
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo chiral catalysts in action: insights into cooperativity and stereoselectivity in proline and cinchona-thiourea dual organocatalysis†
Bangaru
Bhaskararao
 and
Raghavan B.
Sunoj
*
and
Raghavan B.
Sunoj
*
Department of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, India. E-mail: sunoj@chem.iitb.ac.in
First published on 17th September 2018
Abstract
Increasing use of two chiral catalysts in cooperative asymmetric catalysis in recent years raises some fundamental questions on chiral compatibility between the catalysts, modes of activation, and relative disposition of substrates within the chiral environment of the catalysts for effective asymmetric induction. We present molecular insights into a one-pot catalytic Michael reaction cascade between a dicarbonyl compound (7-oxo-7-phenylhept-5-enal) and nitrostyrene, catalyzed by two chiral organocatalysts (proline and cinchona-thiourea), leading to a densely functionalized tetra-substituted cyclohexane product. The density functional theory (SMD(toluene)/M06-2X/6-31G**) computations helped us identify the role of the organocatalytic catalytic dyad in providing a lower energy pathway. The covalent activation of the aldehydic end by (S)-proline results in an enamine, which then adds to the noncovalently activated nitrostyrene in the first Michael addition to give a nitronate anion. The configuration at two of the four chiral centers of the product gets fixed in this step whereas that of the remaining two is determined by intramolecular cyclization between the nitronate and the enone. Important mechanistic features such as (a) a lower energy pathway as compared to a proline-only route for the formation of the syn-enamine and its participation in the first Michael addition and (b) the origin of the preferred prochiral faces in the C–C bond formation are traced to the active involvement of the cinchona-thiourea catalyst in conjunction with proline in each step of the reaction. The true cooperative action by both the catalysts is identified as enabled by a network of hydrogen bonding, and π⋯π stacking between the aryl ring of the cinchona-thiourea catalyst as well as other noncovalent interactions between the catalysts themselves, and that between the catalysts and substrate.
Introduction
The progress in homogeneous catalysis has been quite impressive over the last few decades.1 The majority of such efforts were confined to the use of one catalyst for a given reaction. One of the newer avenues that is beginning to get recognized as an effective tool in asymmetric catalysis is to use multiple catalysts under one-pot reaction conditions.2 The mode of action of each such catalyst is generally proposed to manifest in the form of activation of one of the reacting partners. The domain of cooperative multi-catalysis is gaining considerable momentum at this stage, both in methodological advances as well as in its applications to realize synthetically challenging targets.2In one-pot multi-catalytic reactions, one would typically use one chiral catalyst in combination with an achiral catalyst or some achiral additives.3 However, interesting recent examples are now available that use two chiral catalysts in one-pot reactions.4 The mechanism of various forms of dual chiral catalyst combinations (metal–metal, metal–organo, and organo–organo) continues to remain less understood at this stage. Although there have been some efforts toward unraveling the origin of cooperativity in a few reactions involving two chiral catalysts consisting of a chiral transition metal catalyst and a chiral organocatalyst,5 interesting combinations employing two chiral organocatalysts are not yet reported. The catalyst combination can be of an ion-pair type wherein one cationic complex partners with a counter anion. The concept of asymmetric counterion directed catalysis (ACDC) is a particularly noteworthy advancement on this front.2g Another intriguing avenue in asymmetric multi-catalysis is to use self-assembled catalysts.6
Two of the best-known examples of organocatalysts for asymmetric applications are proline and cinchona-thiourea hybrid catalysts. A natural extension toward harnessing the catalytic features of these organocatalysts is to make them work in concert. Indeed, there are emerging examples wherein proline is employed to activate a pro-nucleophile while cinchona-thiourea provides covalent activation to an electrophilic partner. In one of the most recent demonstrations of modularly designed organocatalysts (MDOs), the Zhao group proposed that a self-assembled catalytic dyad between proline (P) and the cinchona-thiourea derivative (C) is responsible for high stereocontrol in the formation of tetrasubstituted cyclohexanes (Scheme 1(b)).7b It is important to note that highly substituted cyclohexyl structural motifs constitute the tetracyclic core of several biologically active natural products such as lycorine-type alkaloids.8 The concept of combining proline and cinchona-thiourea is extended to other reactions as well.2f
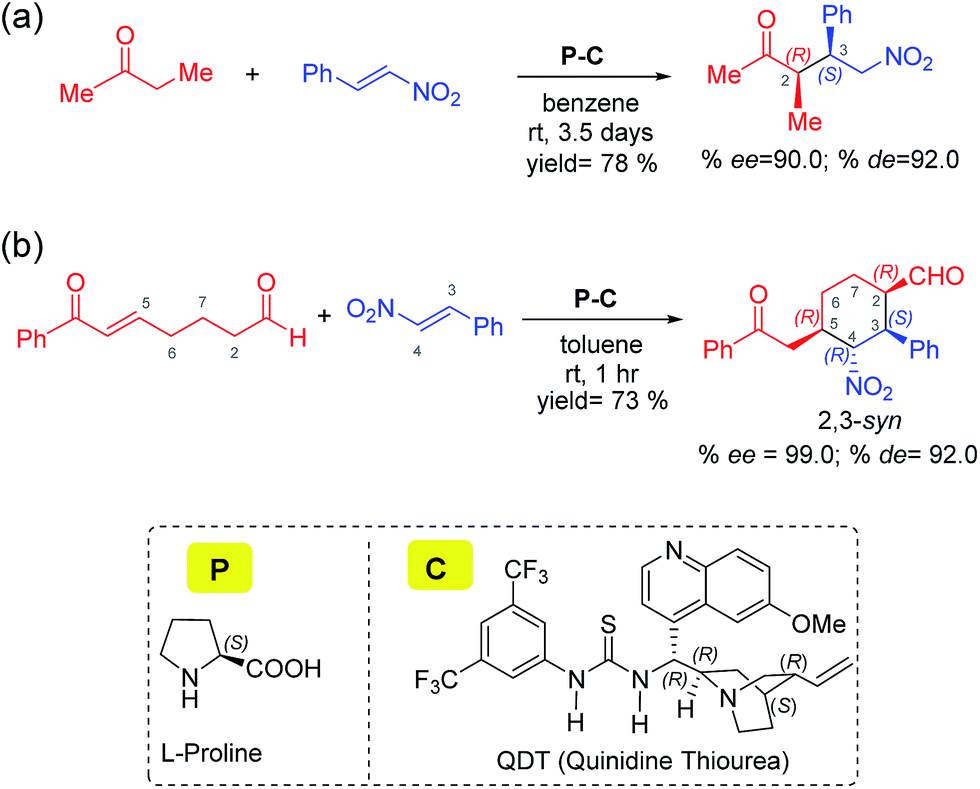 | ||
| Scheme 1 Proline and cinchona-thiourea dual catalytic Michael addition of nitrostyrene (a) to butanone leading to a linear product,7a and (b) to a dicarbonyl compound (7-oxo-7-phenylhept-5-enal) leading to 2,3-syn diastereomers of tetrasubstituted cyclohexanes through a tandem Michael–Michael cascade.7b | ||
Two conceptually similar reactions are shown in Scheme 1, which make use of proline-cinchona dual catalysis.7 The reaction as shown in Scheme 1(a) is a Michael addition with a relatively simpler ketone. A significant extension of the concept of self-assembled catalysis has been reported by the same group wherein a highly diastereodivergent synthesis of tetrasubstituted cyclohexanes could be accomplished (Scheme 1(b)). In this study, a simpler Michael addition reaction is first presented (Scheme 1(a)) to bring improved clarity on the role of catalysts in the generation of vital intermediates such as the enamine as well as to provide molecular insights on the origin of stereoselectivity.
The use of proline as the sole catalyst in Michael reactions generally yielded lower enantioselectivities as compared to other aminocatalyst analogues such as diarylprolinol silyl ether.9 Hence, it is of inherent interest to examine the origin of how parent proline itself could offer excellent enantio- as well as diastereo-selectivities in Michael addition reactions, when employed in conjunction with cinchona-thiourea as the partner catalyst (Scheme 1).10
The impressive developments in the domain of cooperative catalysis opened up more questions on its mechanism, sequence as well as modes of action of catalysts on the substrates. For instance, the question of how in situ self-assembled catalysts activate the substrates and influence the diastereo- and enantio-selectivities is not adequately understood owing to the lack of knowledge on the stereocontrolling transition states. Herein, we intend to shed light on some of these fundamental questions in asymmetric cooperative dual chiral catalysis (ADCC) involving proline and cinchona-thiourea as the catalytic dyad. In addition, certain interesting observations such as the rate enhancements when the catalyst combination is employed as compared to when only one catalyst is used demand knowledge on the kinetic features of this dual catalytic reaction. Similarly, whether or not both catalysts are directly involved in each step, or only in the stereocontrolling step of the mechanism needs careful consideration.
