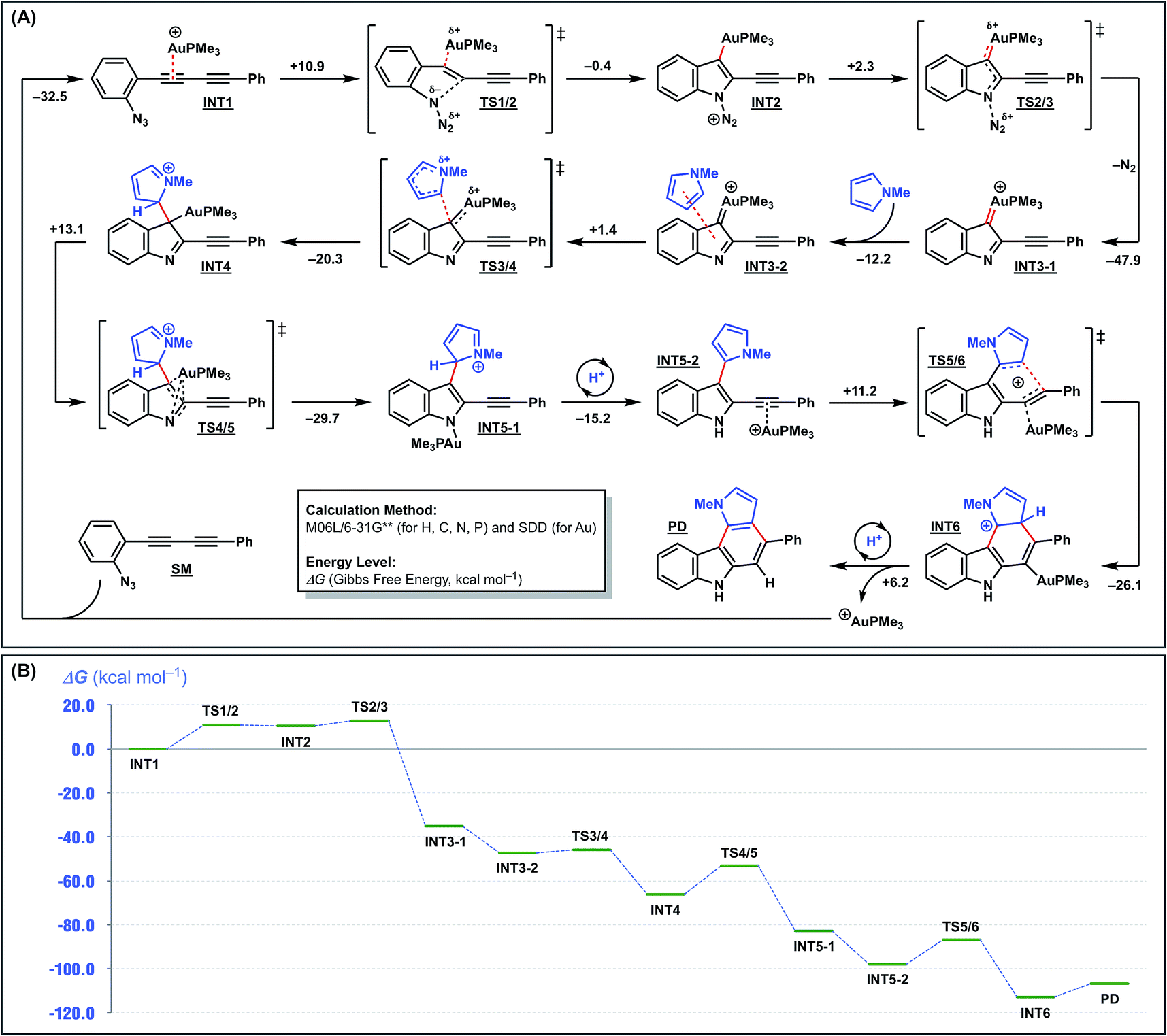 | ||
| Fig. 4 DFT calculations for cyclisation of 1a with N-methylpyrrole [M06L/6-31G** (H, C, N, P) & SDD (Au)]. | ||
Electrochemical investigations of pyrrolo[2,3-c]carbazoles
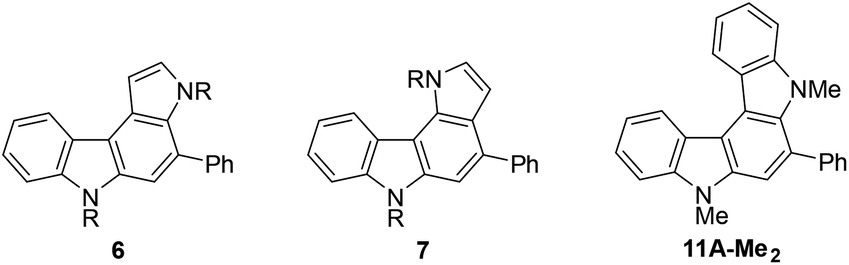
From the viewpoint of the electronic structure, pyrrolo[2,3-c]carbazoles 6 and indolo[2,3-c]carbazoles 11 could be considered as π-fused 1,4-phenylenediamines. Thus, their cation radicals would be generated as persistent species in the π-extended form9c–e,28 of Wurster's blue.29 According to voltammetric analyses in CH2Cl2 (Table 7), the oxidation process of pyrrolo[2,3-c]carbazole 6aF-H was irreversible as in the isomer pyrrolo[3,2-c]carbazole 7aF-H. Substitution with methyl groups at the reactive position (N–H of pyrrole) failed to stabilise the cation radical species, as shown by the irreversible oxidation wave for 6aF-Me2. This was despite the electron-donating nature of the substituents marginally facilitating electrochemical oxidation, as indicated by the less positive oxidation potentials. By fusing the benzene nucleus in 6aF-Me2 to furnish the indolo[2,3-c]carbazole skeleton, the cation radical species could attain enough persistency. 11A-Me2 underwent reversible two-stage one-electron oxidation processes, as shown by the voltammogram (Fig. 5). The redox pathway can be postulated as shown in the scheme, similar to that for 1,4-phenylenediamine.|
