DOI:
10.1039/C8SC03510E
(Edge Article)
Chem. Sci., 2019,
10, 198-207
Molecular design of near-infrared fluorescent Pdots for tumor targeting: aggregation-induced emission versus anti-aggregation-caused quenching†
Received
7th August 2018
, Accepted 3rd October 2018
First published on 4th October 2018
Abstract
Semiconducting polymer dots (Pdots) have recently emerged as a new type of ultrabright fluorescent probe that has been proved to be very useful for biomedical imaging. However, Pdots often suffer from serious fluorescence aggregation-caused quenching (ACQ) especially for near-infrared (NIR) fluorescent Pdots. This article compared two strategies to overcome the ACQ effect in near-infrared emissive Pdot systems: aggregation-induced emission (AIE) and anti-aggregation-caused quenching (anti-ACQ). The results show that the anti-ACQ platform outperforms the AIE system. The fluorescence quantum yield of anti-ACQ-based Pdots can be over 50% and the average per-particle brightness of the Pdots is about 5 times higher than that of the commercially available quantum dots. To help understand why the monomer conformations could greatly affect the optical properties of Pdots, molecular dynamics simulations were performed for the first time in such complicated Pdot systems. To demonstrate applications for in vivo fluorescence imaging, both microangiography imaging on living zebrafish embryos and specific tumor targeting on mice were performed. We anticipate that these studies will pave the way for the design of new highly fluorescent Pdot systems.
Introduction
In vivo biomedical fluorescence imaging has been extensively used to study physiological and pathological models of human diseases, with advantages of its real-time/wide-field acquisition ability, exceptional spatial resolution, and non-invasive visualization. Over the past years, fluorescence imaging has been successfully translated into a clinical imaging modality due to its exciting advances in the development of fluorescence-guided surgery systems in human patients, including the fluorescence-assisted resection and exploration (FLARE) imaging system and commercialized SurgVision platform.1–4 Despite the fast-growing progress in in vivo fluorescence imaging, traditional excitation wavelengths in the visible region (400–700 nm) have hindered its clinical use due to the significant photon attenuation by endogenous molecules.5,6 Compared with the visible light spectrum, the near-infrared (NIR) window (700–1700 nm) can travel several millimetres to centimeters in most mammalian tissues due to less light scattering/absorption, and reduced interference from tissue autofluorescence. However, the currently FDA-approved NIR fluorophores still have major limitations. For example, indocyanine green and methylene blue have no specific targeting ability to specific tissues and their net charges on the chemical structures often cause non-specific adsorption to proteins. Therefore, an increasing amount of research has been devoted to the development of functional NIR fluorescent agents including nanoparticle-based probes.7–15
Recently, semiconducting polymer dots (Pdots) have emerged as a new type of organic fluorescent nanoparticle with extraordinary fluorescence brightness, high photostability, large Stokes shift, and enhanced energy-transfer properties.16–28 As compared to other nanoparticle-based fluorescent probes such as dye-doped polymeric nanoparticles or semiconductor quantum dots, the advantages of Pdots include facile surface functionalization, excellent biocompatibility (surfactant free and no leaching issue of heavy metal elements), and superior absorption cross-sections, making them extremely suitable for biological and medical applications.26,29–41 Despite the many advantages, the exploitation of NIR-emitting Pdots for in vivo bioimaging has been hampered by their low fluorescence quantum yields due to the serious fluorescence aggregation-caused quenching (ACQ) in Pdot form.42,43 Chiu's and our groups have focused on ameliorating the ACQ effect by either reducing the molar concentrations of NIR monomers or introducing steric hindrance on the polymer backbones.44–47 While the low molar concentration of NIR units compromises the photostability of the resulting Pdots,47 the incorporation of bulky π-bridges into the polymer backbones45,47 or the design of hyperbranched/dendronized architectures on the polymers44,46 in an effort to suppress self-quenching remains the main strategy to obtain NIR fluorescent Pdots with both high brightness and good photostability.
Almost all of the aforementioned studies employed light-harvesting fluorene segments as the energy donor in semiconducting polymers but the absorption of polyfluorene (350–400 nm) in the ultraviolet region is less friendly for biological imaging. In our previous studies, we have used fluorene as a bridge extender rather than an energy donor and at the same time tailored fluorene with carboxyl groups on its side chains for specific cellular labeling.47 In this scenario, however, fluorene simply serves as the polymer linker and functional group but barely contributes to steric hindrance. Herein, we designed and synthesized two donor-type monomers with opposite characteristics, a tetraphenylethene (TPE)-based aggregation induced emission (AIE)-active fluorogen and a rigid three-dimensional pentiptycene (Pttc)-based moiety; and then compared their optical performance with the fluorene counterparts. We further performed molecular dynamics simulations on our Pdot systems and then compared them with our experimental results.