Computational methods
Computations were performed using the Gaussian09 (Revision D.01) suite.11 The geometries were optimized in a solvent continuum (toluene or benzene) using the M06-2X hybrid density functional theory12 in conjunction with Pople's 6-31G** basis set. All the stationary points were characterized, as minima or a first-order saddle point (transition state), by evaluating the corresponding Hessian indices. The transition states were verified by examining whether they have a unique imaginary frequency representing the desired reaction coordinate. Intrinsic reaction coordinate (IRC) calculations were additionally carried out to further characterize the true nature of the transition states.13 Additional computations using a larger basis set (6-311+G**) in conjunction with dispersion-corrected functionals (B3LYP-D3 and M06-2X-D3) were carried out on the stereoselectivity determining steps (i.e., the C–C bond formation) (see Tables S3, S10 and S13†). The geometries obtained as the end-points on the either side of the IRC trajectories were subjected to further optimization to identify the reactant and product that arise from the transition state. In particular, the IRC calculations for each transition state shown in the energy profile diagram were performed. For situations where the IRC trajectories in the forward and reverse directions of the transition states led to similar, but different, conformers of the same intermediate, we have used the lower energy conformer in the energy profile diagrams. The effect of a solvent continuum, in toluene, was evaluated using the Cramer–Truhlar continuum solvation model that employs quantum mechanical charge densities of solutes, designated as SMD.14 Graphical representation of the optimized geometries was generated by using CYLView.15 Topological analyses of electron densities were carried out using Bader's Atoms-in-Molecule formalism wherein bond paths and bond critical points are first identified and the corresponding electron densities at the bond critical points are analyzed.16 Analyses of noncovalent interactions were carried out using the noncovalent interaction index (NCI), as proposed by Yang and co-workers.17 The strength of H-bonding interactions in various situations was estimated using Abramov's method that relies on the virial equation.18Results and discussion
The discussions are organized into three major sections for the sake of clarity and better comprehension. These are (i) enamine formation between the aldehydic end of the dicarbonyl compound and proline (or that from butanone), (ii) the C–C bond formation between the enamine and nitrostyrene (first Michael addition), and (iii) intramolecular C–C bond formation within the first Michael adduct leading to a ring closure (second Michael addition) to the syn-cyclohexane as the final product (shown in Scheme 2). First two sections such as (i) enamine formation and (ii) C–C bond formation between the enamine and nitrostyrene are common to both examples shown in Schemes 1(a) and 1(b).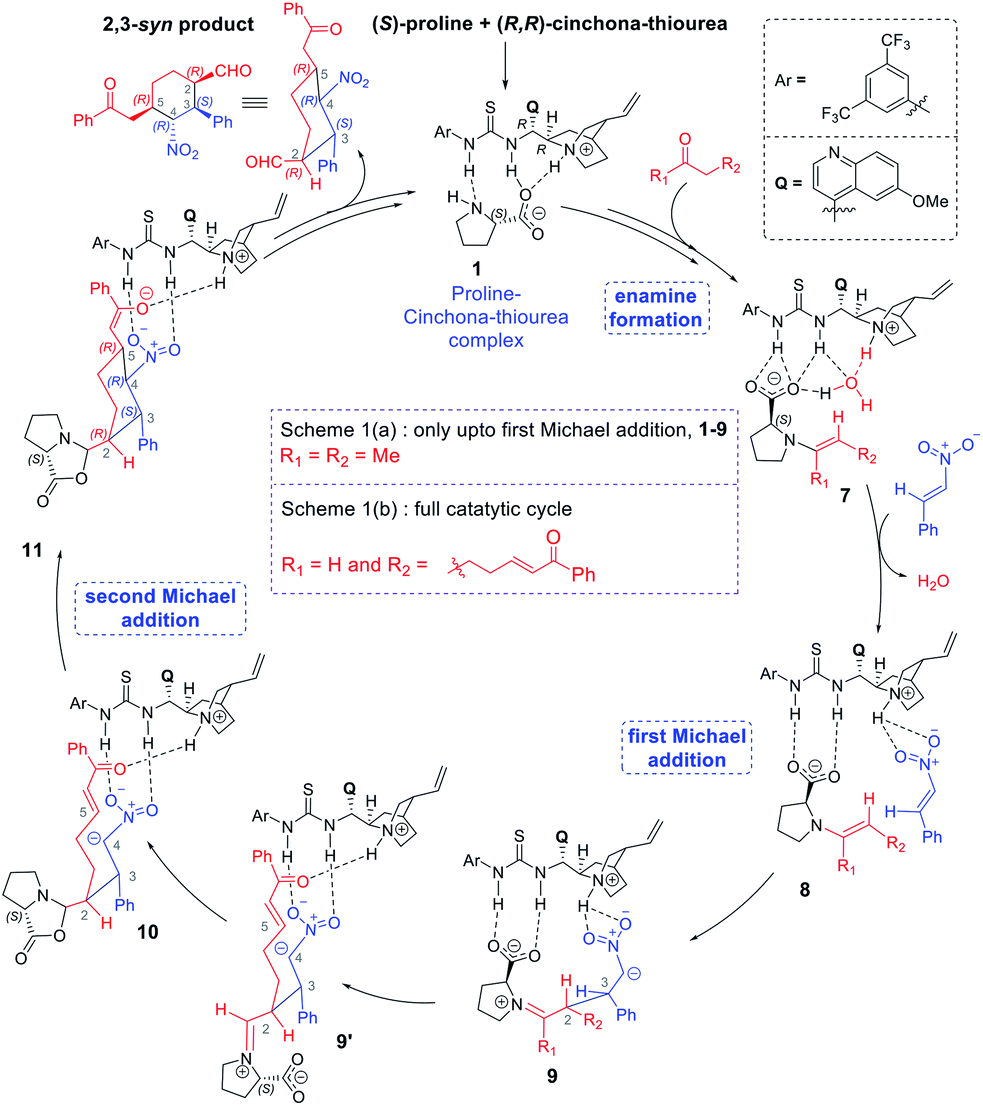 | ||
| Scheme 2 A general mechanism of formation of the tetra-substituted cyclohexyl product obtained using a Michael reaction cascade between a dicarbonyl compound (7-oxo-7-phenylhept-5-enal) and nitrostyrene under dual chiral catalytic conditions provided by proline and cinchona-thiourea. Only the most important intermediates are shown here for brevity. | ||
(i) Enamine formation in the presence of the cinchona-thiourea catalyst
Studies spanning more than a decade have revealed the vital role of enamine intermediates in the stereocontrolling step of proline catalyzed reactions.19 Although various modes of formation of proline enamines, with or without additives that offer hydrogen bonding stabilization (e.g., diaryl urea) have also been a subject of discussion,20 the question of how the presence of another organocatalyst (cinchona-thiourea in the present case) would influence the energetics of enamine formation is not established as yet. In other words, whether a cooperative dual catalytic pathway to the enamine is likely or not warrants attention. To address this question, we have compared the energetics of enamine formation between the dicarbonyl compound and proline, with and without the explicit inclusion of the cinchona-thiourea catalyst.The formation of the enamine involves two major steps: the addition of proline onto the aldehyde moiety of the dicarbonyl compound to form a carbinolamine intermediate and a subsequent dehydration, as shown in Scheme 3. Under one-pot multi-catalytic conditions, the catalysts are expected to be mutually compatible. Mechanistic models considered in this study suggest that both these catalysts can act in concert and can as well accommodate the substrate through a series of noncovalent interactions. The transition state geometries conveyed the presence of an effective hydrogen bonding network between (a) the substrate and the catalysts, and (b) the catalysts themselves. The corresponding Gibbs free energy profile and the optimized geometries are given in Fig. 1.21
