
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransient [3,3] Cope rearrangement of 3,3-dicyano-1,5-dienes: computational analysis and 2-step synthesis of arylcycloheptanes†
Ehsan
Fereyduni
 a,
Jacob N.
Sanders
b,
Gabriel
Gonzalez
a,
K. N.
Houk
*b and
Alexander J.
Grenning
*a
a,
Jacob N.
Sanders
b,
Gabriel
Gonzalez
a,
K. N.
Houk
*b and
Alexander J.
Grenning
*a
aDepartment of Chemistry, University of Florida, P.O. Box 117200, Gainesville, FL 32611, USA. E-mail: grenning@ufl.edu
bDepartment of Chemistry and Biochemistry, University of California – Los Angeles, 607 Charles E. Young Drive East, Box 951569, Los Angeles, CA 90095-1569, USA. E-mail: houk@chem.ucla.edu
First published on 21st September 2018
Abstract
A simple and modular route to arylcycloheptene scaffolds is reported. The two-step route from Knoevenagel adducts and allylic electrophiles is made possible through the design of a Cope rearrangement that utilizes a “traceless” activating group to promote an otherwise thermodynamically unfavorable transformation. Experimentally, the [3,3] rearrangement occurrs transiently at room temperature with a computed barrier of 19.5 kcal mol−1, which ultimately allows for three-component bis-allylation. Ring-closing metathesis delivers the arylcycloheptane and removes the activating group. This report describes the design and optimization of the methodology, scope and mechanistic studies, and computational analysis.
Introduction
Structurally complex natural products are promising leads for treating diseases1 and are commonly acquired by isolation and semisynthesis.2De novo synthesis of complex bioactive molecules and their analogs is a modern synthetic challenge.3,4 The most successful examples address synthetic ideality:5 efficiency, practicality, and scalability. From a drug discovery perspective, modularity needs to also be addressed.We and others are interested in designing practical routes to polycyclic architectures that are simple and efficient from abundant starting material classes.6 Adhering to these requirements will result in routes amenable to target and target-analog synthesis. Inspired by bioactive aryl-cycloheptanes (Fig. 1) which include terpenes (the