Results and discussion
Our aim was to tackle the commonly encountered ACQ problem of Pdots, especially for NIR-fluorescent Pdots due to the rigid and flat scaffolds of NIR semiconducting polymers. We would like to obtain NIR-emitting Pdots with high fluorescence quantum yields by altering the chemical conformations of the donors (i.e. bridge extenders in this work) to circumvent π–π stacking as shown in Scheme 1. We also conducted theoretical calculations of these molecules to understand how the donors affect the optical properties of the resulting Pdots. Their biological applications including in vitro specific cellular imaging and in vivo tumor targeting were also demonstrated.
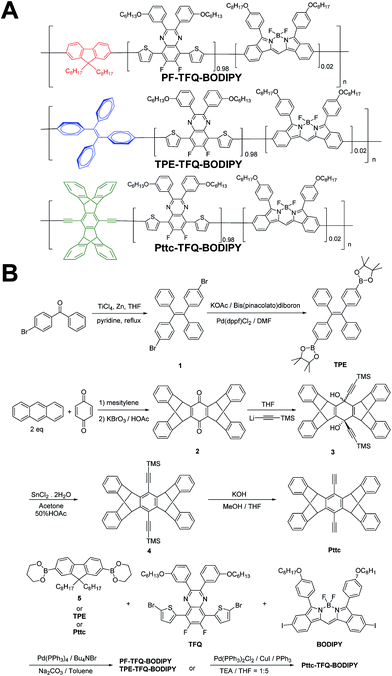 |
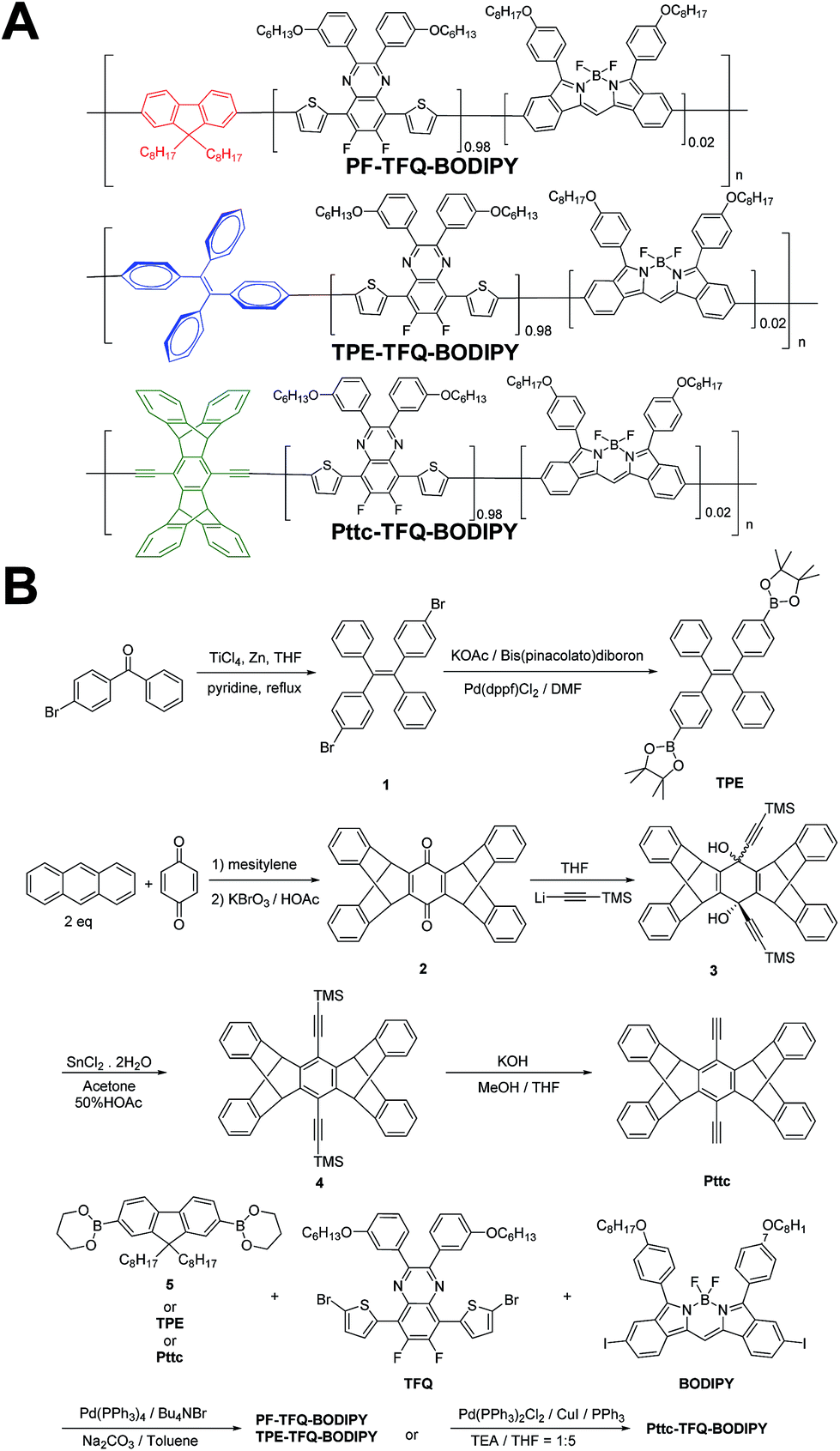
| | Scheme 1 Schematic showing (A) the chemical structures of three NIR semiconducting polymers with different donors and (B) their synthetic routes. | |
Design and synthesis of narrow-band and NIR fluorescent Pdots with different donors
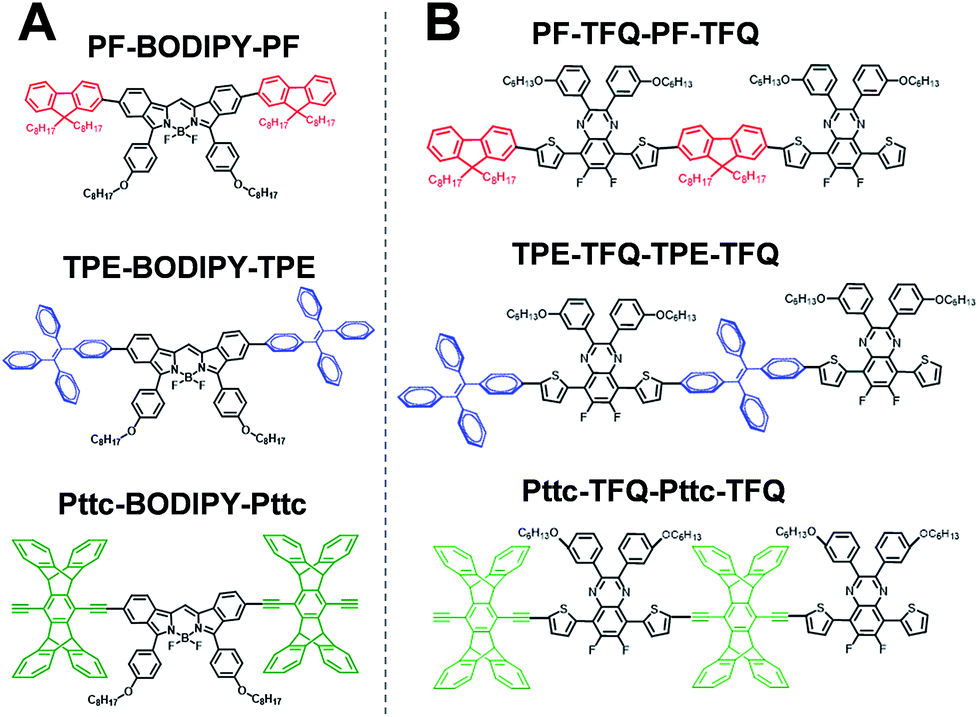
The general synthetic procedures for the monomers and polymers are described in Scheme 1. Herein, we selected BODIPY as our acceptor because of its narrow-band and NIR fluorescence properties, while the monomer TFQ served as our π-bridge owing to the substantial spectral overlap between the TFQ emission (∼660 nm) and the BODIPY absorption (∼700 nm) for efficient energy transfer.45,48 For the choice of donors, fluorene served as the control because it is most commonly used in Pdot-based systems. AIE-active TPE and sterically bulky three-dimensional Pttc donors were synthesized to compare with fluorene. The three donors (i.e., fluorene, TPE, and Pttc) were synthesized according to previously reported studies with modified procedures,42,49–53 while the π-bridge (TFQ) and the NIR acceptor (BODIPY) were synthesized based on our reported methods.45,47 Finally, monomers were polymerized through palladium-catalyzed Suzuki or Sonogashira coupling, yielding three NIR fluorescent semiconducting polymers, PF-TFQ-BODIPY, TEP-TFQ-BODIPY, and Pttc-TFQ-BODIPY.
Preparation and characterization of Pdots
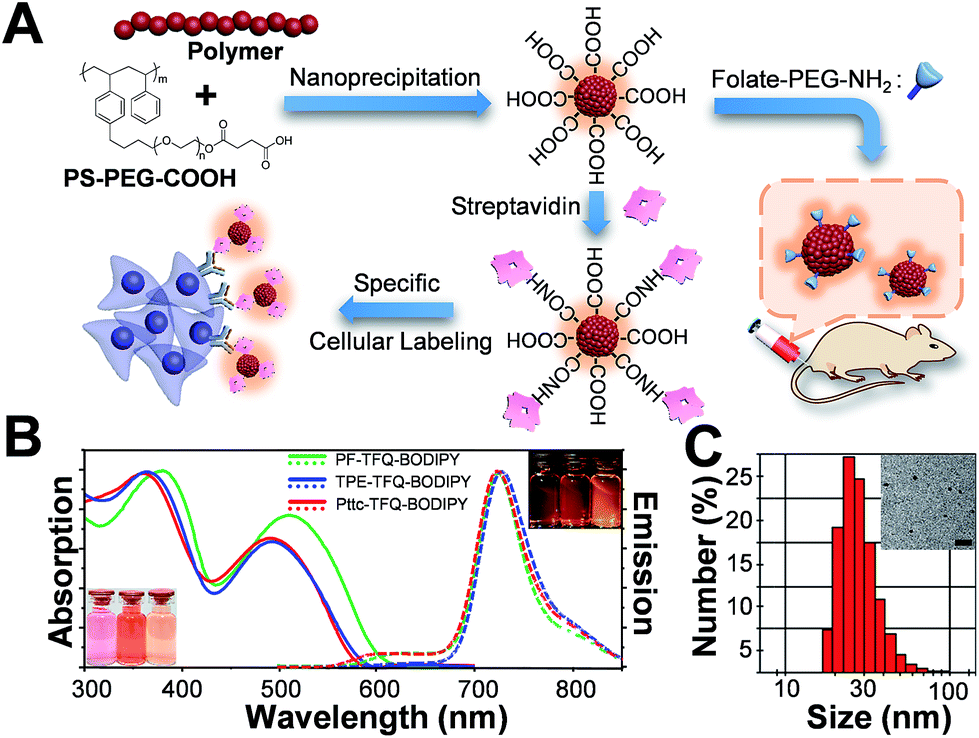
The preparation of Pdots and the subsequent surface functionalization for bioconjugation are illustrated in Fig. 1A. We functionalized the surface of the Pdots with streptavidin or folic acid via an EDC-catalyzed reaction for specific cellular imaging and in vivo tumor targeting, respectively (vide infra). The absorption (solid lines) and emission (dashed lines) spectra of the three Pdots with different types of donors are shown in Fig. 1B. The absorption peaks at 320–430 nm and 450–610 nm derive from fluorene/TPE/Pttc moieties and TFQ segments, respectively. The insets at the bottom-left and upper-right corners of Fig. 1B represent the photographs of aqueous Pdot solutions under ambient light and 365 nm UV light, respectively (from left to right: PF-TFQ-BODIPY, TPE-TFQ-BODIPY, and Pttc-TFQ-BODIPY Pdots). We found that the absorption of PF-TFQ-BODIPY Pdots exhibited a redshift at both peaks as compared to the other two Pdots, which could be attributed to the strong electron-donating ability and/or the high aggregation tendency of fluorene units. For the emission spectra, we found that the fluorescence of the TFQ spacer (ca. 580–650 nm) could be efficiently quenched by BODIPY for all of the three Pdots except for a slight emission shoulder observed at ∼610 nm for Pttc-TFQ-BODIPY Pdots. Because the energy transfer from TFQ to BODIPY can be ascribed to both Förster resonance energy transfer (FRET) and Dexter electron transfer, we speculate that the incomplete fluorescence quenching of TFQ might be attributed to the bulky and rigid scaffold of Pttc that leads to larger separation between bridges and acceptors. We also performed molecular dynamics simulations on these Pdots to validate our assumption (vide infra). The average hydrodynamic diameters of the Pdots were determined by dynamic light scattering (Fig. 1C, Pttc-TFQ-BODIPY as an example) and are summarized in Table 1, in which the average hydrodynamic sizes of the Pdots ranged from 27 to 28 nm in diameter. We also summarized the photophysical properties of the three Pdots in Table 1.
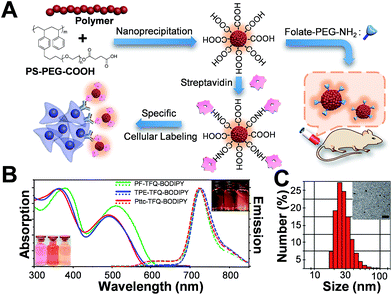 |
| | Fig. 1 (A) Schematic illustration of the preparation of carboxyl-functionalized Pdots through nanoprecipitation and subsequent bioconjugation for in vitro specific cellular imaging and in vivo tumor targeting. (B) Absorption spectra of PF-TFQ-BODIPY Pdots (solid green line), TPE-TFQ-BODIPY Pdots (solid blue line), and Pttc-TFQ-BODIPY (solid red line) in water. Their corresponding emission spectra are shown with dashed lines. The insets in the bottom-left and upper-right corners show the photographs of the Pdot solutions (from left to right: PF-TFQ-BODIPY, TPE-TFQ-BODIPY, and Pttc-TFQ-BODIPY Pdots in sequence) under daylight and under UV light illumination, respectively. (C) Hydrodynamic diameters of Pttc-TFQ-BODIPY Pdots measured by dynamic light scattering. The inset shows the transmission electron microscopy image of Pttc-TFQ-BODIPY Pdots. Scale bar is 100 nm. | |
Table 1 Summary of the optical properties of the Pdots in water
| Copolymers |
λ
absmax
(nm) |
λ
emmax
(nm) |
Size (nm) |
Φ
(%) |
τ
(ns) |
|
Absorption maximum.
Fluorescence maximum.
Fluorescence quantum yield (700–800 nm).