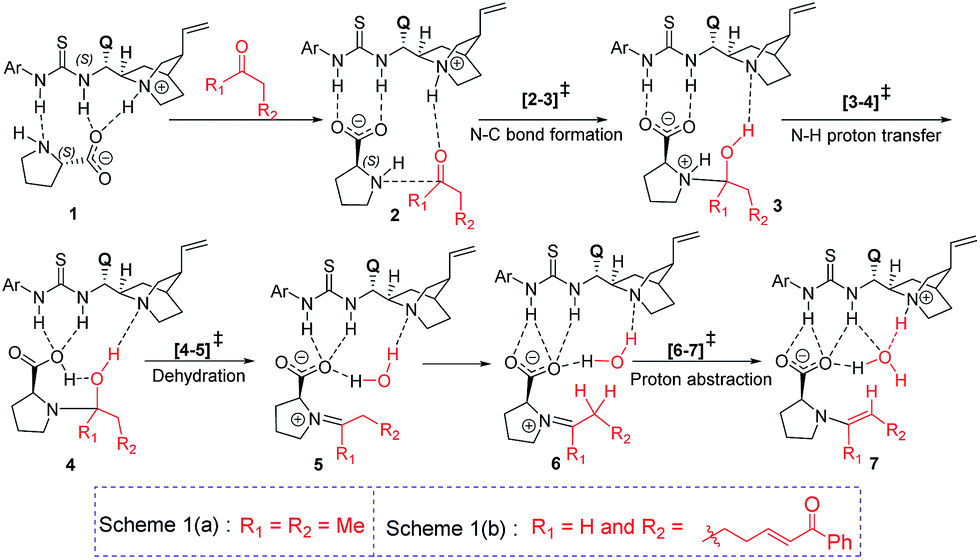 | ||
| Scheme 3 Important elementary steps in the formation of enamine. | ||
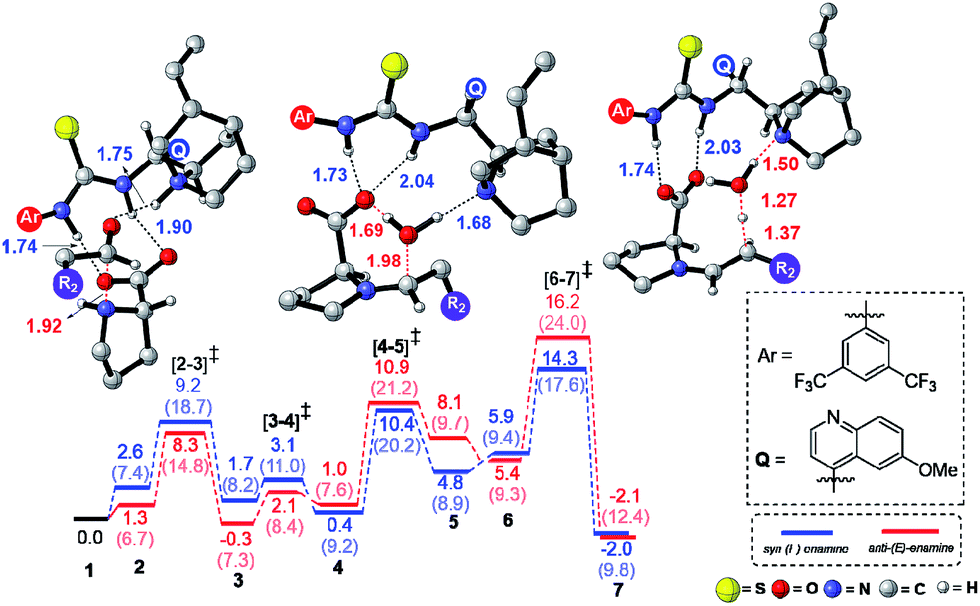 | ||
| Fig. 1 Free energy profile for the formation of the enamine intermediate and the optimized geometries of the key transition states obtained at the SMD(toluene)/M06-2X/6-31G** level of theory for Scheme 1(b). All distances are in angstroms and energies are in kcal mol−1. The free energies for the butanone system (Scheme 1(a)) at the SMD(benzene)/M06-2X/6-31G** level of theory are given in parentheses. | ||
In the conventional transition state model for the N–C bond formation [2–3], the developing alkoxide of the carbonyl group of the substrate develops a hydrogen bonding interaction with the α-carboxylic acid group of catalyst proline.22 In the present dual catalytic scenario, the interaction of proline with the cinchona-thiourea catalyst can lead to an acid–base adduct wherein the deprotonation of the carboxylic acid group enables an effective interaction between the resulting prolinate and the thiourea moiety, as shown in 1 (Scheme 3).23 In the addition of proline to aldehyde, the presence of the quinuclidinium as well as the thiourea N–H groups in the near vicinity can stabilize the incipient alkoxide moiety through a more effective hydrogen bonding pattern than that in simple proline-only mode of catalysis. The transfer of the quinuclidinium proton to the alkoxide oxygen subsequently can give rise to a carbinol amine intermediate 3. The dehydration of the carbinolamine via[4–5]‡ provides an iminium ion. In the most preferred transition state for the iminium to enamine conversion ([6–7]‡), the abstraction of the β-methylene proton by the quinuclidine is assisted by a water molecule (generated in the preceding step, as shown in Fig. 1). Through this step, a prolinate-enamine (7) with a distal protonated quinuclidinium nitrogen will be generated. This is an interesting reactive intermediate compared to the traditional proline–enamines where the incoming electrophile enjoys a vital hydrogen bonding interaction with the α-carboxylic acid group.22 Since the origin of stereoinduction in proline catalysis heavily relies on this particular hydrogen bonding, it is of inherent interest to examine what difference the distal quinuclidinium proton would make in influencing the stereochemically preferred mode of approach of the incoming electrophile.
The important aspect of this dual catalytic pathway is the additional stabilization rendered by the thiourea moiety of the cinchona-thiourea catalyst to all the intermediates and transition states involved in the formation of enamine 7. The computed energetics indicates that the elementary step barriers for the key steps (computed with respect to the respective intermediates for the N–C bond formation, N–H proton transfer, dehydration of carbinol amine, and the conversion of iminium to enamine) due to the active participation of cinchona-thiourea are lower than the proline-only pathway (Scheme 3).24 An elegant corroboration of this additional stabilization of the key intermediates in proline catalysis came in the form of a recent experiment by the Gschwind group using a chemical exchange saturation transfer NMR study.20e In their study, in situ detection of, an otherwise elusive, iminium ion intermediate (derived from proline and an aldehyde) could be accomplished in the presence of diphenylurea.
The above insights suggest a true cooperative action of both catalysts in the formation of the vital enamine intermediate. Another key feature of the enamine formation transition state [6–7]‡, with and without the active participation of the cinchona-thiourea catalyst, is that the preferred enamine formed through the cooperative pathway is a syn enamine as opposed to the standard anti enamine of proline with various pronucleophilic carbonyl compounds (Fig. 1). The anti/syn notation here refers to the position of the enamine double bond with respect to the carboxylic acid. The transition state [6–7]‡ for the syn enamine is lower in energy (1.9 kcal mol−1) than that for the anti enamine formation, so are the barriers for dehydration toward the syn enamine (8.5 kcal mol−1) and for the anti enamine (10.7 kcal mol−1). In the case of butanone with proline, the transition state [6–7]‡ for the syn enamine formation is lower in energy (7.6 kcal mol−1) than that for the anti enamine, so are the barriers for dehydration toward the syn enamine (8.2 kcal mol−1) and for the anti enamine (14.7 kcal mol−1). The overall involvement of cinchona-thiourea in the enamine formation is in the form of a bifunctional mode of catalysis wherein it provides a network of hydrogen bonding to the key intermediates through the thiourea region and also actively engages the quinuclidine moiety for deprotonation. Thus, proline catalysis is cooperatively assisted by the second organocatalyst in the present example.