|
|||
|---|---|---|---|
| Entry | Compound | R | Oxidation potentiala |
| a E/V vs. SCE, CH2Cl2 containing 0.1 M Bu4NPF6, Pt electrode, and 100 mV s−1. Eox = Epa − 0.03 V (for entries 1–5). E(Fc/Fc+) = +0.53 V under similar conditions. b Reversible redox reaction was observed. | |||
| 1 | 6aF-H | H | +0.85 |
| 2 | 6aF-Me2 | Me | +0.79 |
| 3 | 7aF-H | H | +0.73 |
| 4 | 7aF-Me 2 | Me | +0.75 |
| 5 | 11A-Me 2 | — | +0.80b |
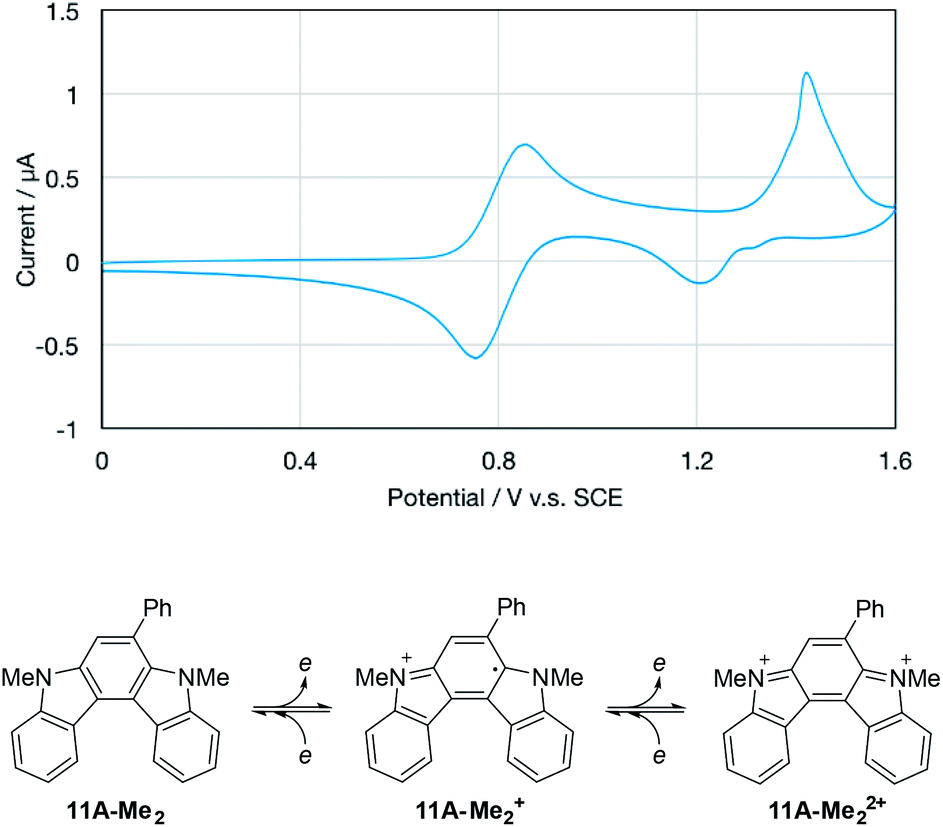 | ||
| Fig. 5 Cyclic voltammogram of 11A-Me2 in CH2Cl2 (upper panel) and the possible redox pathway (lower panel). The irregular peak shape for the second oxidation wave may have been related to partial adsorption of the doubly-charged species on the electrode. | ||
Upon the electrochemical oxidation of 11A-Me2 in CH2Cl2, the colourless solution turned green, which demonstrated its electrochromic nature. A continuous change in ultraviolet-visible-near-infrared (UV-Vis-NIR) absorption was accompanied by several isosbestic points, indicating that 11A-Me2 was cleanly oxidised into the corresponding cation radical species (Fig. 6). Wurster's blue exhibits absorption only in the visible region (λ < 700 nm). Thus, the observed red shift was induced through π-extension by the fusion of the indole rings. The carbazole skeleton gives rise to fluorescence properties,30 so the electrolysis of 11A-Me2 also caused a change in the fluorescence (FL) spectrum [λem 424, 442 (sh) nm in CH2Cl2 (λex 354 nm)]. The steady decrease in fluorescence with increasing electrochemical oxidation time could be rationalised by the non-fluorescent nature of its cation radical. Such dual electrochromism in which changes occur in both UV-Vis-NIR and FL spectra is rare,30 but was also realised in our previous study on benzo[g]indolo[2,3-c]carbazole derivatives,9c–e which were synthesised through a different mode of the gold(I)-catalysed cascade reaction.9a Thus, the gold-catalysed synthesis of annulated carbazoles is a powerful tool for exploring the little developed category of advanced electrochromic systems.
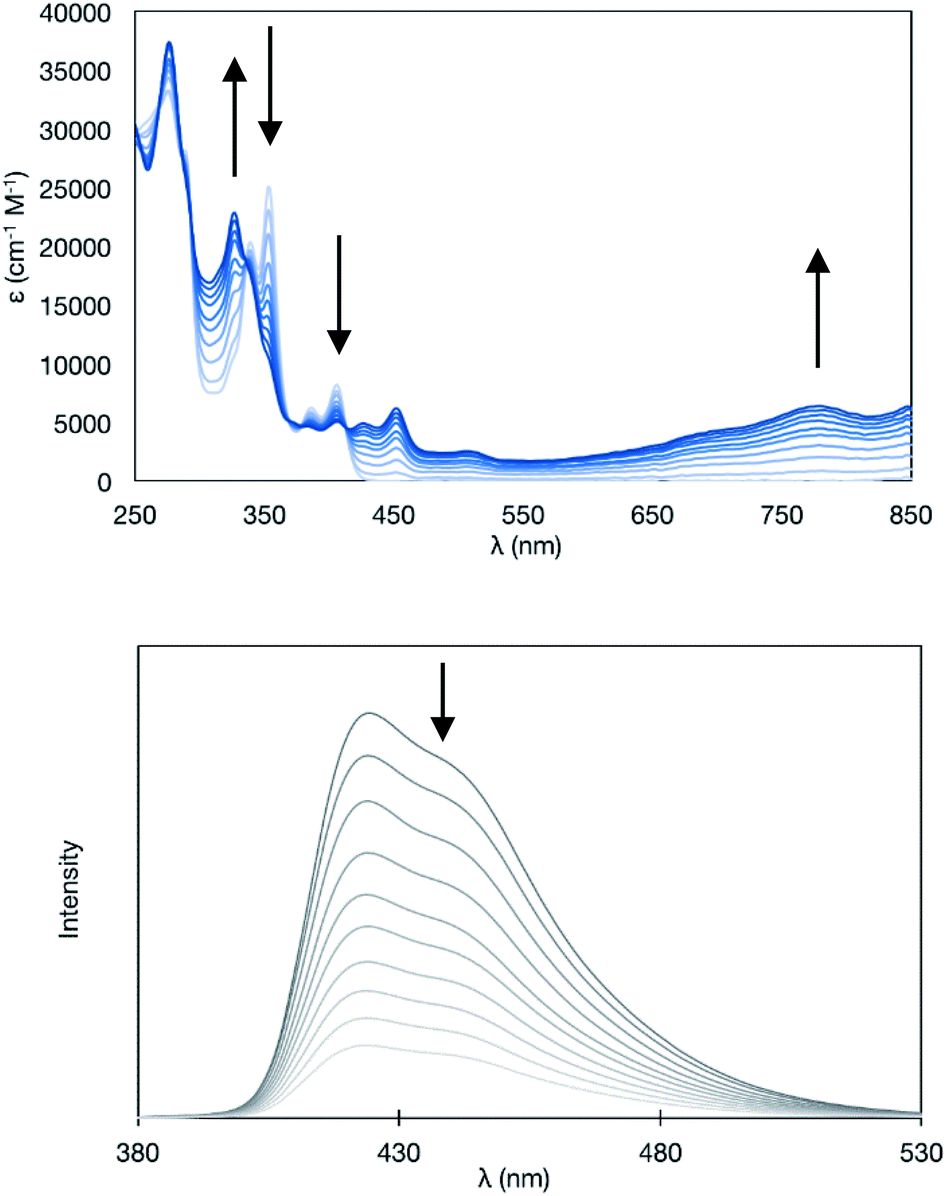 | ||
| Fig. 6 Continuous changes in UV-Vis-NIR (upper panel) and fluorescence (lower panel) spectra upon constant current electrochemical oxidation of 11A-Me2 [in CH2Cl2 (7.1 × 10−6M) containing 0.05 M Bu4NPF6 (20 μA, every 8 min)]. | ||
Conclusions
We have developed a strategy for synthesising aryl-annulated [c]carbazoles through the gold-catalysed cascade cyclisation of azido-diynes. The reaction with electron-rich benzenes such as anisole and xylene gave benzo[c]carbazoles via the functionalisation of two benzene C–H bonds. The use of N-Boc-pyrrole and indoles as a coupling partner regioselectively produced their corresponding heteroaryl-annulated carbazoles, namely pyrrolo[2,3-c]carbazoles and indolo[2,3-c]carbazoles, respectively. The