Fluorescence lifetime.
|
| PF-TFQ-BODIPY |
380, 512 |
724 |
27 |
7 |
1.42 |
| TPE-TFQ-BODIPY |
365, 493 |
725 |
28 |
37 |
1.53 |
| Pttc-TFQ-BODIPY |
361, 490 |
724 |
28 |
51 |
1.53 |
Photophysical properties of the Pdots
We are surprised to find out that the fluorescence quantum yields of PF-TFQ-BODIPY, TPE-TFQ-BODIPY, and Pttc-TFQ-BODIPY Pdots were measured to be 7, 37, and 51%, respectively. The most commonly used donor in Pdot systems, fluorene, appears to have the lowest fluorescence quantum yield, probably due to its easy aggregation characteristic arising from its planar structure. On the other hand, TPE-and Pttc-based Pdots have much higher fluorescence quantum yields as compared to PF-based Pdots. TPE is a well-known AIE luminogen with non-planar, twisted, and rigid conformations.54 The AIE process of TPE is activated by the restriction of intramolecular rotation that blocks the non-radiative decay channel and opens up the radiative pathway.55,56 Therefore, the increase in the fluorescence quantum yield of TPE-based Pdots might be responsible for the turn-on emission upon aggregate formation. In contrast, Pttc-based Pdots could effectively resist the π–π stacking interaction of conjugated polymers due to the bulky three-dimensional pentiptycene moieties, which successfully overcame the ACQ issue. It is interesting to find out that the anti-ACQ unit (i.e., Pttc) outperforms the AIE-active one (i.e., TPE) in the Pdot system. We further examined the phenomena of ACQ (i.e., PF), AIE (i.e., TPE), and anti-ACQ (i.e., Pttc) by controlling their extent of aggregation.
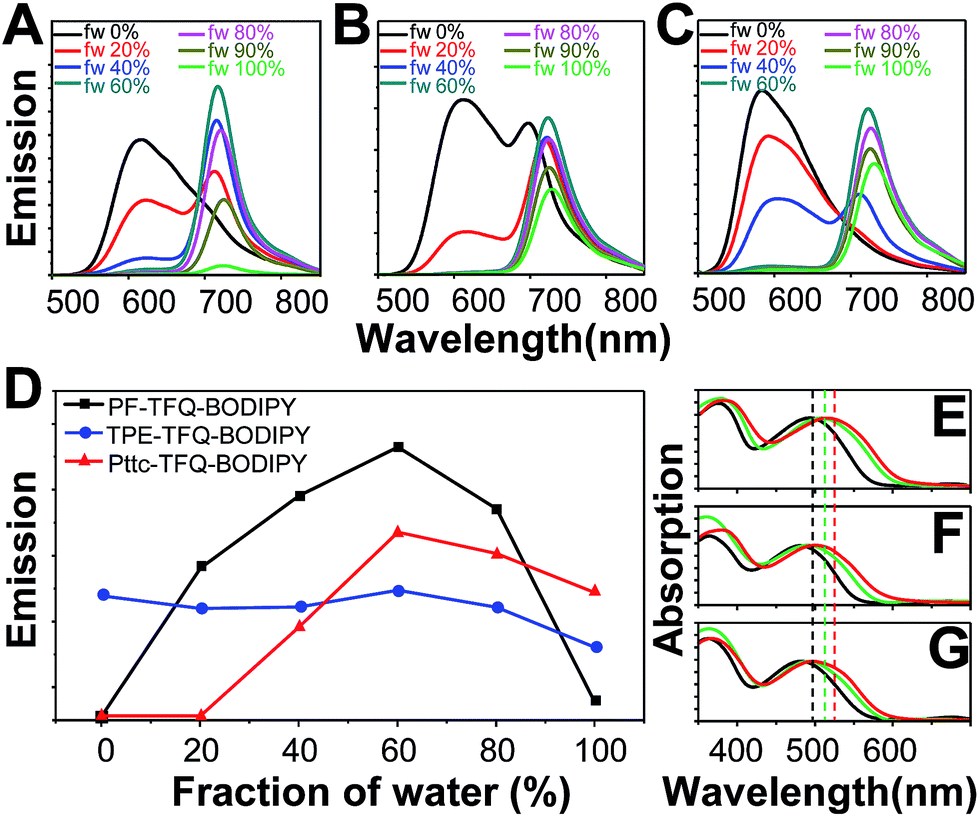
The three conjugated polymers are very soluble in tetrahydrofuran (THF) but completely insoluble in water. We first dissolved these polymers in pure THF separately and then added different amounts of water into the THF solutions. The results are shown in Fig. 2A–C, in which their fluorescence intensities responded distinctly to different volumetric fractions of water. We clearly observe that the fluorescence from TFQ (∼620 nm) decreases gradually as the water fraction increases, accompanied by the increase in the emission of BODIPY (∼724 nm) for all three polymers. This phenomenon can be explained by the hydrophobic effect of the semiconducting polymers, leading to the formation of polymer aggregates and thereby resulting in energy/electron transfer from TFQ to BODIPY. From Fig. 2D, the emission intensity of BODIPY for PF-TFQ-BODIPY increases initially as the water fraction is lower or equal to 60% but starts to decrease drastically once the water fraction is higher than 60%. The former part can be attributed to the aforementioned energy/electron transfer while the latter part is dominated by the ACQ effect. A similar, but milder, phenomenon of fluorescence increase and quenching can be seen for Pttc-TFQ-BODIPY. Interestingly, neither initial fluorescence increase nor obvious AIE behavior is revealed for TPE-TFQ-BODIPY. We also assessed the extent of their π–π stacking interactions by monitoring the absorption band of TFQ under different conditions (i.e., pure CH2Cl2, pure water, and thin film). As displayed in Fig. 2E–G, the absorption peak of TFQ in PF-TFQ-BODIPY exhibits a significant redshift from CH2Cl2 (495 nm) to pure water solution (512 nm), and then to thin films (519 nm), in which the total redshift of 24 nm suggests strong intermolecular stacking among polymers in the solid state.57–59 Modest and small redshifts of 16 nm and 14 nm are observed for TPE-TFQ-BODIPY and Pttc-TFQ-BODIPY, respectively, indicating a lower degree of molecular packing for both polymers in their solid films. To further understand the behavior of the donor from the molecular viewpoint, we then conducted molecular dynamics simulations for these three polymer systems.
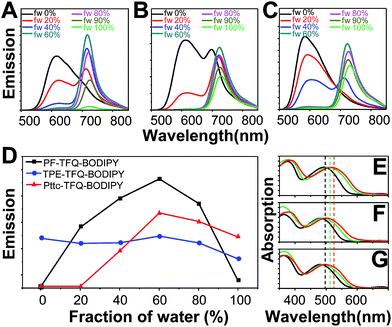 |
| | Fig. 2 Emission spectra of the solutions or suspensions containing (A) PF-TFQ-BODIPY, (B) TPE-TFQ-BODIPY, and (C) Pttc-TFQ-BODIPY polymers in THF/water mixtures with different volumetric fractions of water (fw, vol%). (D) Response of BODIPY fluorescence intensities (∼724 nm) for the three polymers in mixed solvents of THF/water (water fraction, v/v%). Normalized absorption spectra of (E) PF-TFQ-BODIPY, (F) TPE-TFQ-BODIPY, and (G) Pttc-TFQ-BODIPY in pure CH2Cl2 (black lines), Pdot aqueous form (green lines), and film form (red lines). | |
Structure–performance relationship of Pdots from molecular dynamics simulations
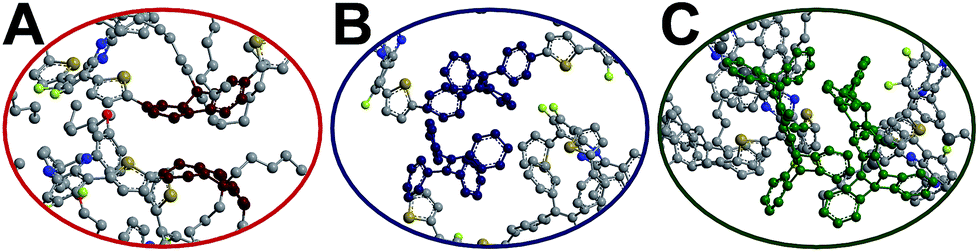
To gain insights into the relationship between the structures of polymer chains and their performance in Pdots, we have conducted a series of molecular dynamics (MD) simulations. The force field was obtained by following the recommended procedure described in previously reported work (further details are included in Methodology and ESI† sections).60 Since the copolymer has no determined sequence, we have simulated the system with small model oligomers such as the donor–bridge–donor–bridge (DBDB) and donor–acceptor–donor (DAD) model oligomers, as shown in Fig. 3. We note that since the Suzuki coupling connects the donor and either the bridge or acceptor, and there is no direct linkage between the bridge and acceptor. The simulated system contains 2500 DBDB oligomers and 50 DAD oligomers, roughly following the stoichiometry of the donor, the bridge, and the acceptor fragments in the synthesized Pdots (Scheme 1A). The structural features of the oligomers were further characterized. In particular, we studied (i) the interruption of energy transfer pathways from the donor to the bridge or the acceptor moieties along the backbone chain, and (ii) non-radiative quenching of BODIPY (the acceptor).
 |
| | Fig. 3 Chemical structure of (A) donor–acceptor–donor (DAD) oligomers and (B) donor–bridge–donor–bridge (DBDB) oligomers in molecular dynamics simulations: PF-TFQ-BODIPY (top), TPE-TFQ-BODIPY(middle), and Pttc-TFQ-BODIPY (bottom). | |
Previous experimental observations and theoretical predictions have revealed that highly distorted conformations of the backbone could lead to an interruption of energy transfer pathways.61–65 To quantify the distorted conformations, we calculated the distribution of torsional angles (θ) of the donor–bridge of DBDB oligomers and donor–acceptor of DAD oligomers. In Fig. 4A, it is seen that the DBDB oligomers in the PF-TFQ-BODIPY system have the largest populations with a highly distorted conformation (θ ∼90°) followed by TPE-TFQ-BODIPY (θ ∼90°). The distribution of the D–B torsional angle in the Pttc-TFQ-BODIPY system is quite even, and it has the lowest amount of the highly distorted conformation and the largest population of the planar conformation. The distributions of the torsional angle indicate that the energy transfer from a donor fragment to an adjacent bridge fragment could occur more easily in Pttc-TFQ-BODIPY than in the other Pdot systems. For DAD oligomers, on the other hand, the population of the highly distorted conformation appears to follow the trend: TPE-TFQ-BODIPY > Pttc-TFQ-BODIPY > PF-TFQ-BODIPY as seen in Fig. 4B. The presence of the highly distorted conformation in either the donor–acceptor moiety or the donor–bridge moiety would break down the energy transfer pathway. Thus, even though the DAD oligomer in PF-TFQ-BODIPY has the least population of the highly distorted conformation, most of the energy transfer is interrupted on the pathway from donor to bridge fragments due to the distorted DBDB oligomers. Similarly, for TPE-TFQ-BODIPY, the interruption of energy transfer may occur frequently in the region between the donor and acceptor. In the case of Pttc-TFQ-BODIPY, the energy transfer pathway would be the most effective due to the good population in near planar geometry in both torsional angle statistics. The relatively flat distribution of the torsional angle for Pttc-TFQ-BODIPY is due to the triple bonds connecting donor fragments with acceptor or bridge fragments, as one such example is shown in the top panel of Fig. 4C.