frondosins,7 liphagal,8 pharbinilic acid9), resveratrol-derivatives (vitisinol C,10 ampelopsin A11), alkaloids12 (ambiguine,13 actinophyllic acid,14 exotine B15), and marketed drugs (irosustat16) and drug leads (the synthetic SIRT1 inhibitor17) (Scheme 1), we hypothesized that 1,5-dienes A and an allylic electrophile B, could be converted to the aryl-cycloheptane scaffold C over, in theory, a simple procedure involving a Cope rearrangement, deconjugative allylation, and ring-closing metathesis (RCM) (Scheme 1).6b,c Notably, 1,5-dienes of type A are prepared by a simple and convergent two-step protocol from ketones, malononitrile, and cinnamyl electrophiles: all abundant starting material classes.6b Unfortunately, in model studies, the Cope rearrangement is not thermodynamically favorable due to styrene-deconjugation.6b
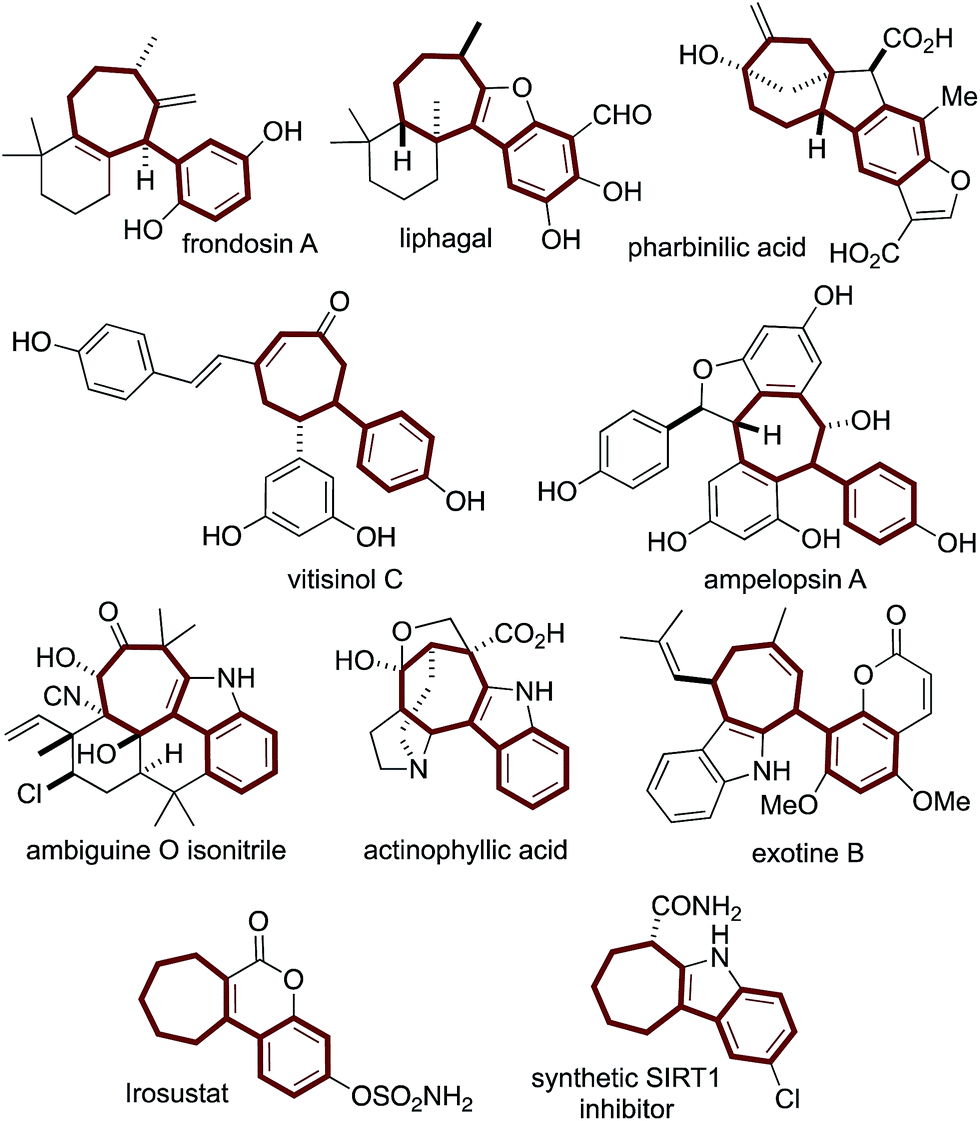 | ||
| Fig. 1 The arylcycloheptane scaffold is common to terpenoid-, resveratrol-, alkaloidal-, and synthetic drug molecules. | ||
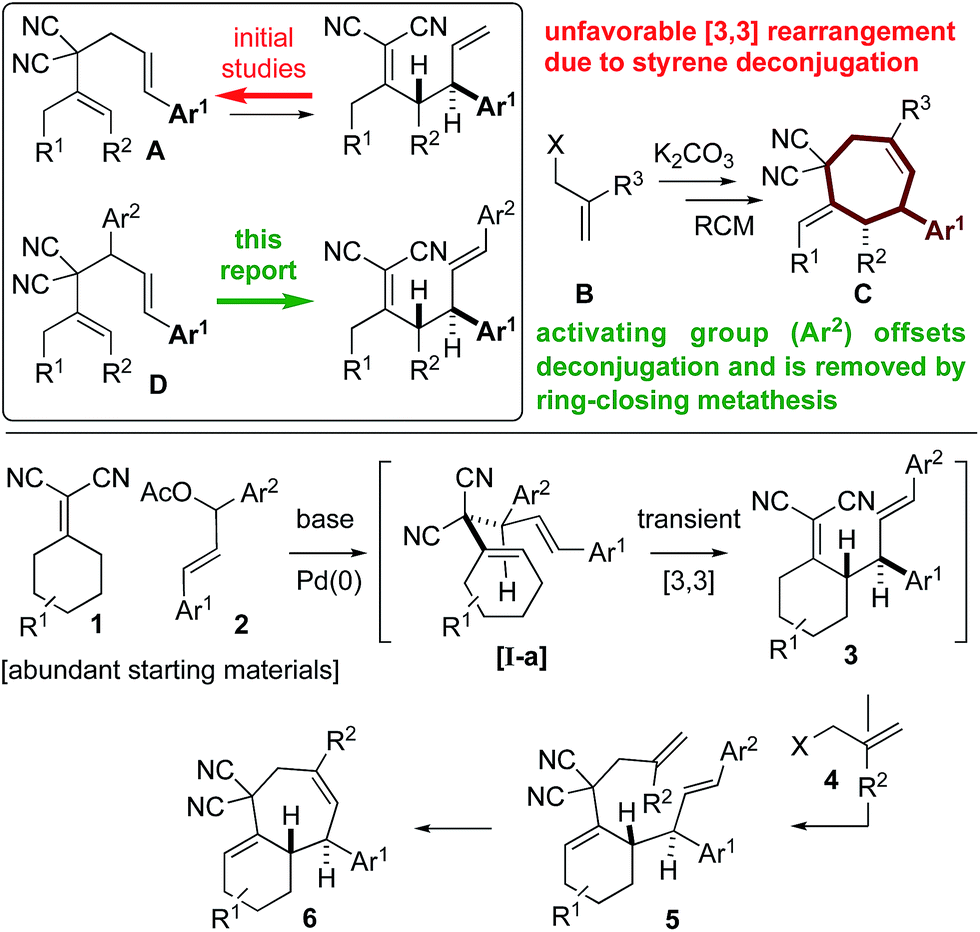 | ||
| Scheme 1 Overcoming thermodynamic limitations of [3,3] Cope rearrangements though the introduction of a “traceless” activating group, which is removed during the RCM step. | ||
To combat the poor conversions observed with 1,5-dienes A, we hypothesized that the 4,6-diaryl-1,5-dienes D would be more reactive toward thermal rearrangement as styrene deconjugation is offset (Scheme 2).6b,18 Furthermore, the exact same products C can be accessed as the [3,3]-promoting aryl group (Ar2) is removed in the ring-closing metathesis step. To reiterate, the additional aryl group serves to provide a driving-force for an otherwise unfavorable [3,3] rearrangement and is “traceless” upon RCM. Herein we report that Knoevenagel adducts 1 and chalcone-derived electrophiles 2 undergo deconjugative alkylation to [I-a] followed by a transient [3,3] rearrangement (unexpectedly occurring at room-temperature with a calculated barrier of 19.5 kcal mol−1) to yield the γ-allylated Knoevenagel adduct 3. Deconjugative alkylation with allylic electrophiles 4 yields the bis-allylated building blocks 5 in one-step from 1, 2, and 4, which undergo facile RCM to aryl-cycloheptenes 6.
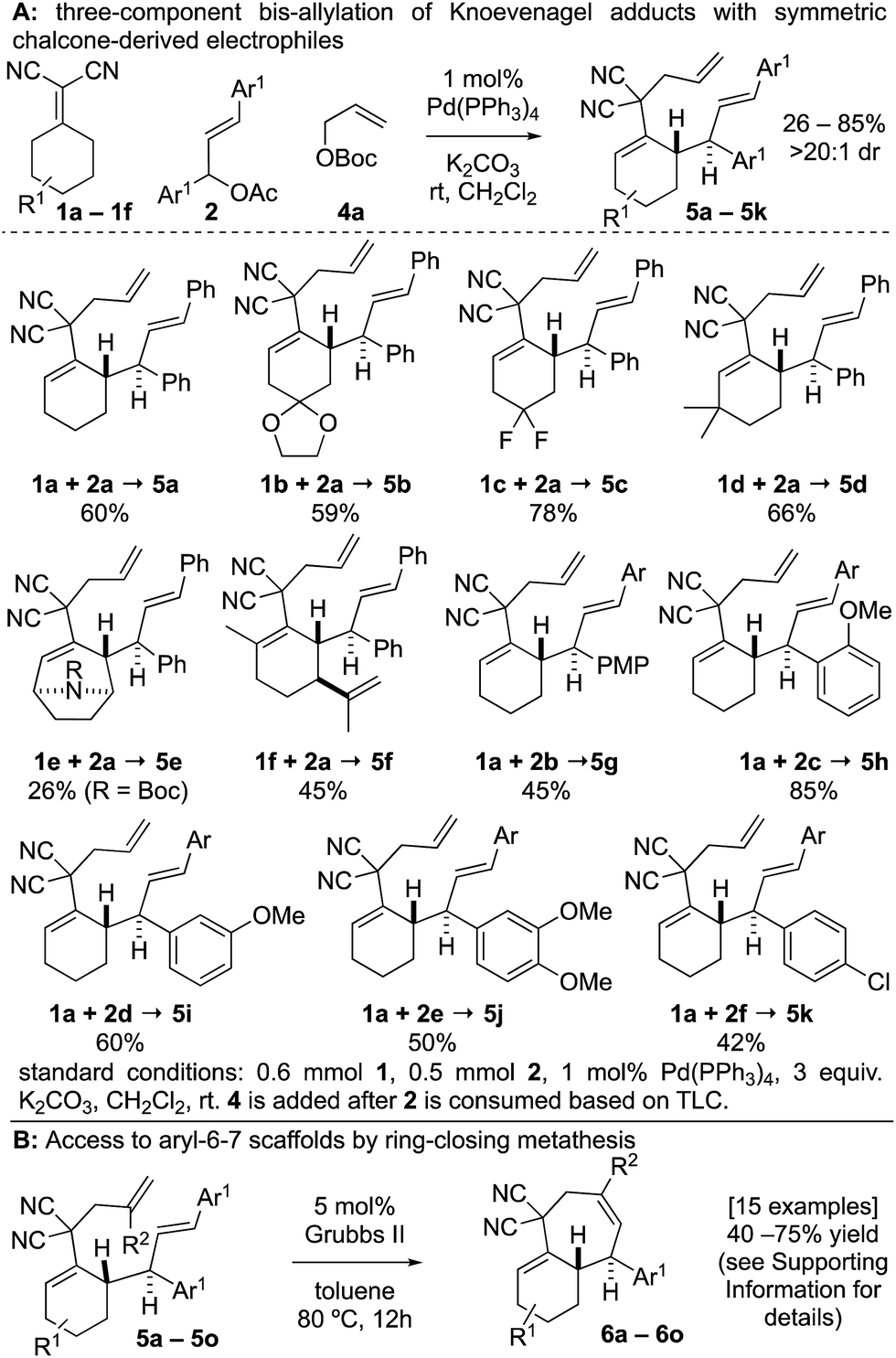 | ||
| Scheme 2 Scope of two-step arylcycloheptene synthesis. | ||
Results and discussion
At the outset of our studies, we examined model Knoevenagel adduct 1a and chalcone-derived electrophile 2a and uncovered that by Pd(0)-catalysis, the molecules couple yielding the γ-allylated scaffold 3a (eqn (1)). The reaction was efficient, diastereoselective, scalable, and the product can be recrystallized. The regioselectivity was also unexpected as alkyl electrophiles (e.g. alkyl halides and allyl acetates/carbonates) tend to yield deconjugative alkylation products.19,20 Thus, this result raises the question as to whether 3 is accessed by a [3,3] rearrangement21 as originally proposed or by a direct γ-allylation mechanism.20 This connectivity is nonetheless welcomed as the high acidity of the Knoevenagel adduct remains unchanged allowing for three-component coupling directly to bis-allylated scaffolds 5a–5k (Scheme 2A) which are routinely converted to arylcycloheptenes by ring-closing metathesis (Scheme 2B).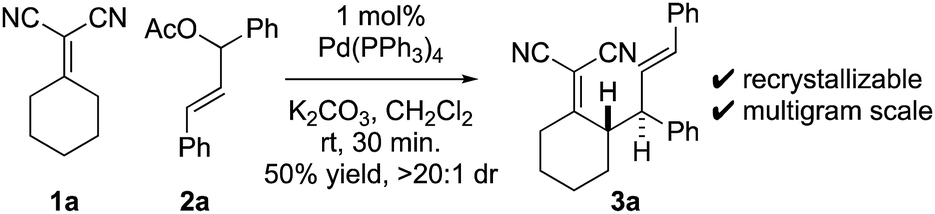 | (1) |
The three-component coupling tolerated a wide array of cyclic Knoevenagel adducts 1a–1f and symmetric chalcone-derived electrophiles 2a–2f (Scheme 2A). The mild reaction is tolerant to ketal (5b), gem-difluoro (5c), carbamate (5e), and alkene functional groups (5f). 5d was prepared by a regioselective deprotonation to initiate the transformation. A variety of substitution patterns on the arene could also be incorporated including p-, o-, m-methoxy (5g–5i), dimethoxy (5j), and p-chloro (5k) substitutions.
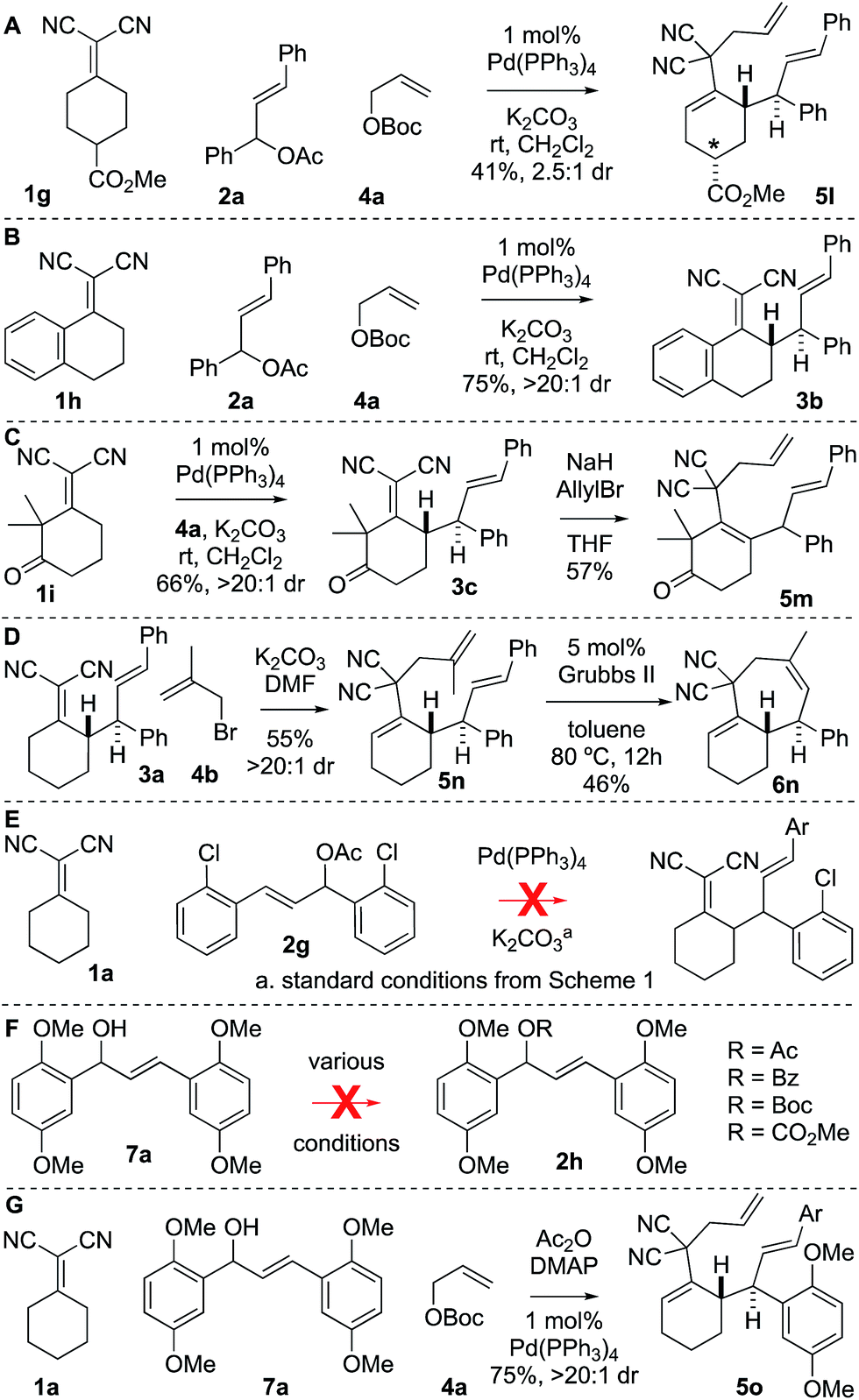
There were several other notable experiments performed related to the scope of the Knoevenagel adduct bis-allylation protocol (Scheme 3). Pro-chiral Knoevenagel adducts with a remote stereocenter (e.g.1g) gave rise to diastereomeric mixtures (Scheme 3A). Also, when examining the tetralone-derived Knoevenagel adduct 1h for three-component bis-alkylation reactivity, only two-component coupling was observed to 3b (Scheme 3B). Similarly, this was observed with Knoevenagel adduct 1i. However, deprotonation of sterically encumbered γ-C–H's can be achieved with NaH as the base (5m, Scheme 3C). The sequence can also be performed with 2-substituted allylic electrophiles ultimately yielding trisubstituted olefins by RCM (Scheme 3D). Next, the electron-deficient chalcone-derivative 2g did not react under the standard conditions (0% conversion), likely due to challenges associated with the oxidative addition step (Scheme 3E). Attempts to make the activated chalcone-derived electrophile 2h were unsuccessful (Scheme 3F). We suspected that the issue might be that acylation is occurring, but the acetate/carbonate is prone to hydrolysis back to the alcohol under standard work-up conditions (extraction conditions, silica gel, etc.). In agreement with this, successful coupling was achieved directly from the alcohol 7a using an in situ acylation strategy (Scheme 3G).