reaction proceeded through the intramolecular nucleophilic attack of azide on the proximal alkyne to form a gold carbenoid species, nucleophilic attack of arenes on the carbenoid, and subsequent 6-endo-dig cyclisation of the introduced arene to the other alkyne. This proposed reaction mechanism was well supported by the results of DFT calculations, competition experiments, and deuterium-labeling experiments. An N,N′-dimethylated derivative of indolo[2,3-c]carbazole showed dual UV-Vis-NIR and fluorescence spectral changes on electrolysis, which demonstrates the potential utility of this reaction in materials chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JSPS KAKENHI [Grant numbers JP15KT0061 (HO), 18H04408 (HO), 18H04376 (TK), 17H05430 (MU), and 17H06173 (MU)], the Platform Project for Supporting Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Japan Agency for Medical Research and Development (AMED), and the Hoansha Foundation. The generous allotment of computational resources (Projects G17022 and G18008) from HOKUSAI GreatWave (RIKEN) is gratefully acknowledged.Notes and references
- For reviews, see: (a) A. W. Schmidt, K. R. Reddy and H.-J. Knölker, Chem. Rev., 2012, 112, 3193–3328 CrossRef CAS PubMed; (b) J.-P. Lellouche, R. R. Koner and S. Ghosh, Rev. Chem. Eng., 2013, 29, 413–437 CAS; (c) A. Venkateswararao and K. R. J. Thomas, Sol. Cell Nanotechnol., 2014, 41–96 CAS.