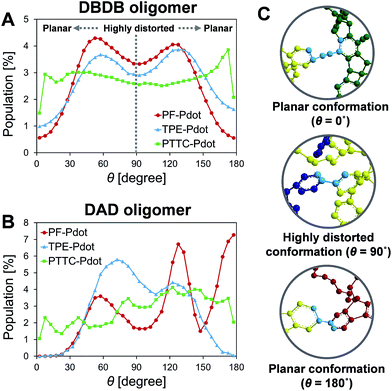 |
| | Fig. 4 Distributions of the torsional angle between (A) the donor and bridge on the DBDB oligomers, and between (B) the donor and acceptor on the DAD oligomers; (C) the representative planar conformations (θ ∼0° or ∼180°) and highly distorted conformation (θ ∼90°), where the torsional angles are marked in light blue color and hydrogen atoms are not shown for clarity. | |
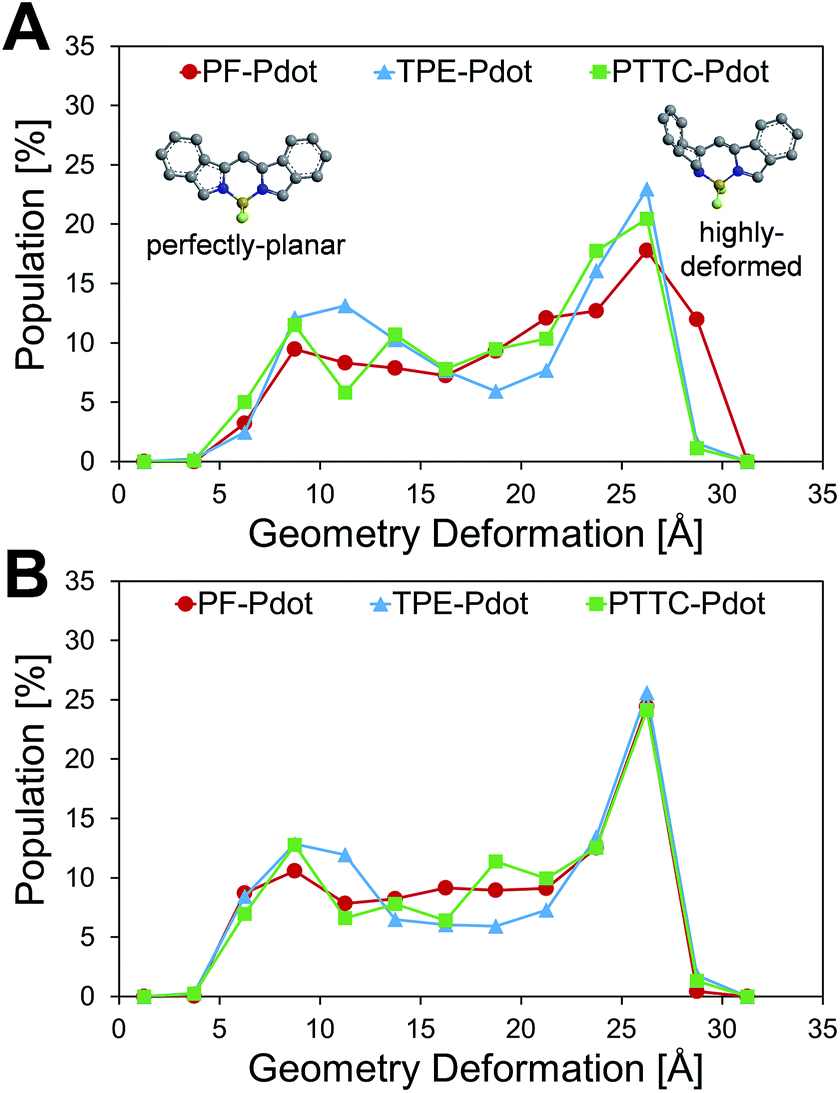
Another important factor affecting the fluorescence quantum yield is the competition between the non-radiative quenching process and light-emitting process. Recently, it has been found that a deformed BODIPY (the acceptor, emitting fragment) promotes a non-radiative quenching process, but a perfect planar BODIPY follows a light-emitting process.66,67 To characterize the structural features, we defined a parameter, DGD, for evaluating the deformation of BODIPY deviating from a perfectly planar BODIPY, where 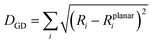 , and Ri and Rplanari are the position of the ith atom on a deformed and a perfectly planar BODIPY, respectively. The atoms used to assess DGD only include the conjugated moiety of BODIPY as shown in the inset of Fig. 5A. A small value of DGD indicates a planar structure, and a high value indicates a highly deformed case. In Table 2, the average deformation parameter (DGD) in PF-TFQ-BODIPY, TPE-TFQ-BODIPY, and Pttc-TFQ-BODIPY is 19.6 ± 6.7, 18.4 ± 6.9, and 18.6 ± 6.6, respectively. In Fig. 5A, a further inspection of DGD distribution shows that BODIPY in PF-TFQ-BODIPY bears more highly deformed conformations (DGD > 30) and fewer less-deformed conformations (DGD < 15) in comparison with TPE-TFQ-BODIPY and Pttc-TFQ-BODIPY. These results imply that BODIPY in PF-TFQ-BODIPY may follow a non-radiative quenching process more easily than TPE-TFQ-BODIPY and Pttc-TFQ-BODIPY. We have also performed a series of calculations with monomer systems where donor, bridge, or acceptor molecules were not linked. The averages and distribution profiles of DGD are almost the same for the three monomer-Pdot systems (see Fig. 5B and Table 2). Our result indicates that the deformation of acceptor BODIPY depends on the linkage with donor molecules. And we believe that the discrepancy between the three Pdot systems becomes more notable with increasing polymer chain length.
, and Ri and Rplanari are the position of the ith atom on a deformed and a perfectly planar BODIPY, respectively. The atoms used to assess DGD only include the conjugated moiety of BODIPY as shown in the inset of Fig. 5A. A small value of DGD indicates a planar structure, and a high value indicates a highly deformed case. In Table 2, the average deformation parameter (DGD) in PF-TFQ-BODIPY, TPE-TFQ-BODIPY, and Pttc-TFQ-BODIPY is 19.6 ± 6.7, 18.4 ± 6.9, and 18.6 ± 6.6, respectively. In Fig. 5A, a further inspection of DGD distribution shows that BODIPY in PF-TFQ-BODIPY bears more highly deformed conformations (DGD > 30) and fewer less-deformed conformations (DGD < 15) in comparison with TPE-TFQ-BODIPY and Pttc-TFQ-BODIPY. These results imply that BODIPY in PF-TFQ-BODIPY may follow a non-radiative quenching process more easily than TPE-TFQ-BODIPY and Pttc-TFQ-BODIPY. We have also performed a series of calculations with monomer systems where donor, bridge, or acceptor molecules were not linked. The averages and distribution profiles of DGD are almost the same for the three monomer-Pdot systems (see Fig. 5B and Table 2). Our result indicates that the deformation of acceptor BODIPY depends on the linkage with donor molecules. And we believe that the discrepancy between the three Pdot systems becomes more notable with increasing polymer chain length.
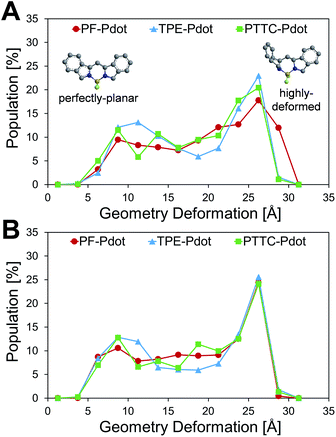 |
| | Fig. 5 Distributions of the geometry deformation of BODIPY (DGD): (A) oligomer models and (B) monomer models, where the inset shows the perfectly planar conformation and the highly deformed conformation of the conjugated moiety of BODIPY. | |
Table 2 Average Geometry Deformation (DGD) of the BODIPY fragment for the oligomer systems and monomer systems
|
|
Monomer |
Oligomer |
| PF-TFQ-BODIPY |
18.1 ± 7.0 |
19.6 ± 6.7 |
| TPE-TFQ-BODIPY |
18.0 ± 7.4 |
18.4 ± 6.9 |
| Pttc-TFQ-BODIPY |
18.4 ± 6.9 |
18.6 ± 6.6 |
Therefore, the high quantum yield of Pttc-TFQ-BODIPY is likely due to easier energy transfer among fragments, and a less deformed structure of BODIPY, while the structural characters for PF-TFQ-BODIPY are opposite, leading to a low energy transfer rate and high probability of quenching in BODIPY. The structural characters in the simulations for TPE-TFQ-BODIPY are in between. The structural features from MD simulations allow us to understand the trend of fluorescence quantum yield for the three Pdots in Table 1.
The red-shift in absorption spectra takes place around 500 nm, which is an overlapped region for all donors, bridges and acceptors. In Fig. 6, it is seen that the chromophore core of PF is a planar moiety with two dangling side chains, which allow strong π–π stacking. In contrast, Pttc is a rather bulky 3-dimensional structure, while TPE has a twisted conformation. The steric hindrances avoid the formation of π–π stacking. Therefore, TPE-TFQ-BODIPY and Pttc-TFQ-BODIPY systems have a similar red-shift from the solution sate to the thin film state (i.e., 16 nm and 14 nm, respectively), while the PF-TFQ-BODIPY system experiences the largest red-shift (i.e., 24 nm) in the absorption.