(ii) (a) Michael addition between butanone and nitrostyrene
The next step is the most important C–C bond formation between the proline enamine and nitrostyrene. There are two crucial aspects to consider in this step; first the mode of activation of the electrophilic nitrostyrene and nucleophilic enamine in the transition state and second is to identify the most preferred prochiral faces that are involved in the bond formation that would dictate the product stereochemistry. As shown in Fig. 2, the nitrostyrene can either develop a hydrogen bonding interaction with the quinuclidinium proton (path-A) or it can engage in two hydrogen bonding interactions with the thiourea region of the catalyst (path-B) as shown in Fig. 3. Path-A is found to be more preferred by 3.3 kcal mol−1 than path-B due to better H-bonding between the substrate (nitrostyrene) and the catalyst (cinchona-thiourea), as shown in Fig. 3. When the activated substrates are held in a well-defined orientation through a series of weak interactions provided by the chiral catalyst, there could be unique facial preferences in the C–C bond formation. For instance, if the addition of the si-face of the enamine to the si-face of nitrostyrene is energetically most preferred, the product configuration would be (2R,3S). On the other hand, a re–re mode of addition would result in the (2S,3R) product. We have examined all such possibilities to be able to identify the stereochemically most preferred C–C bond formation transition state for the Michael addition.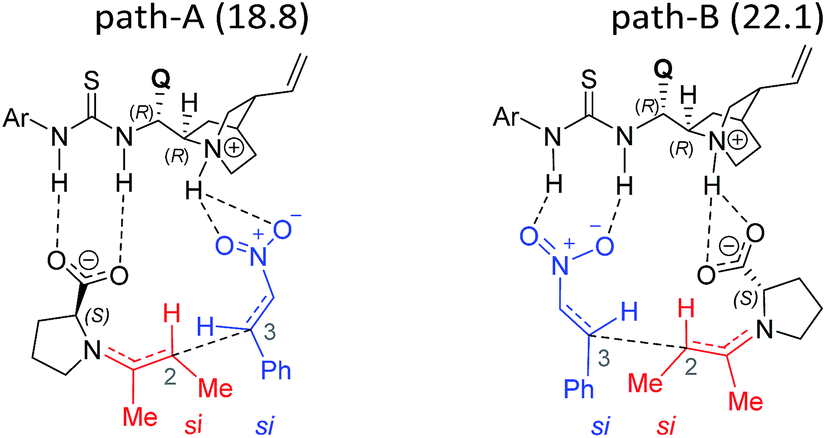 | ||
| Fig. 2 Different modes of the stereocontrolling C–C bond formation transition states [8–9]‡ between the proline syn-(E)-enamine (7) and trans-β-nitrostyrene in the Michael addition for the reaction shown in Scheme 1(a). | ||
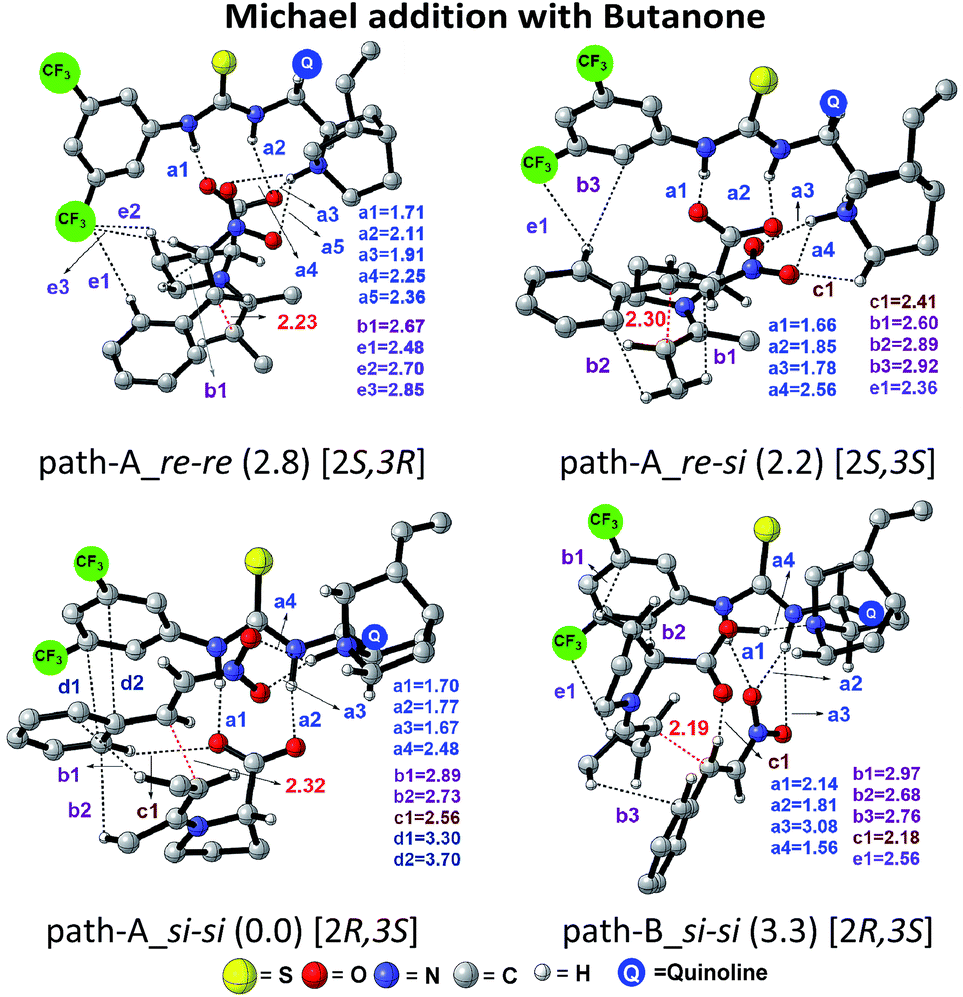 | ||
| Fig. 3 Optimized geometries of the stereocontrolling transition states [8–9]‡ for the C–C bond formation between the enamine and trans-β-nitrostyrene in the presence of the cinchona-thiourea catalyst via path-A and path-B. For improved clarity, only important hydrogen atoms are shown. All distances are in angstroms. Notations a1, a2, a3, … = N–H⋯O; b1, b2, b3 = C–H⋯π; c1 = C–H⋯O; d1, d2 = π⋯π and e1, e2, e3 = C–H⋯F are used for various noncovalent interactions. The configuration of the developing chiral centers in the products is given in square brackets. | ||
In the path-A mode of activation of reactants, the si–si approach is found to be most preferred by virtue of better noncovalent interactions. For instance, key hydrogen bonding interactions between the catalyst and (i) substrate-1 (activated in the form of enamine denoted as a1, a2) and substrate-2 (nitrostyrene, a3, a4) are found to be better in the si–si mode (Fig. 3). In addition, a π⋯π stacking interaction (d1, d2) between nitrostyrene and the aryl ring of the thiourea cinchona catalyst is noted in the lower energy si–si TS. This π⋯π stacking is absent in other stereochemical modes of C–C bond formation TSs. These noncovalent interactions are relatively low in number and efficiency in the higher energy TS such as re–re, thus contributing to the extent of enantioselectivity. The predicted %ee of 98.2 is in good agreement with the experimentally observed value of 90. Similarly, the %de of 95.2 predicted using transition state energies is in concert with the experimental value of 92.0.25
(ii) (b) First Michael reaction between the heptenal derivative and nitrostyrene
The next most important step of the reaction is the C–C bond formation between the proline enamine and nitrostyrene. There are two crucial aspects to consider in this step; first the mode of activation of the electrophilic nitrostyrene and nucleophilic enamine in the transition state and second is to identify the most preferred prochiral faces that are involved in the bond formation, which in turn, would dictate the product stereochemistry. The incoming nitrostyrene can develop a hydrogen bonding interaction either with the quinuclidinium proton or it can engage in two hydrogen bonding interactions with the thiourea moiety of the catalyst.26 We note that the former mode is more preferred by about 2.4 kcal mol−1 than the latter, presumably due to the improved hydrogen bonding interaction with the quinuclidinium proton (Scheme 2).27An important stereochemical feature of this dual catalytic system is worth noting at this stage. The preferred prochiral face provided by the syn enamine is the si-face whereas the anti enamine reacts through its re-face. This is in contrast to the enamine formation by parent proline in the absence of the cinchona-thiourea catalyst, where the anti enamine formation as well as reaction through its re-face is generally more preferred.9 This preference toward the formation of the syn-enamine conformer arises due to a kinetic advantage in the iminium to enamine conversion step (Fig. 1). Effective hydrogen bonding between the N–H groups of the cinchona-thiourea and proline carboxylate in [6–7]‡ makes the syn-enamine more preferred over the corresponding path to the anti-enamine (Fig. 1).
When the activated substrates are held in a well-defined orientation through a series of noncovalent interactions with the chiral catalysts, there could be unique facial preferences in the C–C bond formation (Scheme 2). We have examined many such possibilities to be able to identify the stereochemically most preferred C–C bond formation transition state.28 For instance, if the addition of the si-face of the enamine to the si-face of nitrostyrene is energetically most preferred, the product configuration would be (2R,3S). On the other hand, the transition state for the re–re mode of addition that would result in the (2S,3R) product is found to be 2.1 kcal mol−1 higher in energy. Such a large difference in the transition state energies indicates high enantiocontrol in the first Michael addition leading to a nitronate anion 9. In this first step of Michael addition that determines the configuration of C2 and C3 chiral centers, both enantioselectivity (dictated by the energy difference between transition states for the si–si (0.0) and re–re (2.1) mode of additions) and diastereoselectivity (controlled by modes si–si (0.0) and si–re(3.0)) are predicted to be 94.3 and 98.7% respectively.
Perusal of the geometries of the stereocontrolling transition states, as given in Fig. 4 conveys a few important aspects of catalyst cooperativity as well as on the origin of stereoselectivity. To understand the origin of stereoselectivity in the first Michael addition, we have carefully analyzed the features of the key transition states. In the lowest energy TS ([8–9]‡ in the si–si mode of addition of the enamine to nitrostyrene) a higher number of N–H⋯O (a1, a2, …), C–H⋯π (b1, b2, …), C–H⋯O (c1, c2, …), π⋯π (d1, d2) and C–H⋯F (e1, e2, …) noncovalent interactions are noticed than that in the higher energy re–re and re–si TSs (Fig. 4). In particular, in the si–si TS, the phenyl ring of nitrostyrene participates in an interesting π⋯π interaction with the aryl ring of the cinchona-thiourea catalyst (d1, d2), which is an interaction that is absent in the higher energy re–re TS. In other words, the enantioselectivity that depends on the energy difference between the si–si (0.0) and re–re (2.1) TSs seems to be governed by the efficiency of the π⋯π interaction between the catalyst and the substrate. Interestingly, the diastereoselectivity depends on the differences in C–H⋯π and C–H⋯O interactions, which are higher in number in the case of the si–si (0.0) mode than that in the re–si (3.0). In addition, we have analyzed the origin of stereoselectivity by using the activation strain analysis on the stereocontrolling transition states29 and the role of hydrogen bonding interactions.30
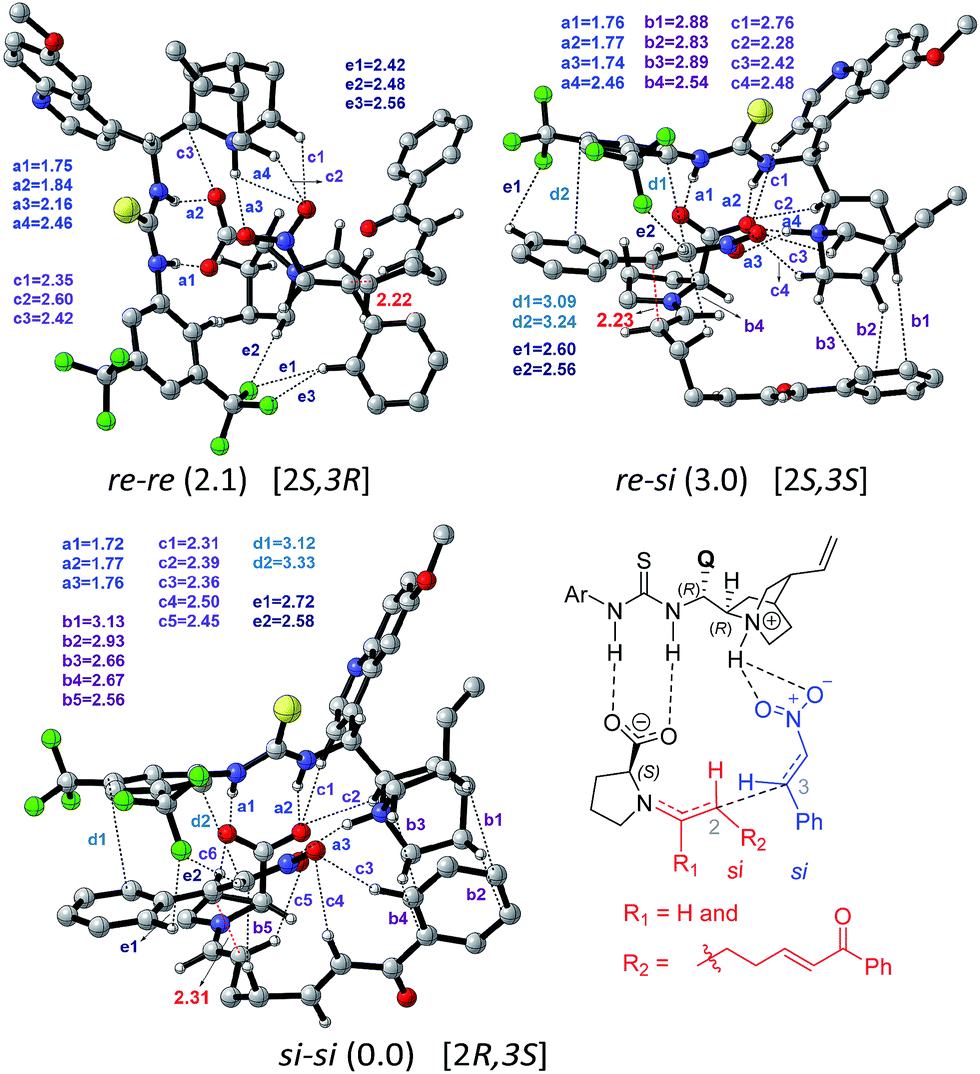 | ||
| Fig. 4 Optimized geometries of the stereocontrolling transition states [8–9]‡ for the C–C bond formation between the syn-(E)-enamine and trans-β-nitrostyrene in the presence of the cinchona-thiourea catalyst via path-A. For improved clarity, only important hydrogen atoms are shown. All distances are in angstroms. Notations a1, a2, a3 = N–H⋯O; b1, b2, b3 = C–H⋯π; c1, c2, c3 = C–H⋯O; d1, d2, d3 = π⋯π and e1, e2, e3 = C–H⋯F are used for various noncovalent interactions. The configuration of the developing chiral centers in the products is given in square brackets. A representative chemdraw is shown for the si-face addition of the enamine to the si-face of nitrostyrene for additional clarity. | ||
(iii) Second and intramolecular Michael reaction
After having established the vital mechanistic features and the origin of stereoselectivity in the first Michael reaction enabled by the proline–cinchona-thiourea catalytic dyad, we turned our attention to the ensuing intramolecular Michael addition leading to the cyclohexyl framework. The key transition state [10–11]‡ for this Michael addition is intramolecular cyclization within the first Michael adduct, involving the activated double bond at the other end of the α,β-unsaturated enone (Scheme 4).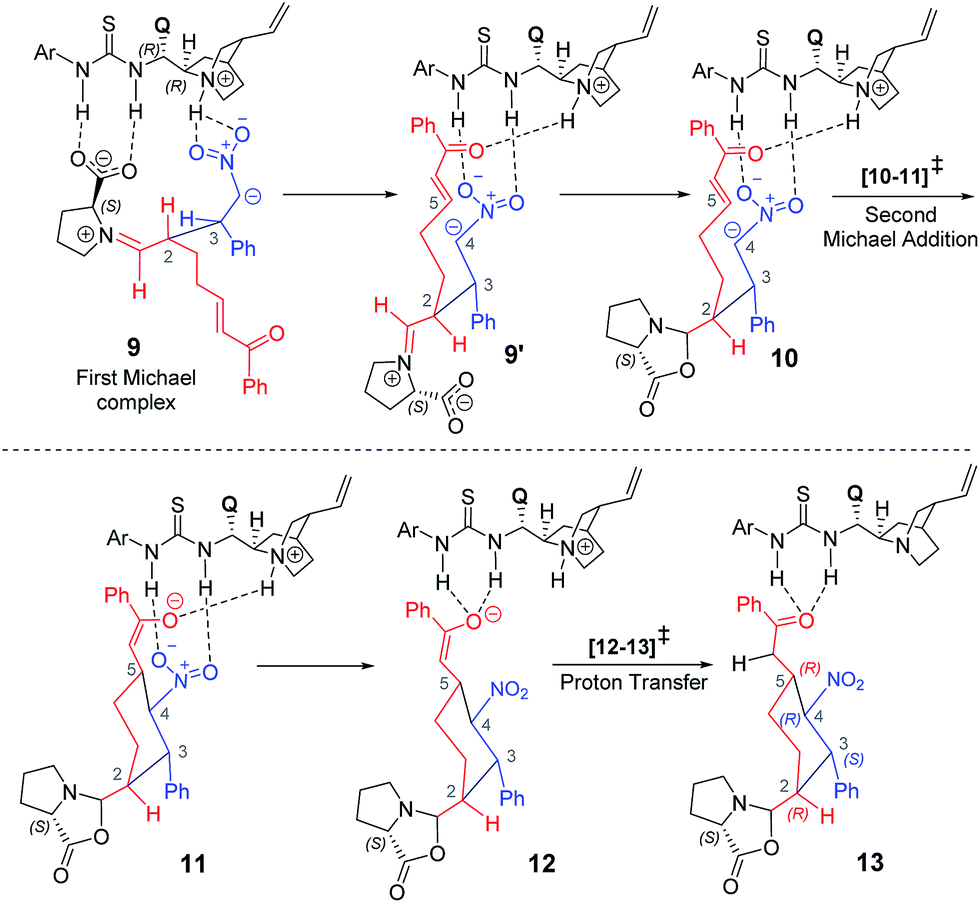 | ||
| Scheme 4 Various steps involved in the formation of the 2,3-syn product through the Michael reaction. | ||
Akin to the first Michael step, two types of activation modes are considered; one in which the styrenyl nitro group is hydrogen bonded to the thiourea moiety and the carbonyl group of the electrophilic enone end of the chain is held in position through a hydrogen bonding with the quinuclidinium N–H.26 In another mode, higher in energy by 4.5 kcal mol−1, the hydrogen bonding is between the carbonyl oxygen and the thiourea at one end whereas the nitro group at the other end interacts with the N–H. Another Michael addition pathway with the same hydrogen bonding pattern as seen in intermediate 9 is also examined. In such a situation, when the acceptor enone lacks hydrogen bonding interaction is found to be of 9.0 kcal mol−1 higher energy as compared to the lowest energy TS 2R,3S_re–re (0.0).
A number of interesting mechanistic insights have emerged in this vital catalytic step. The hydrogen bonding pattern in intermediate 9 can be altered such that the nitro group interacts with the thiourea and the enone carbonyl with the quinuclidinium N–H (9′, as shown in Scheme 4). This change would result in a free carboxylate devoid of hydrogen bonding with the thiourea moiety. An interesting propensity toward immediate cyclization to form an oxazolidinone intermediate (10) is noticed. It is instructive to reckon that the involvement of the oxazolidinone intermediate has been suggested in proline catalysis.8,18,19 An equally important feature of organocatalytic mechanisms of the thiourea-cinchona family of catalysts is the mode of substrate (pro-nucleophile and electrophile) activation. It has been shown that the protonated quinuclidine as well as thiourea can activate the reacting partners, although the mode of activation, among other factors, could vary depending on the nature of the pro-nucleophile involved.27
In the present situation, an oxazolidinone (10) is formed prior to the Michael addition step, when the carboxylate gets disengaged from the thio-urea hydrogen bonding. It can be noticed from the Gibbs free energy profile that formation of 10 is more preferred as compared to 9 (Fig. 5). An alternative possibility, wherein the prolinate is maintained in its iminium form, is 11.7 kcal mol−1 higher in energy.31 In this vital cyclization step, leading to the tetra-substituted cyclohexyl framework of the product, the cinchona-thiourea catalyst is directly involved in the activation of the substrate (Scheme 4). It is also important to note that the configuration of two out of four chiral centers in the product has already been fixed in the first Michael addition step. The decision of what the configuration of C2 and C3 centers would be depends directly on the proline configuration whereas the cinchona impacts the configuration at C4 and C5 (Scheme 1(b)).7b Hence, in the second Michael addition step, thiourea-cinchona can be regarded as the primary catalyst and proline is a secondary catalyst.
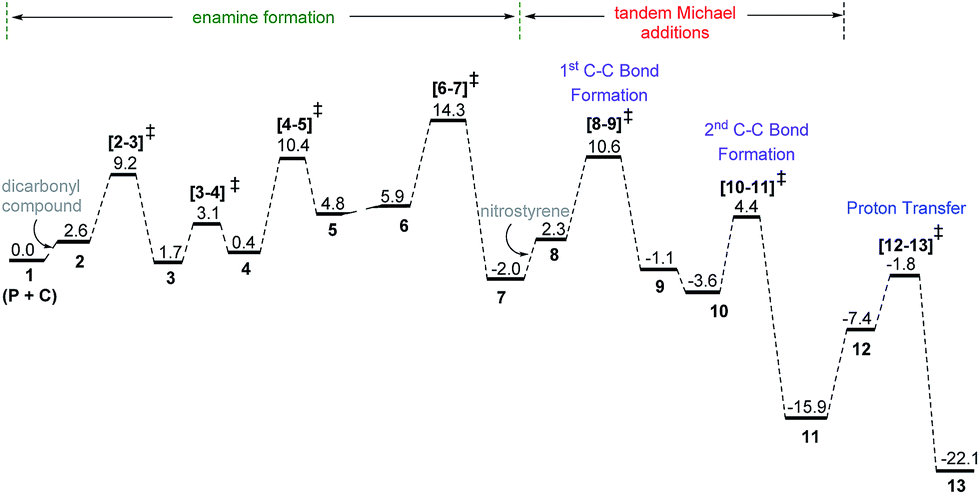 | ||
| Fig. 5 The Gibbs free energy profile obtained at the SMD(toluene)/M06-2X/6-31G** level of theory for the formation of the 2,3-syn product through a dual Michael addition cascade between the dicarbonyl compound and nitrostyrene in the presence of proline (P) and cinchona-thiourea (C) catalyst combination. | ||
As a result of the first and second Michael addition cascade, an enolate intermediate 11 is generated as the Michael adduct (Scheme 4). Another important step at this stage of the reaction is the catalyst regeneration and product release. Here, two possibilities are envisaged depending on the site of protonation; (i) a keto–enol tautomerization if the enolate oxygen gets protonated, or (ii) a protonation at the enolate carbon through intermediate 12. We note that the conversion of 11 to 13 through enolate 12 is an energetically feasible process whereas the keto–enol pathway is much more energy demanding.32
The Gibbs free energy profile for the dual catalytic Michael addition cascade is provided in Fig. 5. A few general features of this dual catalytic reaction can be gathered from the energy profile. Among the three important steps in the overall mechanism (enamine formation and two Michael additions), the formation of the enamine is relatively more energy demanding than the ensuing Michael addition cascade. The application of the energetic span model33 reveals that the enamine formation transition state (from the preceding iminium intermediate, Scheme 4) is the turnover-determining transition state (TDTS) and the penultimate intermediate to the product formation, i.e., oxazolidinone 13, is the turnover-determining intermediate (TDI). Thus, the energetic span (δ) of this catalytic cycle is 14.3 kcal mol−1.34 Overall energetic features provided in Fig. 5 are broadly in concert with the room temperature reaction conditions.
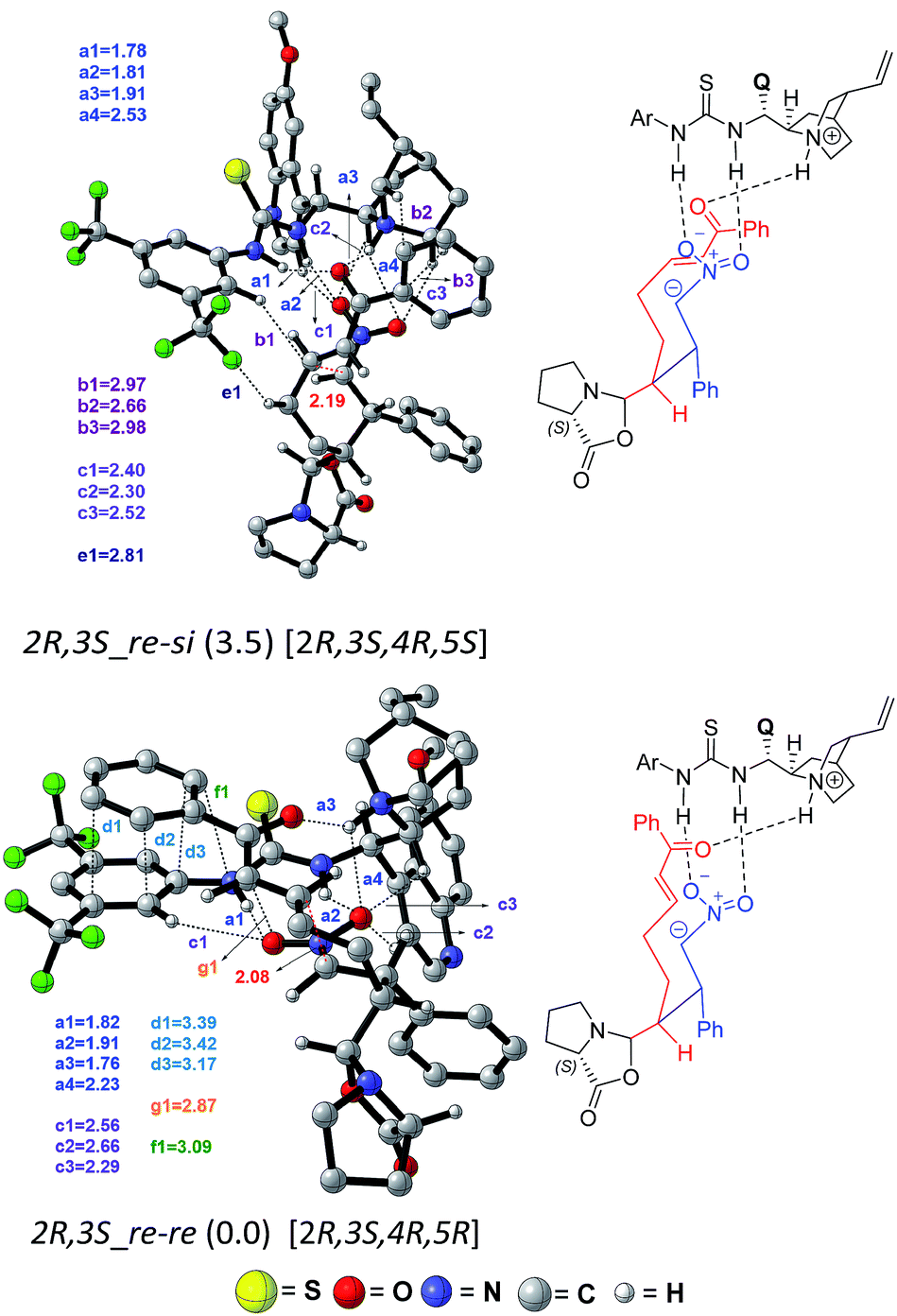 | ||
| Fig. 6 Optimized geometries (and the corresponding Chemdraw structures) of the two lower energy C–C bond formation transition states [10–11]‡ involving different prochiral faces for the second Michael addition obtained at the SMD(toluene)/M06-2X/6-31G** level of theory. Important interaction distances (in angstroms) are shown as a1, a2, a3 = N–H⋯O; b1, b2, b3 = C–H⋯π, and d1, d2, d3 = π⋯π. The absolute configuration of the final product that would arise from each of the transition states is given in square brackets. | ||
In the transition state 2R,3S_re–re, the phenyl ring of the substrate participates in π⋯π stacking (shown using contact distances d1, d2, d3) with the aryl ring of the cinchona-thiourea catalyst (Fig. 6). This catalyst–substrate interaction is absent in the higher energy TS 2R,3S_re–si, suggesting that the additional π⋯π interaction makes a vital contribution to the stabilization of the lower energy transition state that corresponds to the predicted diastereomer. The enantioselectivity, computed on the basis of the energy difference between TSs 2R,3S_re–re (0.0) and 2S,3R_si–si (4.1), is due primarily to more efficient π⋯π (d1, d2, d3) and C–H⋯O (c1, c2, c3) noncovalent interactions in the lower energy TS.35
The lowest energy TS corresponds to the experimentally observed product stereoisomer with a (2R,3S,4R,5R) configuration. Similarly, the computed diastereoselectivity (99.4%) is found to be in good agreement with the experimental value of 92.0%. We also note that the predicted enantioselectivity (>99%) is in near-quantitative agreement with the experimental ee of >99.0%.36 Such good agreements with the experimentally observed stereoselectivities can be regarded a test of our transition state model for this cooperative dual catalytic reaction. One could endeavor to fine-tune the intramolecular interactions noted in the transition states by suitable modifications to the catalysts as well as the choice of other substrate pairs for the same reaction.
The analysis of the geometric features of the stereocontrolling transition states conveys the presence of interesting π⋯π interactions between the catalyst and substrates that can influence the relative energies between these transition states. To examine how significant is the π⋯π interaction in the stereochemically most important transition states, we have re-optimized [8–9]‡ and [10–11]‡ respectively for the first and second Michael addition steps, using modified catalysts and/or substrates. The change in the relative energy ordering is found to be most prominent (a) when the 3,5-trifluoromethyl aryl substituents of the thiourea moiety are changed to 4-nitro aryl, (b) upon replacing the phenyl of nitrostyrene with a methyl group, and (c) when the aryl group of the enone is changed to a methyl group. All these three types of changes resulted in a decrease in the extent of predicted enantio- and diastereo-selectivities in both the first and second Michael addition steps.37
In addition to our detailed study on the origin of stereoinduction presented in the above sections, another interesting aspect of this dual catalytic reaction is also examined. The initially formed 2,3-syn diastereomer at room temperature (1 h) was noted to undergo a complete conversion to the 2,3-anti product upon warming the reaction mixture to 40 °C for 9 h7b While more details can be gleaned from the ESI,† the most essential feature that we wish to highlight here is that both the catalysts are involved in this epimerization as well.38 The primary role is played by cinchona-thiourea and proline renders additional stabilization through a passive hydrogen bonding interaction relatively away from the site of reaction. The mechanistic insights presented in this article are likely to serve an improved guideline toward exploiting the potential dual chiral catalysis using a wider range of substrates and other compatible combinations between organocatalysts.
Conclusions
Density functional theory (M06-2X inclusive of SMD continuum solvation) computations have revealed important molecular details on the mechanism and origin of stereoselectivity in a dual organocatalytic Michael addition cascade between a dicarbonyl compound (heptenal bearing an enone at one end and aldehydic moiety at the other end) and nitrostyrene leading to an important densely substituted cyclohexyl motif. In the most preferred pathway, both chiral organocatalysts, namely, (S)-proline and (8R,9R)-cinchona-thiourea, have been found to be involved in each step of the reaction. A new cooperative catalytic pathway for the formation of the enamine (between proline and heptenal) with explicit participation of cinchona-thiourea has been identified, which is energetically more preferred over the conventional mode in the absence of cinchona-thiourea. The major impact of the cinchona-thiourea catalyst manifests in the form of a network of hydrogen bonding with the dicarbonyl compound leading to a syn-enamine, which is in contrast to the proline-only mechanism where an anti-enamine is energetically more favored. The stereochemistry of the new chiral center in the first Michael addition depends on the syn (offers the si face) or anti (offers the re face) enamine involved in the C–C bond formation with nitrostyrene and hence the influence of the cinchona-thiourea is significant. In the most preferred mode, addition of the si-face of the enamine to the si-face of nitrostyrene leads to a nitronate intermediate with a 2R,3S configuration. In the second Michael addition, the addition of the re-face of nitronate to the re-face of the enone is found to be the most likely cyclization mode. The Michael addition cascade thus leads to a tetra substituted 2,3-syn cyclohexane product with a (2R,3S,4R,5R) configuration, in full accordance with the experimental observation. The precise control of the stereochemical outcome of this relatively complex reaction is found to originate from an effective network of noncovalent interactions between the catalysts and substrates as well as those between the chiral catalysts themselves. In the first Michael addition, the (si–si) mode of addition is the most preferred one due to a critical π⋯π stacking interaction between the catalyst and the substrate (i.e., the stacking of the aryl ring of cinchona-thiourea on the phenyl ring of the nitrostyrene). Favorable interaction between the chiral catalysts, that appears in the form of an effective hydrogen bonding network between the thiourea moiety and proline carboxylate (as well as with the nitro group of the substrate), also contributes toward lowering the energy of the si–si transition state. In the ring closing intramolecular Michael addition, the most preferred transition state exhibits some pivotal catalyst–substrate interactions; (a) π⋯π stacking between the aryl ring of the cinchona-thiourea with the phenyl ring of the α,β-unsaturated carbonyl end of the substrate, and (b) hydrogen bonding between the cinchona thiourea moiety and the carbonyl group of the enone as well as that of the nitro group of nitrostyrene. The mechanistic insights presented in this manuscript suggest that a good number of asymmetric transformations of chiral organocatalyts could be made to work in conjunction with chiral cinchona-thiourea.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous computing time from SpaceTime supercomputing at IIT Bombay is acknowledged. B. B. is grateful to UGC-New Delhi for a Senior Research Fellowship. R. B. S acknowledges the Council of Scientific and Industrial Research (CSIR), New Delhi for funding through 02(0052)/12/EMR-II.Notes and references
- (a) P. J. Walsh and M. C. Kozlowski, Fundamentals of Asymmetric Catalysis, University Science Books, Sausalito, CA, 2008 Search PubMed; (b) B. List, Asymmetric Organocatalysis, Springer, Heidelberg, Germany, 2009 CrossRef; (c) P. I. Dalko, Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications, Wiley-VCH, Weinheim, Germany, 2013 CrossRef.
- (a) G. Jindal, H. K. Kisan and R. B. Sunoj, ACS Catal., 2015, 5, 480–503 CrossRef CAS; (b) Multicatalyst System in Asymmetric Catalysis, ed. J.Zhou, Wiley, NJ, Hoboken, 2014 Search PubMed; (c) S. Piovesana, D. M. S. Schietroma and M. Bella, Angew. Chem., Int. Ed., 2011, 50, 6216–6232 CrossRef CAS PubMed; (d) S. Afewerki and A. Córdova, Chem. Rev., 2016, 116, 13512–13570 CrossRef CAS PubMed; (e) L. Lin and X. Feng, Chem.–Eur. J., 2017, 23, 6464–6482 CrossRef CAS PubMed; (f) M. Bihani and J. C.-G. Zhao, Adv. Synth. Catal., 2017, 359, 534–575 CrossRef CAS; (g) M. Mahlau and B. List, Angew. Chem., Int. Ed., 2013, 52, 518–533 CrossRef CAS PubMed.
- (a) E. M. Vogl, H. Groger and M. Shibasaki, Angew. Chem., Int. Ed., 1999, 38, 1570–1577 CrossRef CAS; (b) L. Hong, W. Sun, D. Yang, G. Li and R. Wang, Chem. Rev., 2016, 116, 4006–4123 CrossRef CAS PubMed; (c) A. Moran, A. C. Hamilton, C. Bo and P. Melchiorre, J. Am. Chem. Soc., 2013, 135, 9091–9098 CrossRef CAS PubMed.
- (a) S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627–5639 CrossRef CAS PubMed; (b) X. Huo, R. He, X. Zhang and W. Zhang, J. Am. Chem. Soc., 2016, 138, 11093–11096 CrossRef CAS PubMed; (c) S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065–1068 CrossRef CAS PubMed; (d) T. Sandmeier, S. Krautwald, H. F. Zipfel and E. M. Carreira, Angew. Chem., Int. Ed., 2015, 54, 14363–14367 CrossRef CAS PubMed; (e) X. Jiang, J. J. Beiger and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 87–90 CrossRef CAS PubMed; (f) F. A. Cruz and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 5627–5639 CrossRef PubMed; (g) X. Jiang, P. Boehm and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140(4), 1239–1242 CrossRef CAS PubMed.
- (a) B. Bhaskararao and R. B. Sunoj, J. Am. Chem. Soc., 2015, 137, 15712–15722 CrossRef CAS PubMed; (b) B. Bhaskararao and R. B. Sunoj, ACS Catal., 2017, 7, 6675–6685 CrossRef CAS; (c) S. Tribedi, C. M. Hadad and R. B. Sunoj, Chem. Sci., 2018, 9, 6126–6133 RSC.
- (a) M. L. Clarke and J. A. Fuentes, Angew. Chem., Int. Ed., 2007, 46, 930–933 CrossRef CAS PubMed; (b) D. Uraguchi, Y. Ueki and T. Ooi, Science, 2009, 326, 120–123 CrossRef CAS PubMed; (c) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed; (d) K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef CAS PubMed.
- (a) T. Mandal and C.-G. Zhao, Angew. Chem., Int. Ed., 2008, 47, 7714–7717 CrossRef CAS PubMed; (b) N. K. Rana, H. Huang and J. C.-G. Zhao, Angew. Chem., Int. Ed., 2014, 53, 7619–7623 CrossRef CAS PubMed.