- For recent examples, see: (a) C. Jiao, K.-W. Huang, J. Luo, K. Zhang, C. Chi and J. Wu, Org. Lett., 2009, 11, 4508–4511 CrossRef CAS PubMed; (b) A. D. Hendsbee, J.-P. Sun, W. K. Law, H. Yan, I. G. Hill, D. M. Spasyuk and G. C. Welch, Chem. Mater., 2016, 28, 7098–7109 CrossRef CAS; (c) S. M. Kim, J. H. Yun, S. H. Han and J. Y. Lee, J. Mater. Chem. C, 2017, 5, 9072–9079 RSC.
- For recent reviews, see: (a) N. Yoshikai and Y. Wei, Asian J. Org. Chem., 2013, 2, 466–478 CrossRef CAS; (b) J. Roy, A. K. Jana and D. Mal, Tetrahedron, 2012, 68, 6099–6121 CrossRef CAS.
- For recent examples of aryl-annulated [c]carbazole syntheses, see: (a) M. E. Budén, V. A. Vaillard, S. E. Martin and R. A. Rossi, J. Org. Chem., 2009, 74, 4490–4498 CrossRef PubMed; (b) T. Chatterjee, G. Roh, M. A. Shoaib, C.-H. Suhl, J. S. Kim, C.-G. Cho and E. J. Cho, Org. Lett., 2017, 19, 1906–1909 CrossRef CAS PubMed.
- For our recent studies on carbazole synthesis, see: (a) T. Watanabe, S. Ueda, S. Inuki, S. Oishi, N. Fujii and H. Ohno, Chem. Commun., 2007, 4516–4518 RSC; (b) T. Watanabe, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2009, 74, 4720–4726 CrossRef CAS PubMed; (c) T. Takeuchi, S. Oishi, T. Watanabe, H. Ohno, J. Sawada, K. Matsuno, A. Asai, N. Asada, K. Kitaura and N. Fujii, J. Med. Chem., 2011, 54, 4839–4846 CrossRef CAS PubMed.
- (a) S. K. Ghosh, B.-C. Kuo, H.-Y. Chen, J.-Y. Li, S.-D. Liu and H. M. Lee, Eur. J. Org. Chem., 2015, 4131–4142 CrossRef CAS; (b) X.-Q. Feng, F. Zhang, X.-P. He, G.-R. Chen, X.-Y. Wu and F. Sha, RSC Adv., 2016, 6, 75162–75165 RSC; (c) F. Yu, D. Li, Y. Wei, R.-M. Kang and Q.-X. Guo, Tetrahedron, 2018, 74, 1965–1972 CrossRef CAS.
- (a) Y. Nagase, H. Shirai, M. Kaneko, E. Shirakawa and T. Tsuchimoto, Org. Biomol. Chem., 2013, 11, 1456–1459 RSC; (b) J. K. Tan, M. Mathiew, S. Nayak and P. W. H. Chan, Chem.–Asian J., 2017, 12, 1475–1479 CrossRef CAS PubMed.
- (a) R. Y. Huang, P. T. Franke, N. Nicolaus and M. Lautens, Tetrahedron, 2013, 69, 4395–4402 CrossRef CAS; (b) P. Raju and A. K. Mohanakrishnan, Eur. J. Org. Chem., 2016, 4361–4371 CrossRef CAS.