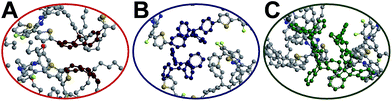 |
| | Fig. 6 Snapshot of the chromophore core of the oligomer pairs in (A) PF-TFQ-BODIPY, (B) TPE-TFQ-BODIYP, and (C) Pttc-TFQ-BODIPY, where hydrogen atoms are not shown for clarity. | |
With the aid of MD simulations, we are now able to elucidate the emission data shown in Fig. 2D. For PF-TFQ-BODIPY and Pttc-TFQ-BODIPY under good solvent conditions (0% volume fraction of water), a polymer undergoes dynamic intramolecular motions, such as rotation and vibration, which provides a possible way to non-radiatively quench its excited states and results in the absence of light-emission at 700–800 nm. As the fraction of water is increased to 60%, it may approach the theta condition for both systems, where the polymers behave like ideal chains, which slows down intramolecular motions and promotes the fluorescence quantum yield to the maximum. Further increase of the water fraction leads to the so-called ACQ effect in PF-TFQ-BODIPY. However, when Pttc-TFQ-BODIPY begins to form aggregates, its bulky steric hindrance prevents a strong face-to-face π–π stacking interaction, which is regarded as the anti-ACQ effect.68,69 For TPE-TFQ-BODIPY, on the other hand, the fluorescence quantum yield remains at the same level with a minor fluctuation as the solvent quantity is changed. It is worth noting that the monomer TPE has a special property of AIE.70,71 Therefore, the moderate emission quantum yield of TPE-TFQ-BODIPY at a high water content is understandable. The relatively insensitive quantum yield with water content in solution seemingly indicates that the photophysics of TPE is altered in the polymer such that the rotation of the free phenyl groups may still be somewhat hindered in the solvated polymer, leading to relatively good emission with low water content in the solvent.
Single-particle fluorescence brightness comparison
Single-particle brightness is a significant parameter to assess the performance of a new type of fluorescent probe. In theory, the fluorescence brightness can be estimated by the product of the fluorescence quantum yield and peak absorption cross-section. Thus, we summarized the related optical properties of the Pdots in Table 3 and then compared them with commonly used NIR fluorescent dyes and inorganic quantum dots. The per-particle absorption cross-sections (σ) of the Pdots were determined from the UV-visible absorbance of known Pdot concentrations (Beer's law), while their absolute fluorescence quantum yields (Φ) were measured directly by using an integrating sphere unit. The data shown in Table 3 indicate that the Pdots exhibit extraordinary fluorescence brightness, that is 1–4 orders of magnitude higher than that of conventional organic NIR dyes. As compared to inorganic quantum dots, our Pdots reveal fluorescence brightness 1.5–7 times higher than that of Qdot705 quantum dots. Among these three Pdot systems, Pttc-TFQ-BODIPY Pdots show the highest brightness due to their remarkable fluorescence quantum yield. Besides, these Pdots possess a very narrow emission bandwidth with a full width at half-maximum of ∼44 nm, which is comparable to those of small organic dyes. In case the probes are excited with a conventional 473 nm laser, Pttc-TFQ-BODIPY Pdots are ∼7 times brighter than Qdot 705 quantum dots (values are displayed inside the brackets in Table 3).
Table 3 Photophysical data of BODIPY-based NIR Pdots in water compared with other water-soluble typical NIR dyes
| Fluorescent probes |
λ
absmax (nm) |
λ
emmax (nm) |
f
whm
(nm) |
ε
max
(M−1 cm−1) |
σ
(cm2) |
Φ (%) |
Brightnessh (σ × Φ) (cm2) |
|
Data from the ATTO-TEC catalog.
Data from C. Bouteiller, G. Clavé, A. Bernardin, B. Chipon, M. Massoneau, P.-Y. Renard, A. Romieu. Bioconjugate Chem. 2007, 18, 1303–1317.
Data from Invitrogen (Thermo Fisher Scientific).
Pdots in deionized water.
Full width at half maximum.
Mean extinction coefficient.
Mean absorption cross-section.
Mean fluorescence brightness.
|
| ATTO740a |
740 |
764 |
43 |
1.20 × 105 |
4.58 × 10−16 |
10 |
4.58 × 10−17 |
| Cy 5.5b |
674 |
694 |
44 |
1.95 × 105 |
7.45 × 10−16 |
23 |
1.71 × 10−16 |
| NIR7.0–2b |
777 |
798 |
50 |
1.20 × 105 |
4.58 × 10−16 |
2.5 |
1.15 × 10−17 |
| Qdot 705c |
405 (473) |
707 |
72 |
8.30 (3.60) × 106 |
3.17 (1.38) × 10−14 |
82 |
2.60 (1.13) × 10−14 |
| PF-TFQ-BODIPY |
512 (473) |
723 |
45 |
1.43 (1.14) × 108 |
5.46 (4.35) × 10−13 |
7 |
3.82 (3.05) × 10−14 |
| TPE-TFQ-BODIPYd |
493 (473) |
727 |
44 |
1.01 (0.95) × 108 |
3.86 (3.61) × 10−13 |
37 |
1.43 (1.33) × 10−13 |
| Pttc-TFQ-BODIPYd |
490 (473) |
724 |
44 |
1.01 (0.97) × 108 |
3.86 (3.71) × 10−13 |
51 |
1.97 (1.89) × 10−13 |
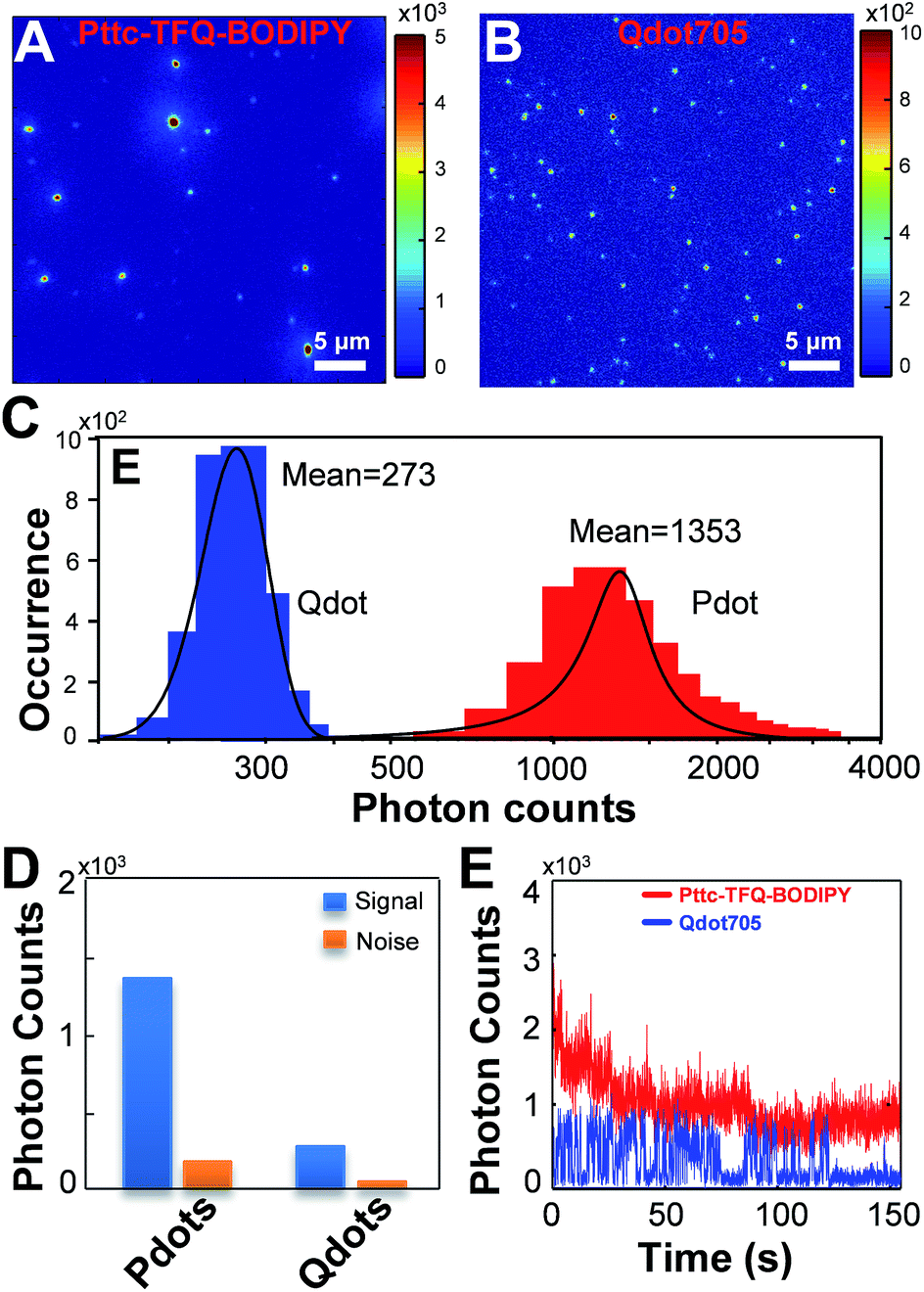
To experimentally confirm the above-mentioned values, we performed single-particle fluorescence imaging on both Pttc-TFQ-BODIPY Pdots and Qdot 705 quantum dots. The results in Fig. 7 reveal that the average per-particle brightness of Pttc-TFQ-BODIPY is about 5 times higher than that of Qdot 705, in which the intensity histograms are collected based on the analysis of hundreds to thousands of nanoparticles for both samples. Single-particle photobleaching trajectories (Fig. 7E) indicate that the Pdots are photostable under laser excitation (1 mW) for 150 s. For Qdot 705, no emission could be seen after 120 s. Moreover, Qdot 705 nanocrystals exhibit well-known fluorescence intermittency (blinking) behavior, while the Pdots emit stable and continuous fluorescence. The bright and nonblinking fluorescence is of importance for single-particle imaging applications.