 | ||
| Scheme 3 Other studies related to the Knoevenagel adduct bis-allylation protocol. | ||
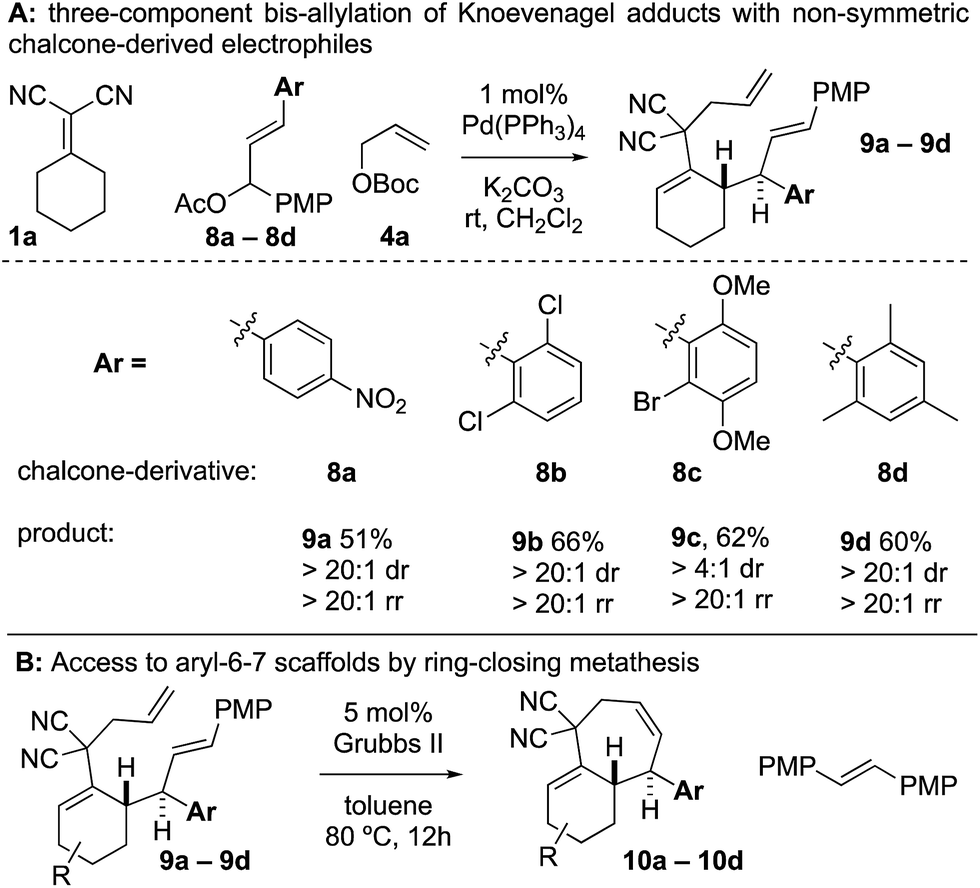
When examining non-symmetric chalcone-derived electrophiles 8a–8d with Knoevenagel adduct 1a, it was uncovered that diastereo- and regioselective transformation to the bis-allylated products 9a–9d could be achieved (Scheme 4). The electrophiles bore a p-methoxyphenyl (PMP) and a variable arene (p-nitrophenyl (8a), 2,6-dichloro (8b), 2-bromo-3,6-dimethoxyphenyl (8c), and 2,4,6-trimethylphenyl (8d)). In all cases, the variable arene was installed at the allylic position. Thus, upon ring-closing metathesis, the PMP-group was removed and the variable-aryl-cycloheptenes 10a–10d were prepared.
 | ||
| Scheme 4 Exploration of nonsymmetric chalcone-derivatives in arylcycloheptane synthesis. | ||
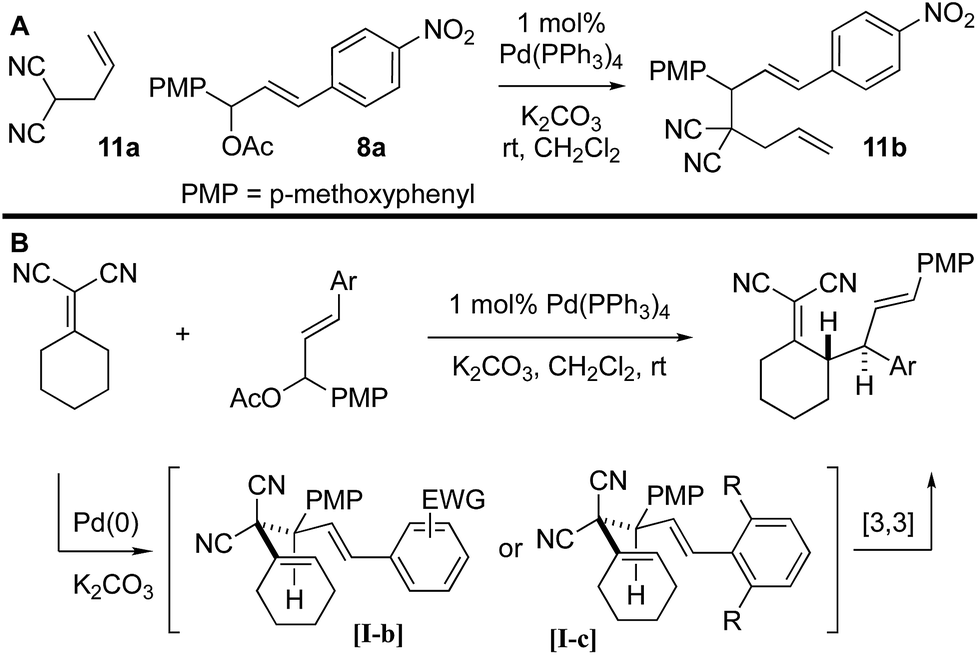
It was not clear whether the mechanism of the initial coupling between the Knoevenagel adduct and the chalcone-derived allylic electrophile occurs by the originally conceived deconjugative alkylation/transient [3,3] rearrangement sequence or by a direct γ-allylation mechanism. The regioselectivity observed in Scheme 4 suggests that the reaction is proceeding by low-barrier Cope rearrangement (occurring at room-temperature). This is surprising as related 3,3-dicyano-1,5-dienes do not undergo rearrangement until heated >120 °C.6b Furthermore, Cope rearrangements occurring at room temperature usually bear a strain element22 or are “oxy-Cope” substrates.23 Consider the following data: (a) allyl malononitrile 11a reacts with 7a to yield product 11b where the PMP group, not the p-nitrophenyl group, is at the allylic position (Scheme 6A). This result is opposite to the connectivity in 9a (Scheme 5). (b) The regioselectivity of allylation with 8b–8d is such that the sterically bulky arene is at the allylic position on the bis-allylated building blocks 9b–9d. As shown in Scheme 5B, we suggest that deconjugative alkylation occurs first yielding the 1,5-dienes [I-a/b]. This transformation is either electronically [I-b] or sterically [I-c] driven (or both). Cope rearrangement then yields the γ-allylated product with connectivity that matches the products from bis-allylation (Scheme 4).
 | ||
| Scheme 5 γ-Allylation occurs by a transient Cope rearrangement. | ||
To probe whether a room-temperature Cope rearrangement of 4,6-diaryl-3,3-dicyano-1,5-dienes was reasonable,24 we performed computational studies for the synthesis of 3a (Fig. 2). Density functional theory calculations showed that the Cope rearrangement was exergonic by 8.6 kcal mol−1 and had an unusually low free energy barrier of 19.5 kcal mol−1, corresponding to a half-life of 23 seconds at room temperature. To explain the facility of this Cope rearrangement at room temperature, we investigated bond lengths and atomic charges in the transition state (Fig. 1). The transition state revealed substantial dissociative character (2.38 Å and 2.46 Å for the breaking and forming bonds respectively) and significant charge separation, with a stabilized negative charge (−0.34e) alpha to the two nitrile groups and stabilized positive charges (+0.13e and +0.12e) at the two benzylic positions. The ability of the nitrile groups and phenyl groups to stabilize each transition state fragment via conjugation accounts for the low barrier of this Cope rearrangement. All structures were optimized at the M06-2X/6-31+G(d) level of theory with single-point energy corrections computed at the M06-2X-D3/6-311++G(2d,2p) level of theory with dichloromethane CPCM solvent; partial charges were computed via NBO analysis and hydrogen atom charges were summed into the neighboring heavy atom.
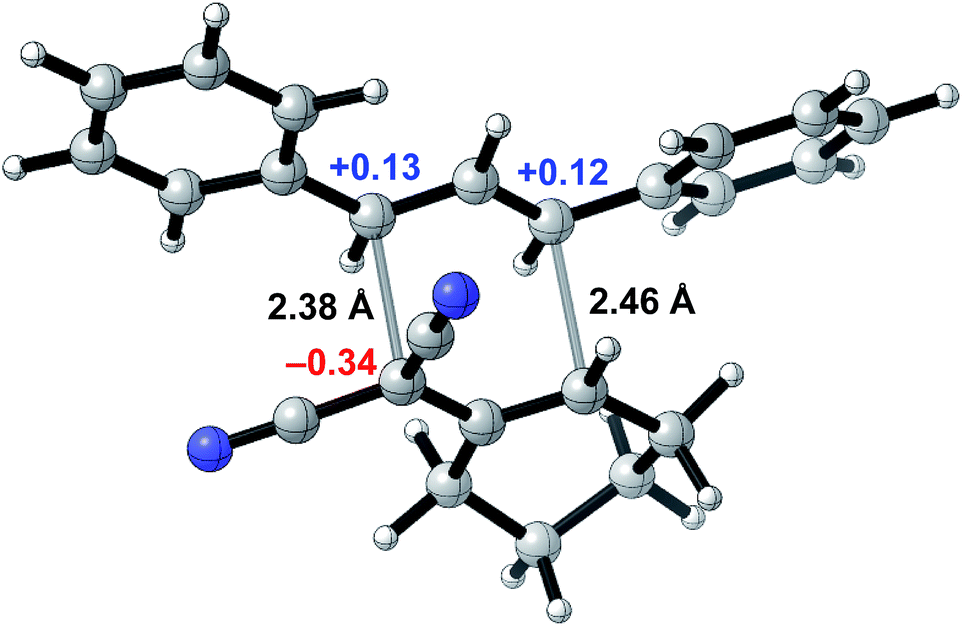 | ||
| Fig. 2 Transition state for the Cope rearrangement leading to the formation of 3a computed via density functional theory. | ||
Another beneficial consequence of the Cope rearrangement being exceedingly mild is we can begin to prepare cycloheptene scaffolds bearing an embedded Meldrum's acid moiety in lieu of the gem-dinitrile (Scheme 6). Meldrum's acid moieties are excellent handles for functional group interconversion.25 This was previously not possible because the standard Cope rearrangements of this type occur at temperatures >120 °C and Meldrum's acid derivatives tend to decompose to ketene, CO2, and acetone at temperatures lower than this.26 As a preliminary example of this, we coupled the Meldrum's acid–isovaleraldehyde Knoevenagel adduct 12a and the chalcone-derived alcohol directly to 12b by a one-pot alcohol activation, Pd-catalyzed deconjugative allylation-transient-Cope sequence, and alkylidene reduction. In two additional simple steps the Meldrum's acid embedded arylcycloheptene 12c is accessed.
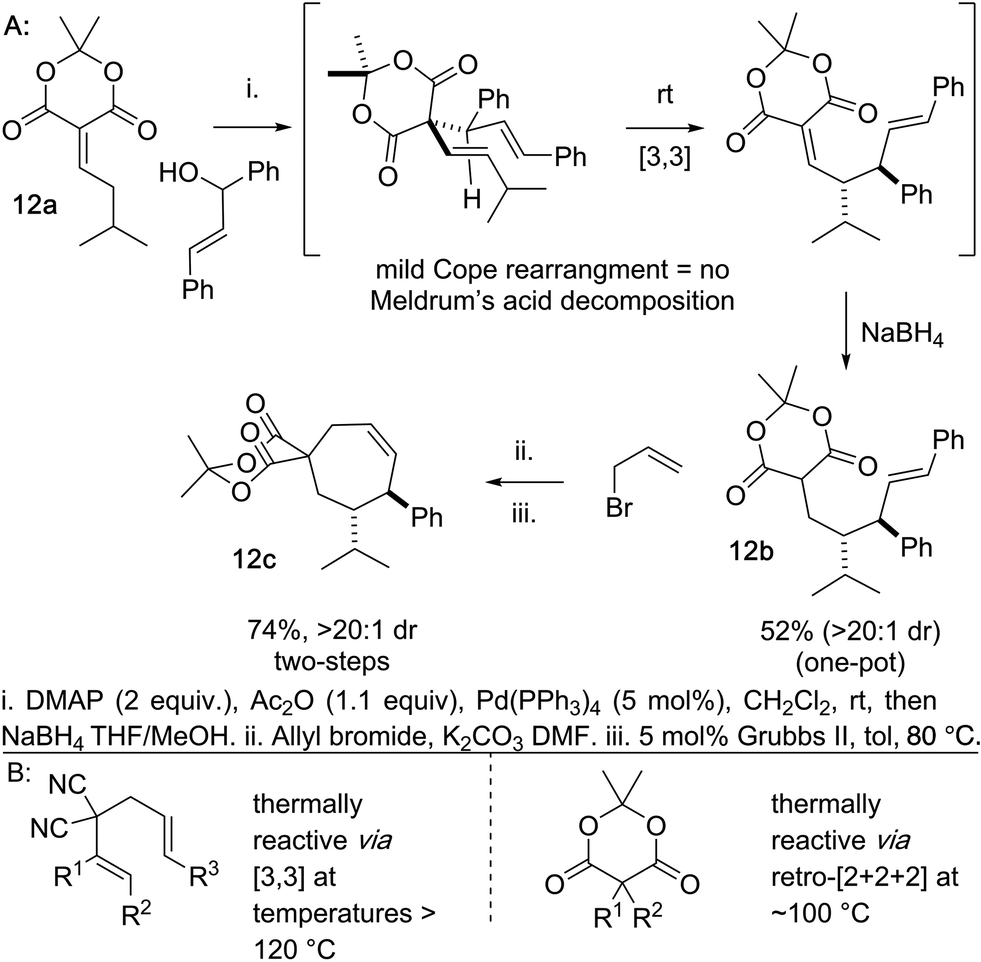 | ||
| Scheme 6 (A) Meldrum's acid-derived Knoevenagel adducts can undergo deconjugative alkylation/Cope rearrangement. (B) This would not possible with higher energy Cope rearrangements because Meldrum's acid is unstable. | ||
Conclusions
Inspired by arylcycloheptane architectures, we have uncovered a two-step route to such core scaffolds that is amenable to structural change due to the concise synthetic sequence from abundant starting material classes. The route is made possible through the development of a [3,3] promoting group strategy for driving forward otherwise thermodynamically unfavorable Cope rearrangements. The surprisingly low-barrier Cope rearrangement, occurring transiently with a calculated barrier of 19.5 kcal mol−1, ultimately enabled a one-pot bis-allylation protocol to be developed for Knoevenagel adducts. Future studies include improving the scope, applying the method in target/target-analog synthesis, and rendering the strategy enantioselective. The [3,3] “promoting group strategy” described herein may be a general manifold for promoting many other unfavorable [3,3] rearrangements. We plan to examine this broadly too.