- For our studies on annulated [c]carbazoles, see: (a) K. Hirano, Y. Inaba, K. Takasu, S. Oishi, Y. Takemoto, N. Fujii and H. Ohno, J. Org. Chem., 2011, 76, 9068–9080 CrossRef CAS PubMed; (b) M. Taguchi, Y. Tokimizu, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2015, 17, 6250–6253 CrossRef CAS PubMed; see also: (c) T. Suzuki, Y. Tokimizu, Y. Sakano, R. Katoono, K. Fujiwara, S. Naoe, N. Fujii and H. Ohno, Chem. Lett., 2013, 42, 1001–1003 CrossRef CAS; (d) T. Suzuki, Y. Sakano, Y. Tokimizu, Y. Miura, R. Katoono, K. Fujiwara, N. Yoshioka, N. Fujii and H. Ohno, Chem.–Asian J., 2014, 9, 1841–1846 CrossRef CAS PubMed; (e) T. Suzuki, W. Nojo, Y. Sakano, R. Katoono, Y. Ishigaki, H. Ohno and K. Fujiwara, Chem. Lett., 2016, 45, 720–722 CrossRef CAS.
- Quite recently, the synthesis of aryl-annulated [c]carbazoles via a silver-mediated amination/cyclisation/aromatisation cascade was reported: X. Fan, L.-Z. Yu, Y. Wei and M. Shi, Org. Lett., 2017, 19, 4476–4479 CrossRef CAS PubMed.
- For carbazole syntheses based on rhodium-catalysed [2 + 2 + 2] cycloisomerisation of alkynes, see: B. Witulski and C. Alayrac, Angew. Chem., Int. Ed., 2002, 41, 3281–3284 CrossRef CAS PubMed.
- For selected reviews, see: (a) H. Ohno, Isr. J. Chem., 2013, 53, 869–882 CrossRef CAS; (b) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed; (c) D. Pflästerer and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331–1367 RSC; (d) Y. Li, W. Li and J. Zhang, Chem.–Eur. J., 2017, 23, 467–512 CrossRef CAS PubMed.
- (a) A. M. Asiria and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 4471–4503 RSC; (b) S. M. Abu Sohel and R.-S. Liu, Chem. Soc. Rev., 2009, 38, 2269–2281 RSC.
- (a) Y. Matsuda, S. Naoe, S. Oishi, N. Fujii and H. Ohno, Chem.–Eur. J., 2015, 21, 1463–1467 CrossRef CAS PubMed; (b) N. Hamada, Y. Yoshida, S. Oishi and H. Ohno, Org. Lett., 2017, 19, 3875–3878 CrossRef CAS PubMed.
- For the pioneering work, see: (a) D. J. Gorin, N. R. Davis and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 11260–11261 CrossRef CAS PubMed; for a review, see: (b) P. W. Davies and M. Garzón, Asian J. Org. Chem., 2015, 4, 694–708 CrossRef CAS.
- (a) A. Wetzel and F. Gagosz, Angew. Chem., Int. Ed., 2011, 50, 7354–7358 CrossRef CAS PubMed; (b) B. Lu, Y. Luo, L. Liu, L. Ye, Y. Wang and L. Zhang, Angew. Chem., Int. Ed., 2011, 50, 8358–8362 CrossRef CAS PubMed.
- For related studies based on azide-derived gold carbenoids, see: (a) Z.-Y. Yan, Y. Xiao and L. Zhang, Angew. Chem., Int. Ed., 2012, 51, 8624–8627 CrossRef CAS PubMed; (b) C. Gronnier, G. Boissonnat and F. Gagosz, Org. Lett., 2013, 15, 4234–4237 CrossRef CAS PubMed; (c) N. Li, T. Wang, L. Gong and L. Zhang, Chem.–Eur. J., 2015, 21, 3585–3588 CrossRef CAS PubMed; (d) C. Shu, Y.-H. Wang, B. Zhou, X.-L. Li, Y.-F. Ping, X. Lu and L.-W. Ye, J. Am. Chem. Soc., 2015, 137, 9567–9570 CrossRef CAS PubMed; (e) Y. Wu, L. Zhu, Y. Yu, X. Luo and X. Huang, J. Org. Chem., 2015, 80, 11407–11416 CrossRef CAS PubMed; (f) N. Li, X.-L. Lian, Y.-H. Li, T.-Y. Wang, Z.-Y. Han, L. Zhang and L.-Z. Gong, Org. Lett., 2016, 18, 4178–4181 CrossRef CAS PubMed; (g) C. Shu, Y.-H. Wang, C.-H. Shen, P.-P. Ruan, X. Lu and L.-W. Ye, Org. Lett., 2016, 18, 3254–3257 CrossRef CAS PubMed; (h) G. H. Lonca, C. Tejo, H. L. Chan, S. Chiba and F. Gagosz, Chem. Commun., 2017, 53, 736–739 RSC; (i) J. Cai, B. Wu, G. Rong, C. Zhang, L. Qiu and X. Xu, Org. Lett., 2018, 20, 2733–2736 CrossRef CAS PubMed; for oxidative cyclisation of azido-diynes for the synthesis of fused quinolines and indoles, see: (j) W.-B. Shen, Q. Sun, L. Li, X. Liu, B. Zhou, J.-Z. Yan, X. Lu and L.-W. Ye, Nat. Commun., 2017, 8, 1748 CrossRef PubMed.