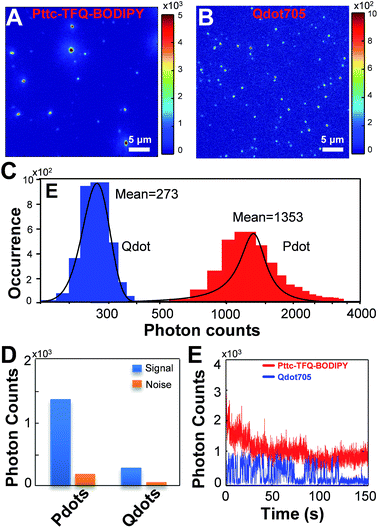 |
| | Fig. 7 Single-particle fluorescence images of (A) Pttc-TFQ-BODIPY Pdots and (B) commercial Qdot 705 quantum dots. Their corresponding (C) intensity distributions and (D) intensity histograms of signal to noise. (E) Single-particle photobleaching trajectories of Pttc-TFQ-BODIPY (red line) and Qdot705 (blue line) under a 473 nm laser (1 mW). All of the images were acquired with a 473 nm laser under the same experimental conditions. | |
Surface functionalization of the Pdots for specific cellular labeling
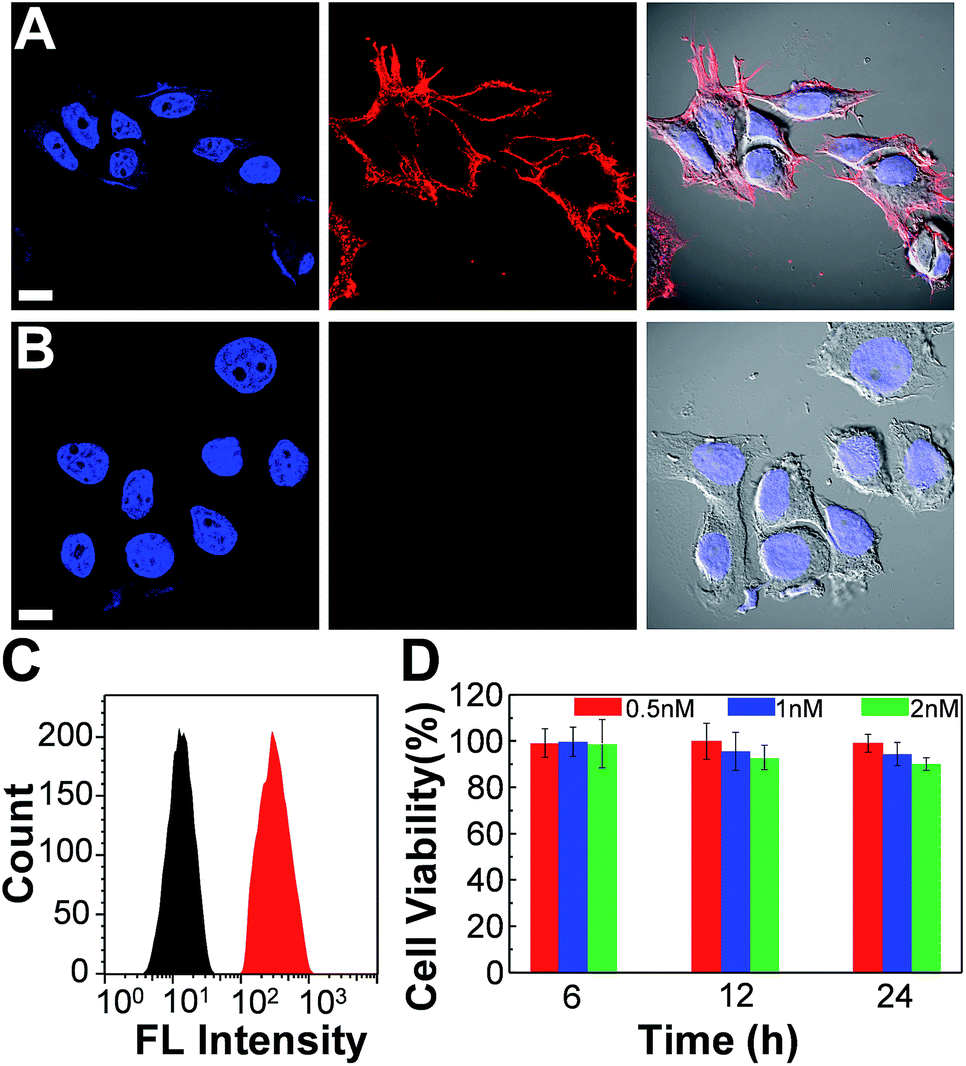
We further employed the Pdots for specific cellular labeling to demonstrate their biological uses. To endow the Pdots with labeling ability, we first conjugated streptavidin with carboxyl-functionalized Pdots via 1-ethyl-3-[3-(dimethylamino)propyl]-carbodiimide hydrochloride-(EDC)-catalyzed coupling (Scheme 1A). After bioconjugation, the Pdot–streptavidin conjugates together with biotinylated CD326 EpCAM antibodies were incubated with MCF-7 cells, allowing the Pdots to target the membrane proteins of MCF-7 cells. Fig. 8A shows that the Pdots could be specifically targeted onto the surfaces of MCF-7 cells in the presence of the biotinylated antibody. In contrast, minimal nonspecific adsorption could be detected for the negative control sample (Fig. 8B, in the absence of antibodies). Moreover, flow cytometry measurements were executed to further confirm the specificity of Pdot–streptavidin conjugates. As shown in Fig. 8C, a good separation between the Pdot-labeled cells (red line, in the presence of antibodies) and the negative control samples (black line, in the absence of antibodies) was clearly seen, in which the results are consistent with the images from confocal microscopy. In addition to specific cellular labeling, the cytotoxicity of Pdots is also an important criterion for an ideal fluorescent probe. Therefore, in vitro cytotoxicity studies were performed by using an MTT assay, as shown in Fig. 8C. The results suggest that these Pdots have a minimal cytotoxic effect on cells, even at a high concentration of 2 nM. It is worth mentioning that the typical concentration of the Pdots we use for cellular imaging is less than 1 nM, which is made possible by their superior fluorescence brightness.
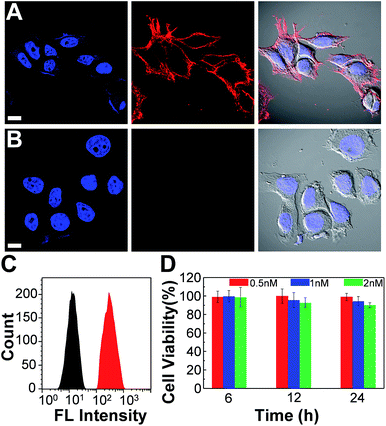 |
| | Fig. 8 Confocal fluorescence images of MCF-7 cells labeled with Pdot–streptavidin conjugates and the cytotoxicity along with flow cytometry results. (A) Blue fluorescence (left panel) is from nuclear counterstain Hoechst 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 580 and red fluorescence (middle panel) is from Pdot–streptavidin. The right panel displays the bright-view image overlaid with blue and red fluorescence. (B) Images of negative control samples in which the cells were incubated with Pdot–streptavidin conjugates but in the absence of biotinylated primary antibodies. The scale bars are 30 μm. (C) Flow cytometry measurements of Pdot-labeled MCF-7 cells. The black line shows the fluorescence intensity distribution of the negative control sample in which primary biotin anti-human CD326 EpCAM antibodies were absent. The red line displays the result of Pdot–streptavidin labeled cells in the presence of biotinylated antibodies. (D) The impact of the Pdots on the cell viability after 6, 12, and 24 h of incubation time at different concentrations. The results were determined by using MTT assays. 580 and red fluorescence (middle panel) is from Pdot–streptavidin. The right panel displays the bright-view image overlaid with blue and red fluorescence. (B) Images of negative control samples in which the cells were incubated with Pdot–streptavidin conjugates but in the absence of biotinylated primary antibodies. The scale bars are 30 μm. (C) Flow cytometry measurements of Pdot-labeled MCF-7 cells. The black line shows the fluorescence intensity distribution of the negative control sample in which primary biotin anti-human CD326 EpCAM antibodies were absent. The red line displays the result of Pdot–streptavidin labeled cells in the presence of biotinylated antibodies. (D) The impact of the Pdots on the cell viability after 6, 12, and 24 h of incubation time at different concentrations. The results were determined by using MTT assays. | |
In vivo microangiography imaging in zebrafish and tumor targeting in mice
We also evaluated the in vivo toxicity of the Pdots by use of the zebrafish model because zebrafish embryos are great for statistical analysis. The results shown in Fig. S2† reveal that zebrafish can grow normally in the presence of the Pdots (2–4 nM). Both in vitro and in vivo toxicity tests demonstrate that the Pdots are highly biocompatible and are promising for advanced biological applications. By taking advantage of the optically transparent properties of zebrafish, we further performed in vivo microangiography imaging on live zebrafish by injecting Pttc-TFQ-BODIPY Pdots into the sinus venosus of the zebrafish larva. As displayed in Fig. 9A, we found that the Pdots (red fluorescence) could distribute immediately to the vascular systems with negligible non-specific adsorption on the blood vessel walls (green fluorescence from eGFP-expressing endothelial cells).
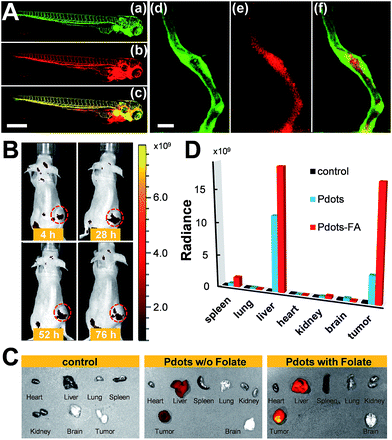 |
| | Fig. 9 (A) Zebrafish microangiography by injection of Pttc-TFQ-BODIPY Pdots into 3 days post fertilization Tg(kdrl:eGFP) zebrafish embryos. The global view of zebrafish vessel and Pdot images is shown in left panels (a–c), and a close view of the trunk vasculature (intersegmental vessel, ISV) is shown in the right panels (d–f). Green emission is from endothelial cells expressing eGFP, while red emission is from the Pdots. Scale bars represent 600 μm in (a–c) and 20 μm in (d–f), respectively. (B) Time-dependent in vivo bright-view overlaid with fluorescence imaging of the mouse intravenously injected with folate-functionalized Pdots. (C) Overlap images of fluorescence images and bright-view images of different organs from the mice at 76 h after tail-vein injection of saline (left), bare Pdots (middle), and folate-functionalized Pdots (right). (D) Comparison of biodistribution of the three Pdot probes in the resected organs. Black columns: saline treated mouse; light blue columns: bare Pdot treated mouse; red columns: Pdot–folate treated mouse. | |
To translate our Pdots into the mouse model for tumor targeting, we first functionalized the surface of the Pdots with folic acid (Fig. S3†) and then injected the Pdot–folate conjugates into the mouse via the tail vein. For the control and comparative samples, two mice were administrated with saline and bare Pdots (i.e., without folate modification), respectively. Whole-body fluorescence imaging of the Pdot-injected mice was performed under an IVIS imaging system and then monitored for 76 h to evaluate their long-term tracking ability. As shown in Fig. 9B, we found that the fluorescence of the tumor became apparent within 4 h, indicating the successful accumulation of the Pdots in the tumor. Due to the merits of NIR fluorescence and the ultrahigh brightness of the Pdots, the strong fluorescent signal from the Pdots underneath the skin can be directly seen using the IVIS system without anatomizing the mouse, which is critical for long-term tumor monitoring. Moreover, we were surprised to find out that strong fluorescence could still be visualized even after 76 h post-injection. This phenomenon suggests that the polyethylene glycol (PEG) units on the Pdots can efficiently eliminate the adsorption of the Pdots on blood proteins to prolong the blood-circulation time and thus prevent the Pdots from rapid renal clearance and/or hepatic clearance. The mice receiving different agents (i.e., saline, bare Pdots, and Pdot–folate conjugates) were then euthanized to study the biodistribution of the Pdots in different organs. For the control sample injected with saline, no fluorescence signal could be detected as shown in Fig. 9C. For both mice treated with bare Pdots and Pdot–folate conjugates, the Pdots were dominantly accumulated in the tumor, liver, and spleen. Quantitative fluorescence intensities of the Pdots in different organs were also analyzed as shown in Fig. 9D. It clearly shows that the functionalization of folic acid groups on the surface of the Pdots highly enhances the retention of Pdot probes at the tumor site. This result can be attributed to the aid of the receptor-mediated endocytosis of tumor cells (SKOV-3 cancer cells in this case with overexpression of folate receptors), aside from the typical enhanced permeability and retention effect of tumors. The prominent fluorescence of the livers in both Pdot-treated mice is probably due to the uptake of the Pdots by the reticuloendothelial system. These results demonstrate the promising potential of Pdots as a contrast agent for fluorescence-guided surgery or other related clinical applications.
Conclusions
In summary, we have successfully synthesized narrow-band NIR fluorescent Pdots with AIE or anti-ACQ properties and then compared their optical performance theoretically and experimentally. Both results indicate that the introduction of bulky frameworks could efficiently suppress the ACQ effect, resulting in an improved quantum yield of over 7 times higher than that of the planar structure, or ∼1.4 times higher than that of the AIE-based conformation. Molecular dynamics simulation suggests that the bulky steric hindrance prevents the strong face-to-face π–π stacking interaction of polymer backbones inside the Pdot matrix and outperforms the influence of AIE-active fluorogens. To demonstrate their bioconjugation ability, in vitro specific cellular imaging as well as in vivo tumor targeting in mice by using these Pdots were also realized. We believe that these results can provide valuable information for the future molecular design of semiconducting polymers in Pdot or nanoparticle-related fields.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the support from the Ministry of Science and Technology (105-2113-M-110-012-MY3), National Chiao Tung University, and National Sun Yat-sen University. Animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of NHRI. Seven-week-old female BALB/c nude mice were purchased from the National Laboratory Animal Center, and housed under specific pathogen-free conditions. Mice were obtained from the National Laboratory Animal Center (Taipei, Taiwan). All the mice were housed under specific pathogen-free conditions with moderate humidity and temperature at the Laboratory Animal Center of the National Health Research Institutes (NHRI). All animal experimental procedures followed published guidelines approved by the NHRI's Institutional Animal Care and Use Committee.
Notes and references
- A. V. DSouza, H. Lin, E. R. Henderson, K. S. Samkoe and B. W. Pogue, J. Biomed. Opt., 2016, 21, 080901 CrossRef.
- S. L. Troyan, V. Kianzad, S. L. Gibbs-Strauss, S. G. Matsui, R. Oketokoun, L. Ngo, A. Khamene, F. Azar and J. V. Frangioni, Ann. Surg. Oncol., 2009, 16, 2943 CrossRef.
- J. S. D. Mieog, S. L. Troyan, M. Hutteman, K. J. Donohoe, J. R. v. d. Vorst, A. Stockdale, G.-J. Liefers, H. S. Choi, S. L. Gibbs-Strauss, H. Putter, S. Gioux, P. J. K. Kuppen, Y. Ashitate, C. W. G. M. Löwik, V. T. H. B. M. Smit, R. Oketokoun, L. H. Ngo, C. J. H. v. d. Velde, J. V. Frangioni and A. L. Vahrmeijer, Ann. Surg. Oncol., 2011, 18, 2483 CrossRef.
- M. Hutteman, J. S. D. Mieog, J. R. v. d. Vorst, G. J. Liefers, H. Putter, C. W. G. M. Löwik, J. V. Frangioni, C. J. H. v. d. Velde and A. L. Vahrmeijer, Breast Cancer Res. Treat., 2011, 127, 163 CrossRef CAS.
- Y. Jiang and K. Pu, Adv. Biosyst., 2018, 1700262 CrossRef.
- G. Hong, A. L. Antaris and H. Dai, Nature Biomed. Eng., 2017, 1, 0010 CrossRef.
- S. W. Bae, W. Tan and J.-I. Hong, Chem. Commun., 2012, 48, 2270 RSC.
- M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Chem. Soc. Rev., 2014, 43, 4243 RSC.
- Y. Jilin, M. C. Estévez, J. E. Smith, K. Wang, X. He, L. Wang and T. Weihong, Nano Today, 2007, 2, 44 Search PubMed.
- J. E. Lee, N. Lee, H. Kim, J. Kim, S. H. Choi, J. H. Kim, T. Kim, I. C. Song, S. P. Park, W. K. Moon and T. Hyeon, J. Am. Chem. Soc., 2010, 132, 552 CrossRef CAS.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. L. Breton and V. Préat, J. Controlled Release, 2012, 161, 505 CrossRef CAS.
- Y. Yang, F. An, Z. Liu, X. Zhang, M. Zhou, W. Li, X. Hao, C.-s. Lee and X. Zhang, Biomaterials, 2012, 33, 7803 CrossRef CAS.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435 CrossRef CAS.
- R. C. Somers, M. G. Bawendi and D. G. Nocera, Chem. Soc. Rev., 2007, 36, 579 RSC.
- X. Michalet, F. F. Pinaud, L. A. Bentolila, J. M. Tsay, S. Doose, J. J. Li, G. Sundaresan, A. M. Wu, S. S. Gambhir and S. Weiss, Science, 2005, 307, 538 CrossRef CAS.
- C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, ACS Nano, 2008, 2, 2415 CrossRef CAS PubMed.
- Z. Tian, J. Yu, C. Wu, C. Szymanski and J. McNeill, Nanoscale, 2010, 2, 1999 RSC.
- K.-Y. Pu and B. Liu, Adv. Funct. Mater., 2011, 21, 3408 CrossRef CAS.
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086 CrossRef CAS.
- Y.-H. Chan and P.-J. Wu, Part. Part. Syst. Charact., 2015, 32, 11 CrossRef CAS.
- K. Li and B. Liu, Chem. Soc. Rev., 2014, 43, 6570 RSC.
- K. Pu, A. J. Shuhendler, J. V. Jokerst, J. Mei, S. S. Gambhir, Z. Bao and J. Rao, Nat. Nanotechnol., 2014, 9, 233 CrossRef CAS.
- K. Pu, N. Chattopadhyay and J. Rao, J. Controlled Release, 2016, 240, 312 CrossRef CAS.
- X. Lim, Nature, 2016, 531, 26 CrossRef CAS.
- L. Feng, C. Zhu, H. Yuan, L. Liu, F. Lv and S. Wang, Chem. Soc. Rev., 2013, 43, 6620 RSC.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102 CAS.
- J. Yu, Y. Rong, C.-T. Kuo, X.-H. Zhou and D. T. Chiu, Anal. Chem., 2017, 89, 42 CrossRef CAS PubMed.
- H.-S. Peng and D. T. Chiu, Chem. Soc. Rev., 2015, 44, 4699 RSC.
- Y. Guo, Y. Li, Y. Yang, S. Tang, Y. Zhang and L. Xiong, ACS Appl. Mater. Interfaces, 2018, 10, 20884 CrossRef CAS.
- F. Ye, C. Wu, Y. Jin, Y.-H. Chan, X. Zhang and D. T. Chiu, J. Am. Chem. Soc., 2011, 133, 8146 CrossRef CAS.
- Y. Rong, C. Wu, J. Yu, X. Zhang, F. Ye, M. Zeigler, M. E. Gallina, I.-C. Wu, Y. Zhang, Y.-H. Chan, W. Sun, K. Uvdal and D. T. Chiu, ACS Nano, 2013, 7, 376 CrossRef CAS.
- S.-Y. Kuo, H.-H. Li, P.-J. Wu, C.-P. Chen, Y.-C. Huang and Y.-H. Chan, Anal. Chem., 2015, 87, 4765 CrossRef CAS.
- C.-T. Kuo, A. M. Thompson, M. E. Gallina, F. Ye, E. S. Johnson, W. Sun, M. Zhao, J. Yu, I.-C. Wu, B. Fujimoto, C. C. DuFort, M. A. Carlson, S. R. Hingorani, A. L. Paguirigan, J. P. Radich and D. T. Chiu, Nat. Commun., 2016, 7, 11468 CrossRef CAS.