- (a) A. G. Schultz, Acc. Chem. Res., 1990, 23, 207–213 CrossRef CAS; (b) J. R. Lewis, Nat. Prod. Rep., 1994, 11, 329–332 RSC; (c) Y. Wang, H. Lu and P. –F. Xu, Acc. Chem. Res., 2015, 48, 1832–1844 CrossRef CAS PubMed.
- (a) B. List, P. Pojarliev and H. J. Martin, Org. Lett., 2001, 3, 2423–2425 CrossRef CAS PubMed; (b) B. List, Tetrahedron, 2002, 58, 5573–5590 CrossRef CAS; (c) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Angew. Chem., Int. Ed., 2005, 44, 4212–4215 CrossRef CAS PubMed; (d) D. Seebach, A. K. Beck, D. M. Badine, M. Limbach, A. Eschenmoser, A. M. Treasurywala, R. Hobi, W. Prikoszovich and B. Linder, Helv. Chim. Acta, 2007, 90, 425–471 CrossRef CAS; (e) A. K. Sharma and R. B. Sunoj, J. Org. Chem., 2012, 77, 10516–10524 CrossRef CAS PubMed.
- (a) Y. Wang, R.-G. Han, Y.-L. Zhao, S. Yang, P.-F. Xu and D. J. Dixon, Angew. Chem., Int. Ed., 2009, 48, 9834–9838 CrossRef CAS PubMed; (b) S. Parera, D. Sinha, N. K. Rana, V. Trieu-Do and J. C.-G. Zhao, J. Org. Chem., 2013, 78, 10947–10953 CrossRef PubMed; (c) H. Huang, S. Konda and J. C.-G. Zhao, Angew. Chem., Int. Ed., 2016, 55, 2213–2216 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- (a) C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS; (b) C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS; (c) S. Santoro, M. Kalek, G. Huang and F. Himo, Acc. Chem. Res., 2016, 49, 1006–1018 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- C. Y. Legault, CYLview, 1.0 b, Université de Sherbrooke, Quebec Canada, 2009, (https://www.cylview.org) Search PubMed.
- (a) R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS; (b) AIM2000 Version 2.0, Buro fur Innovative Software, SBK-Software, Bielefeld, Germany, 2002 Search PubMed; (c) C. F. Matta and R. J. Boyd. Quantum theory of atoms in molecules: Recent progress in theory and application, Wiley-VCH, Weinheim, 2007 CrossRef.
- (a) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed; (b) J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J. –P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- (a) Y. A. Abramov, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1997, 53, 264–272 CrossRef; (b) E. Espinosa, E. Molins and C. Lectome, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS; (c) P. v. R. Schleyer, Chem. Rev., 2005, 105, 3433–3435 CrossRef CAS; (d) J. S. Grabowski, Chem. Rev., 2011, 111, 2597–2625 CrossRef PubMed.
- (a) A. K. Sharma and R. B. Sunoj, Angew. Chem., Int. Ed., 2010, 49, 6373–6377 CrossRef CAS PubMed; (b) P. H.-Y. Cheong, C. Y. Legault, J. M. Um, N. Çelebi-Ölçüm and K. N. Houk, Chem. Rev., 2011, 111, 5042–5137 CrossRef CAS PubMed.
- (a) P. Renzi, J. Hioe and R. M. Gschwind, Acc. Chem. Res., 2017, 50, 2936–2948 CrossRef CAS PubMed; (b) M. H. Haindl, J. Hioe and R. M. Gschwind, J. Am. Chem. Soc., 2015, 137, 12835–12842 CrossRef CAS PubMed; (c) J. Burés, A. Armstrong and D. G. Blackmond, Chem. Sci., 2012, 3, 1273–1277 RSC; (d) N. El-Hamdouni, X. Companyo, R. Rios and A. Moyano, Chem.–Eur. J., 2010, 16, 1142–1148 CrossRef CAS PubMed; (e) N. Lokesh, A. Seegerer, J. Hioe and R. M. Gschwind, J. Am. Chem. Soc., 2018, 140, 1855–1862 CrossRef CAS PubMed.
- Details of the mechanism of proline enamine formation in the absence of cinchona-thiourea is provided in Schemes S4–S5, Fig. S4 in the ESI.†.
- (a) C. Allemann, R. Gordillo, F. R. Clemente, P. H.-Y. Cheong and K. N. Houk, Acc. Chem. Res., 2004, 37, 558–569 CrossRef CAS PubMed; (b) R. B. Sunoj, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 920–931 CAS.
- (a) A number of different possibilities for the catalyst dyad are identified within a narrow energy difference. See Fig. S5 in the ESI.†; (b) Certain NMR chemical shift values in parent proline are known to change upon mixing it with the cinchona-thiourea catalyst. Disappearance of the carboxylic acid peak at δ 11.4 ppm, upon mixing with cinchona-thiourea, is particularly noteworthy. See ref. 7a..
- All the elementary step barriers for the formation the proline enamine intermediate, with and without cinchona-thiourea, as well as the details of conformation analyses are provided in Tables S4–S6 and Figs. S6–S7 in the ESI.†.
- Further details of Michael addition between butanone and nitrostyrene are provided in Fig. S1–S3, Schemes S1–S3 and Tables S1–S3 in the ESI.†.
- A number of different activation modes of nitrostyrene by the thiourea family of organocatalysts in single-catalyst reactions were proposed earlier. See (a) T. Okino, Y. Hoashi, T. Furukawa, X. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119–125 CrossRef CAS PubMed; (b) A. Hamza, G. Schubert, T. Soos and Ì. Papai, J. Am. Chem. Soc., 2006, 128, 13151–13160 CrossRef CAS PubMed; (c) P. Hammar, T. Marcelli, H. Hiemstra and F. Himo, Adv. Synth. Catal., 2007, 349, 2537–2548 CrossRef CAS; (d) J.-L. Zhu, Y. Zhang, C. Liu, A.-M. Zheng and W. Wang, J. Org. Chem., 2012, 77, 9813–9825 CrossRef CAS PubMed; (e) A. Sengupta and R. B. Sunoj, J. Org. Chem., 2012, 77, 10525–10536 CrossRef CAS PubMed.
- More details of comparison of different activation modes of nitrostyrene in the first Michael addition are shown in Fig. S8 and Table S7 in the ESI.†.
- Conformational analysis is provided in more detail as shown in Fig. S9 and Table S8 in the ESI.† In addition, we have considered Boltzmann distribution across different conformers of the stereocontrolling transition states, and the predicted enantio- and diastereo-selectivities exhibited a very similar pattern to what we predicted using the lowest energy transition states as shown in Table S9.†.
- (a) F. M. Bickelhaupt, J. Comput. Chem., 1999, 20, 114–128 CrossRef CAS; (b) W.-J. van Zeist and F. M. Bickelhaupt, Org. Biomol. Chem., 2010, 8, 3118–3127 RSC; (c) F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed; (d) The origin of the energy difference between the stereocontrolling transition states is also analyzed using the activation strain analysis. We note that the total distortion in the catalysts and substrate in the lowest energy si-si TS is lower than that found in the other modes. See Fig. S10 and Table S11 in the ESI† for additional details..
- (a) The contributions due to the hydrogen bonding interactions probed using the Abrovamov equation (ref 18) that makes use of the electron densities (ρ) at the bond critical points, Laplacian of ρ and kinetic energy (G) as obtained from the atoms in molecules (AIM) analysis. The estimated hydrogen bond energies in the si-si TS are more by 0.9 (re-si), 2.6 (si-re), and 9.1 kcal/mol (re-re) compared to other modes. For additional details see Table S12 in the ESI.†; (b) AIM2000 Version 2.0, Buro fur Innovative Software, SBK-Software, Bielefeld, Germany, 2002 Search PubMed; (c) C. F. Matta and R. J. Boyd. Quantum Theory of Atoms in Molecules: Recent Progress in Theory and Application, Wiley-VCH, Weinheim, 2007 CrossRef.
- For additional details see Fig. S11 in the ESI.†.
- A water assisted keto-enol tautomerization [(11–13) H2O]‡ is found to be 20.0 kcal/mol higher in energy than the protonation at the enolate carbon [12–13]‡. More details are provided in Fig. S12 in the ESI.†.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- More details of energetic span calculations are provided in Fig. S13 in the ESI.†.
- Detailed mapping of noncovalent interactions in the higher energy 2S,3R_si-si TS is provided in Fig. S14 in the ESI.†.
- The application of the activation strain model revealed a lesser distortion in the cinchona-thiourea catalyst and the reactant (nitronate) in the lower energy transition state (2R,3S_re-re) as compared to the other higher energy analogues responsible for the formation of different stereoisomers (2R,3S_re-si and 2S,3R_si-si). More details are provided in Table S14 in the ESI.†.
- For more details see Tables S15–S17 in the ESI.†.
- For more details of epimerization, see Scheme S6 and Fig. S15 in the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized geometries, additional schemes, figures, and tables are provided as part-A and Cartesian coordinates of all stationary points are provided as part-B. See DOI: 10.1039/c8sc03078b |
| This journal is © The Royal Society of Chemistry 2018 |