- For pioneering studies on the gold-catalysed hydroarylation of alkynes, see: (a) A. Fürstner and V. Mamane, J. Org. Chem., 2002, 67, 6264–6267 CrossRef; (b) V. Mamane, P. Hannen and A. Fürstner, Chem.–Eur. J., 2004, 10, 4556–4575 CrossRef CAS PubMed; (c) E. Soriano and J. Marco-Contelles, Organometallics, 2006, 25, 4542–4553 CrossRef CAS; for a recent theoretical study, see: (d) V. M. Lau, W. C. Pfalzgraff, T. E. Markland and M. W. Kanan, J. Am. Chem. Soc., 2017, 139, 4035–4041 CrossRef CAS PubMed.
- A portion of this study for the total synthesis of dictyodendrins has already been disclosed as a preliminary communication: J. Matsuoka, Y. Matsuda, Y. Kawada, S. Oishi and H. Ohno, Angew. Chem., Int. Ed., 2017, 56, 7444–7448 CrossRef CAS PubMed.
- (a) A. Padwa, D. J. Austin, Y. Gareau, J. M. Kassir and S. L. Xu, J. Am. Chem. Soc., 1993, 115, 2637–2647 CrossRef CAS; for a recent review, see: (b) K. S. Sindhu, A. P. Thankachan, P. S. Sajitha and G. Anilkumar, Org. Biomol. Chem., 2015, 13, 6891–6905 RSC.
- The substituent effect at the terminal position is a subject of future investigation..
- For the C2-selective addition of pyrrole in the gold-catalyzed reaction of alkynes, see: S. Naoe, Y. Suzuki, K. Hirano, Y. Inaba, S. Oishi, N. Fujii and H. Ohno, J. Org. Chem., 2012, 77, 4907–4916 CrossRef CAS PubMed.
- In single crystals of 7aF-Me2, two crystallographically independent molecules, both of which were disordered at the phenyl group..
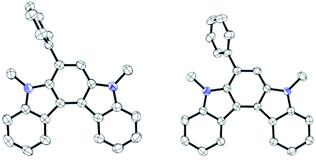
- Recrystallisation of 11A-Me2 from CH2Cl2 gave four crystallographically independent molecules in the single crystal lattices, two of which are shown below. As expected, the indolo[2,3-c]carbazole core had a planar structure in all four molecules.
 .
. - (a) S. Pratihar and S. Roy, J. Org. Chem., 2010, 75, 4957–4963 CrossRef CAS PubMed; (b) T. A. Nigst, M. Westermaier, A. R. Ofial and H. Mayr, Eur. J. Org. Chem., 2008, 2369–2374 CrossRef CAS; for the nucleophilic reaction of N-Boc-pyrrole at the 2-position, see: (c) J. Tang, J.-J. Yue, F.-F. Tao, G. Grampp, B.-X. Wang, F. Li, X.-Z. Liang, Y.-M. Shen and J.-H. Xu, J. Org. Chem., 2014, 79, 7572–7582 CrossRef CAS PubMed.