- K. Sun, Y. Tang, Q. Li, S. Yin, W. Qin, J. Yu, D. T. Chiu, Y. Liu, Z. Yuan, X. Zhang and C. Wu, ACS Nano, 2016, 10, 6769 CrossRef CAS.
- K. Chang, Z. Liu, X. Fang, H. Chen, X. Men, Y. Yuan, K. Sun, X. Zhang, Z. Yuan and C. Wu, Nano Lett., 2017, 17, 4323 CrossRef CAS.
- Y. Han, X. Li, H. Chen, X. Hu, Y. Luo, T. Wang, Z. Wang, Q. Li, C. Fan, J. Shi, L. Wang, Y. Zhao, C. Wu and N. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 21200 CrossRef CAS.
- L. Wu, I.-C. Wu, C. C. DuFort, M. A. Carlson, X. Wu, L. Chen, C.-T. Kuo, Y. Qin, J. Yu, S. R. Hingorani and D. T. Chiu, J. Am. Chem. Soc., 2017, 139, 6911 CrossRef CAS.
- Y. Lyu, D. Cui, H. Sun, Y. Miao, H. Duan and K. Pu, Angew. Chem., Int. Ed., 2017, 56, 9155 CrossRef CAS.
- C. Xie, X. Zhen, Y. Lyu and K. Pu, Adv. Mater., 2017, 29, 1703693 CrossRef.
- Y. Jiang, P. K. Upputuri, C. Xie, Y. Lyu, L. Zhang, Q. Xiong, M. Pramanik and K. Pu, Nano Lett., 2017, 17, 4964 CrossRef CAS.
- C.-C. Fang, C.-C. Chou, Y.-Q. Yang, T. Wei-Kai, Y.-T. Wang and Y.-H. Chan, Anal. Chem., 2018, 90, 2134 CrossRef CAS PubMed.
- C.-P. Chen, Y.-C. Huang, S.-Y. Liou, P.-J. Wu, S.-Y. Kuo and Y.-H. Chan, ACS Appl. Mater. Interfaces, 2014, 6, 21585 CrossRef CAS.
- I.-C. Wu, J. Yu, F. Ye, Y. Rong, M. E. Gallina, B. S. Fujimoto, Y. Zhang, Y.-H. Chan, W. Sun, X.-H. Zhou, C. Wu and D. T. Chiu, J. Am. Chem. Soc., 2015, 137, 173 CrossRef CAS.
- L. Chen, L. Wu, J. Yu, C.-T. Kuo, T. Jian, I.-C. Wu, Y. Rong and D. T. Chiu, Chem. Sci., 2017, 8, 7236 RSC.
- H.-Y. Liu, P.-J. Wu, S.-Y. Kuo, C.-P. Chen, E.-H. Chang, C.-Y. Wu and Y.-H. Chan, J. Am. Chem. Soc., 2015, 137, 10420 CrossRef CAS.
- R. Du, S. Cui, Z. Sun, M. Liu, Y. Zhang, Q. Wu, C. Wu, F. Guo and L. Zhao, Chem. Commun., 2017, 53, 8612 RSC.
- C.-S. Ke, C.-C. Fang, J.-Y. Yan, P.-J. Tseng, J. R. Pyle, C.-P. Chen, S.-Y. Lin, J. Chen, X. Zhang and Y.-H. Chan, ACS Nano, 2017, 11, 3166 CrossRef CAS PubMed.
- X. Zhang, J. Yu, Y. Rong, F. Ye, D. T. Chiu and K. Uvdal, Chem. Sci., 2013, 4, 2143 RSC.
- J. Wang, J. Mei, W. Yuan, P. Lu, A. Qin, J. Sun, Y. Ma and B. Z. Tang, J. Mater. Chem., 2011, 21, 4056 RSC.
- W. Bai, Z. Wang, J. Tong, J. Mei, A. Qin, J. Z. Sun and B. Z. Tang, Chem. Commun., 2015, 51, 1089 RSC.
- J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864 CrossRef CAS.
- J. Bouffard and T. M. Swager, Macromolecules, 2008, 41, 5559 CrossRef CAS.
- K. R. Ghosh, S. K. Saha, J. P. Gao and Z. Y. Wang, Chem. Commun., 2014, 50, 716 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- Z. He, C. Ke and B. Z. Tang, ACS Omega, 2018, 3, 3267 CrossRef CAS.
- M. R. Busireddy, V. N. R. Mantena, N. R. Chereddy, B. Shanigaram, B. Kotamarthi, S. Biswas, G. D. Sharma and J. R. Vaidya, Phys. Chem. Chem. Phys., 2016, 18, 32096 RSC.
- S. Li, J. Yan, C.-Z. Li, F. Liu, M. Shi, H. Chen and T. P. Russell, J. Mater. Chem. A, 2016, 4, 3777 RSC.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170 CrossRef CAS.
- N. E. Jackson, K. L. Kohlstedt, B. M. Savoie, M. O. d. l. Cruz, G. C. Schatz, L. X. Chen and M. A. Ratner, J. Am. Chem. Soc., 2015, 137, 6254 CrossRef CAS.
- K. M. Gaab and C. J. Bardeen, J. Phys. Chem. A, 2004, 108, 10801 CrossRef CAS.
- S. Habuchi, H. Fujita, T. Michinobu and M. Vacha, J. Phys. Chem. B, 2011, 115, 14404 CrossRef CAS PubMed.
- E. Hennebicq and C. Deleener, J. Chem. Phys., 2006, 125, 054901 CrossRef PubMed.
- I. Hwang and G. D. Scholes, Chem. Mater., 2011, 23, 610 CrossRef CAS.
- C. I. Wang, C. H. Hsu and C. C. Hua, Phys. Chem. Chem. Phys., 2017, 19, 20818 RSC.
- A. Prlj, A. Fabrizio and C. Corminboeuf, Phys. Chem. Chem. Phys., 2016, 18, 32668 RSC.
- A. Prlj, L. Vannay and C. Corminboeuf, Helv. Chim. Acta, 2017, 100, e1700093 CrossRef.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962 CrossRef.
- R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, NMR spectra, and additional information as noted in the text. See DOI: 10.1039/c8sc03510e |
| ‡ Both authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Chia-Hsien
Liao
a,
Chun-Nien
Yao
b,
Tsai-Jhen
Kuo
a,
Ming-Ho
Liu
a,
Chao-Ping
Hsu
c,
Shu-Yi
Lin
b,
Chang-Yi
Wu
d,
Joseph R.
Pyle
e,
Jixin
Chen
b,
Chia-Hsien
Liao
a,
Chun-Nien
Yao
b,
Tsai-Jhen
Kuo
a,
Ming-Ho
Liu
a,
Chao-Ping
Hsu
c,
Shu-Yi
Lin
b,
Chang-Yi
Wu
d,
Joseph R.
Pyle
e,
Jixin
Chen

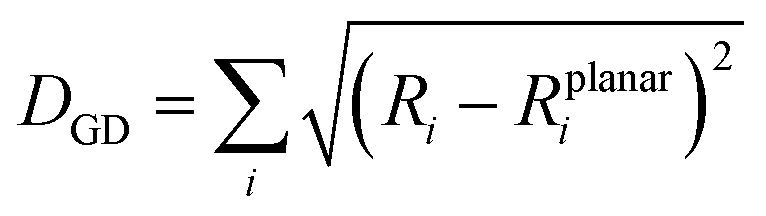 , and Ri and Rplanari are the position of the ith atom on a deformed and a perfectly planar BODIPY, respectively. The atoms used to assess DGD only include the conjugated moiety of BODIPY as shown in the inset of Fig. 5A. A small value of DGD indicates a planar structure, and a high value indicates a highly deformed case. In Table 2, the average deformation parameter (DGD) in PF-TFQ-BODIPY, TPE-TFQ-BODIPY, and Pttc-TFQ-BODIPY is 19.6 ± 6.7, 18.4 ± 6.9, and 18.6 ± 6.6, respectively. In Fig. 5A, a further inspection of DGD distribution shows that BODIPY in PF-TFQ-BODIPY bears more highly deformed conformations (DGD > 30) and fewer less-deformed conformations (DGD < 15) in comparison with TPE-TFQ-BODIPY and Pttc-TFQ-BODIPY. These results imply that BODIPY in PF-TFQ-BODIPY may follow a non-radiative quenching process more easily than TPE-TFQ-BODIPY and Pttc-TFQ-BODIPY. We have also performed a series of calculations with monomer systems where donor, bridge, or acceptor molecules were not linked. The averages and distribution profiles of DGD are almost the same for the three monomer-Pdot systems (see Fig. 5B and Table 2). Our result indicates that the deformation of acceptor BODIPY depends on the linkage with donor molecules. And we believe that the discrepancy between the three Pdot systems becomes more notable with increasing polymer chain length.
, and Ri and Rplanari are the position of the ith atom on a deformed and a perfectly planar BODIPY, respectively. The atoms used to assess DGD only include the conjugated moiety of BODIPY as shown in the inset of Fig. 5A. A small value of DGD indicates a planar structure, and a high value indicates a highly deformed case. In Table 2, the average deformation parameter (DGD) in PF-TFQ-BODIPY, TPE-TFQ-BODIPY, and Pttc-TFQ-BODIPY is 19.6 ± 6.7, 18.4 ± 6.9, and 18.6 ± 6.6, respectively. In Fig. 5A, a further inspection of DGD distribution shows that BODIPY in PF-TFQ-BODIPY bears more highly deformed conformations (DGD > 30) and fewer less-deformed conformations (DGD < 15) in comparison with TPE-TFQ-BODIPY and Pttc-TFQ-BODIPY. These results imply that BODIPY in PF-TFQ-BODIPY may follow a non-radiative quenching process more easily than TPE-TFQ-BODIPY and Pttc-TFQ-BODIPY. We have also performed a series of calculations with monomer systems where donor, bridge, or acceptor molecules were not linked. The averages and distribution profiles of DGD are almost the same for the three monomer-Pdot systems (see Fig. 5B and Table 2). Our result indicates that the deformation of acceptor BODIPY depends on the linkage with donor molecules. And we believe that the discrepancy between the three Pdot systems becomes more notable with increasing polymer chain length.