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the College of Liberal Arts and Sciences and the Department of Chemistry at the University of Florida for start-up funds. We thank the Mass Spectrometry Research and Education Center and their funding source: NIH S10 OD021758-01A1. J. N. S. acknowledges the support of the National Institute of General Medical Sciences of the National Institutes of Health under Award Number F32GM122218. Computational resources were provided by the UCLA Institute for Digital Research and Education (IDRE).Notes and references
- (a) D. J. Newman and G. M. Cragg, Nat. Prod., 2016, 79, 629 CrossRef CAS PubMed; (b) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed.
- B. Ganem and R. R. Franke, J. Org. Chem., 2007, 72, 3981 CrossRef CAS PubMed.
- For a recent editorial pertaining to complex molecules synthesis see: P. S. Baran, J. Am. Chem. Soc., 2018, 140, 4751 CrossRef CAS PubMed.
- For recent reviews of complex molecule synthesis see: (a) D. J. Jansen and R. A. Shenvi, Future Med. Chem., 2014, 6, 1127 CrossRef CAS PubMed; (b) D. Urabe, T. Asaba and M. Inoue, Chem. Rev., 2015, 115, 9207 CrossRef CAS PubMed; (c) Z. G. Brill, M. L. Condakes, C. P. Ting and T. J. Maimone, Chem. Rev., 2017, 117, 11753 CrossRef CAS PubMed.
- T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75, 4657 CrossRef CAS PubMed.
- (a) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40 CrossRef CAS PubMed; (b) O. Lahtigui, F. Emmetiere, W. Zhang, L. Jirmo, S. Toledo-Roy, J. C. Hershberger, J. M. Macho and A. J. Grenning, Angew. Chem., Int. Ed., 2016, 55, 15792 CrossRef CAS PubMed; (c) S. K. Scott and A. J. Grenning, Angew. Chem., Int. Ed., 2017, 56, 8125 CrossRef CAS PubMed; (d) E. Fereyduni and A. J. Grenning, Org. Lett., 2017, 19, 4130 CrossRef CAS PubMed; (e) W. S. Kim, K. Du, A. Eastman, R. P. Hughes and G. C. Micalizio, Nat. Chem., 2017, 10, 70 CrossRef PubMed; (f) R. Wildermuth, K. Speck, F.-L. Haut, P. Mayer, B. Karge, M. Brönstrup and T. Magauer, Nat. Commun., 2017, 8, 2083 CrossRef PubMed; (g) E. E. Robinson and R. J. Thomson, J. Am. Chem. Soc., 2018, 140, 1956 CrossRef CAS PubMed; (h) N. R. Cichowicz, W. Kaplan, Y. Khomutnyk, B. Bhattarai, Z. Sun and P. Nagorny, J. Am. Chem. Soc., 2015, 137, 14341 CrossRef CAS PubMed; (i) M. Christiaens, J. Hullaert, K. Van Hecke, D. Laplace and J. M. Winne, Chem.–Eur. J., 2018 DOI:10.1002/chem.201803248.
- (a) Y. F. Hallock, J. H. Cardellina II and M. R. Boyd, Nat. Prod. Lett., 1998, 11, 153 CrossRef CAS; (b) A. D. Patil, A. J. Freyer, L. Killmer, P. Offen, B. Carte, A. J. Jurewicz and R. K. Johnson, Tetrahedron, 1997, 53, 5047 CrossRef CAS.
- (a) A. R. Pereira, W. K. Strangman, F. Marion, L. Feldberg, D. Roll, R. Mallon, I. Hollander and R. J. Andersen, J. Med. Chem., 2010, 53, 8523 CrossRef CAS PubMed; (b) F. Marion, D. E. Williams, B. O. Patrick, I. Hollander, R. Mallon, S. C. Kim, D. M. Roll, L. Feldberg, R. Van Soest and R. J. Andersen, Org. Lett., 2006, 8, 321 CrossRef CAS PubMed.
- K. H. Kim, S. U. Choi, M. W. Son, S. Z. Choi, J. Clardy and K. R. Lee, J. Nat. Prod., 2013, 76, 1376 CrossRef CAS PubMed.
- (a) Y. H. Choi, M. Y. Yoo, C. W. Choi, M.-R. Cha, G. H. Yon, D. Y. Kwon, Y. S. Kim, W.-K. Park and S. Y. Ryu, Planta Med., 2009, 75, 537 CrossRef CAS PubMed; (b) Y.-L. Huang, W.-J. Tsai, C.-C. Shen and C.-C. Chen, J. Nat. Prod., 2005, 68, 217 CrossRef CAS PubMed.
- M. Ohyama, T. Tanaka, T. Ito, M. Iinuma, K. F. Bastow and K.-H. Lee, Bioorg. Med. Chem. Lett., 1999, 9, 3057 CrossRef CAS PubMed.
- E. Stempel and T. Gaich, Acc. Chem. Res., 2016, 49, 2390 CrossRef CAS PubMed.
- S. Mo, A. Krunic, G. Chlipala and J. Orjala, J. Nat. Prod., 2009, 72, 894 CrossRef CAS PubMed.
- A. R. Carroll, E. Hyde, J. Smith, R. J. Quinn, G. Guymer and P. I. Forster, J. Org. Chem., 2005, 70, 1096 CrossRef CAS PubMed.
- B.-Y. Liu, C. Zhang, K.-W. Zeng, J. Li, X.-Y. Guo, M.-B. Zhao, P.-F. Tu and Y. Jiang, Org. Lett., 2015, 17, 4380 CrossRef CAS PubMed.
- (a) M. P. Thomas and B. V. L. Potter, J. Med. Chem., 2015, 58, 7634 CrossRef CAS PubMed; (b) M. J. Reed, A. Purohit, L. W. L. Woo, S. P. Newman and B. V. L. Potter, Endocr. Rev., 2005, 26, 171 CrossRef CAS PubMed.
- A. D. Napper, J. Hixon, T. McDonagh, K. Keavey, J.-F. Pons, J. Barker, W. T. Yau, P. Amouzegh, A. Flegg and E. Hamelin, et al. , J. Med. Chem., 2005, 48, 8045 CrossRef CAS PubMed.
- (a) W.-B. Liu, N. Okamoto, E. J. Alexy, A. Y. Hong, K. Tran and B. M. Stoltz, J. Am. Chem. Soc., 2016, 138, 5234 CrossRef CAS PubMed; (b) M. Bos and E. Riguet, Chem. Commun., 2017, 53, 4997 RSC.
- (a) A. C. Cope and K. E. Hoyle, J. Am. Chem. Soc., 1941, 63, 733 CrossRef CAS; (b) R. B. Grossman and M. A. Varner, J. Org. Chem., 1997, 62, 5235 CrossRef CAS; (c) H. Nakamura, H. Iwama, M. Ito and Y. Yamamoto, J. Am. Chem. Soc., 1999, 121, 10850 CrossRef CAS; (d) S. R. Waetzig, D. K. Rayabharapu, J. D. Weaver and J. A. Tunge, Angew. Chem., Int. Ed., 2006, 45, 4977 CrossRef CAS PubMed; (e) P. Vertesaljai, P. V. Navaratne and A. J. Grenning, Angew. Chem., Int. Ed., 2016, 55, 317 CrossRef CAS PubMed.
- (a) T. B. Poulsen, C. Alemparte and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 11614 CrossRef CAS PubMed; (b) D. Xue, Y.-C. Chen, Q.-W. Wang, L.-F. Cun, J. Zhu and J.-G. Deng, Org. Lett., 2005, 7, 5293 CrossRef CAS PubMed; (c) T. B. Poulsen, M. Bell and K. A. Jørgensen, Org. Biomol. Chem., 2006, 4, 63 RSC.
- (a) A. C. Cope, K. E. Hoyle and D. Heyl, J. Am. Chem. Soc., 1941, 63, 1843 CrossRef CAS; (b) M. Hiersemann and T. Jaschinski, Selected Diastereoselective Reactions. Diastereoface-Differentiating Claisen, Cope, and [2,3]-Wittig Rearrangements in Contemporary Natural Product Synthesis, in Compr. Chirality, Elsevier B.V., 2012, vol. 2, p. 625 Search PubMed; (c) E. A. Ilardi, C. E. Stivala and A. Zakarian, Chem. Soc. Rev., 2009, 38, 3133 RSC.
- (a) S. Krueger and T. Gaich, Beilstein J. Org. Chem., 2014, 10, 163 CrossRef PubMed; (b) T. Hudlicky, R. Fan, J. W. Reed and K. G. Gadamasetti, Org. React., 1992, 41, 1 CAS.
- L. A. Paquette, Tetrahedron, 1997, 53, 13971 CrossRef CAS.
- We hypothesized that a Pd-catalyzed mechanism was also feasible. For examples of metal-catalyzed [3,3] rearrangements see: (a) L. E. Overman and F. M. Knoll, J. Am. Chem. Soc., 1980, 102, 865 CrossRef CAS; (b) R. J. Felix, D. Weber, O. Gutierrez, D. J. Tantillo and M. R. Gagné, Nat. Chem., 2012, 4, 405 CrossRef CAS PubMed.
- (a) A. S. Ivanov, Chem. Soc. Rev., 2008, 37, 789–811 RSC; (b) H. McNab, Chem. Soc. Rev., 1978, 7, 345–358 RSC.
- A. M. Dumas and E. Fillion, Acc. Chem. Res., 2010, 43, 440–454 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03057j |
| This journal is © The Royal Society of Chemistry 2018 |