- D. Pflästerer, S. Schumacher, M. Rudolph and A. S. K. Hashmi, Chem.–Eur. J., 2015, 21, 11585–11589 CrossRef PubMed.
- C–H insertion is often observed in gold-carbenoid chemistry, see: (a) M. R. Fructos, T. R. Belderrain, P. de Frémont, N. M. Scott, S. P. Nolan, M. M. Díaz-Requejo and P. J. Pérez, Angew. Chem., Int. Ed., 2005, 44, 5284–5288 CrossRef CAS PubMed; (b) S. Bhunia and R.-S. Liu, J. Am. Chem. Soc., 2008, 130, 16488–16489 CrossRef CAS PubMed; (c) L. Cui, G. Zhang, Y. Peng and L. Zhang, Org. Lett., 2009, 11, 1225–1228 CrossRef CAS PubMed; (d) Y. Horino, T. Yamamoto, K. Ueda, S. Kuroda and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 2809–2811 CrossRef CAS PubMed; (e) A. S. K. Hashmi, I. Braun, P. Nösel, J. Schädlich, M. Wieteck, M. Rudolph and F. Rominger, Angew. Chem., Int. Ed., 2012, 51, 4456–4460 CrossRef CAS PubMed; (f) L. Ye, Y. Wang, D. H. Aue and L. Zhang, J. Am. Chem. Soc., 2012, 134, 31–34 CrossRef CAS PubMed; (g) S. Bhunia, S. Ghorpade, D. B. Huple and R.-S. Liu, Angew. Chem., Int. Ed., 2012, 51, 2939–2942 CrossRef CAS PubMed; (h) F. Pan, S. Liu, R.-K. Lin, Y.-F. Yu, J.-M. Zhou and L.-W. Ye, Chem. Commun., 2014, 50, 10726–10729 RSC; (i) Y. Wang, M. Zarca, L.-Z. Gong and L. Zhang, J. Am. Chem. Soc., 2016, 138, 7516–7519 CrossRef CAS PubMed; (j) J. E. M. N. Klein, G. Knizia, L. N. dos Santos Comprido, J. Kästner and A. S. K. Hashmi, Chem.–Eur. J., 2017, 23, 16097–16103 CrossRef CAS PubMed.
- (a) T. Suzuki, T. Tsuji, T. Ohkubo, A. Okada, Y. Obana, T. Fukushima, T. Miyashi and Y. Yamashita, J. Org. Chem., 2001, 66, 8954–8960 CrossRef CAS PubMed; (b) T. Suzuki, Y. Tsubata, Y. Obana, T. Fukushima, T. Miyashi, H. Kawai, K. Fujiwara and K. Akiyama, Tetrahedron Lett., 2003, 44, 7881–7884 CrossRef CAS.
- (a) C. Wurster and R. Sendtner, Ber. Dtsch. Chem. Ges., 1897, 12, 1803 CrossRef; (b) L. Michaelis, M. P. Shubert and S. Granick, J. Am. Chem. Soc., 1939, 61, 1981–1992 CrossRef CAS; (c) J. R. Bolton, A. Carringon and J. Santos-Veiga, Mol. Phys., 1962, 5, 615–619 CrossRef CAS; (d) J. Steigman and W. Cronkright, J. Am. Chem. Soc., 1970, 92, 6736–6743 CrossRef CAS.
- (a) M. Luo, H. Shadnia, G. Qian, X. Du, D. Yu, D. Ma, J. S. Wright and Z. Y. Wang, Chem.–Eur. J., 2009, 15, 8902–8908 CrossRef CAS PubMed; (b) Y. Ishigaki, H. Kawai, R. Katoono, K. Fujiwara, H. Higuchi, H. Kikuchi and T. Suzuki, Can. J. Chem., 2017, 95, 243–252 CrossRef CAS; (c) M. Walesa-Chorab and W. G. Skene, ACS Appl. Mater. Interfaces, 2017, 9, 21524–21531 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data are provided. CCDC 1860899 and 1860900 (6aF-Me2), 1860901 and 1860902 (7aF-Me2), and 1860904 and 1860905 (11A-Me2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03525c |
| ‡ Y. K. and S.O. contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |