
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of aryl-thioglycopeptides through chemoselective Pd-mediated conjugation†
David
Montoir
a,
Mehdi
Amoura
b,
Zine El Abidine
Ababsa
a,
T. M.
Vishwanatha
b,
Expédite
Yen-Pon
a,
Vincent
Robert
c,
Massimiliano
Beltramo
c,
Véronique
Piller
b,
Mouad
Alami
 a,
Vincent
Aucagne
*b and
Samir
Messaoudi
*a
a,
Vincent
Aucagne
*b and
Samir
Messaoudi
*a
aBioCIS, Univ. Paris-Sud, CNRS, Univ. Paris-Saclay, Châtenay-Malabry, France. E-mail: samir.messaoudi@u-psud.fr; Tel: +33 0146835887
bCentre de Biophysique Moléculaire, CNRS, Orléans, France. E-mail: aucagne@cnrs-orleans.fr; Tel: +33 0238255577
cUMR Physiologie de la Reproduction et des Comportements, INRA, CNRS, Univ. Tours, IFCE, Nouzilly, France
First published on 20th September 2018
Abstract
We describe herein a Pd-catalyzed methodology for the thioglycoconjugation of iodoaryl peptides and aminoacids. This operationally simple process occurs under semi-aqueous conditions and displays wide substrate scope. The strategy has been successfully applied to both the thioglycosylation of unprotected peptides and the generation of thioglyco-aminoacid building blocks, including those suitable for solid phase peptide synthesis. To demonstrate the broad potential of this technique for late stage functionalization, we successfully incorporated challenging unprotected β-S-GlcNAc- and α-S-GalNAc-derivatives into very long unprotected peptides. This study opens the way to new applications in chemical biology, considering the well-recognized advantages of S-glycosides over O-glycosides in terms of resistance towards both enzymatic and chemical degradation.
Thioglycosides are attractive substitutes for O-glycosides as they are resistant to enzymatic cleavage and less susceptible to chemical degradation.1 In the past three decades, there has been a significant growth in the synthesis of thioanalogues of naturally-occurring O-glycosides such as oligosaccharides2 and glycolipids.3 Most of the tested thioglycosides showed enhanced biological activities, presumably arising from their resistance to glycosidases combined with an isosteric relationship between thioether and ether bonds. In this context, S-linked glycopeptides have emerged as promising tools for the biological study of O-glycosylated peptides and proteins and are recognized as very good structural mimics.4,5 As proofs-of-concept, the thioglycoside analogue of the post-translational modification (PTM) O-β-D-N-acetylglucosamine (GlcNAc), attached to a cysteine residue instead of serine, was used to examine the effect of O-GlcNAcylation on casein kinase II6 and α-synuclein,4 through the total synthesis of the full length proteins. In similar ways, thioanalogues of the tumor-associated Tn antigen, D-N-acetylgalactosamine (GalNAc) α-O-linked to serine or threonine, have been synthesized for structural immunology studies,5 and thioanalogues of O-β-GlcNAc-containing antimicrobial peptide glycocin F showed improved bacteriostatic activities.7 Cysteine S-glycosylation has also been recently identified as a naturally occurring PTM in several bacteriocins7,8 and was lately found in mammalian proteins.9
From a synthetic chemistry point of view, glycosyl thiols have become useful building blocks for the synthesis of S-linked glyco-aminoacids, glycopeptides and glycoproteins, exploiting the exceptional nucleophilicity of the thiol group. Glycosyl thiols can react chemoselectively through conjugate addition,10 nucleophilic substitution of halogenides,11 ring opening of aziridines12 and cyclic sulfamidates,13 or disulfide formation followed by a desulfurative rearrangement.14 Conversely, reaction of the thiol group of cysteine with a carbohydrate derivative through nucleophilic substitution of glycosyl bromides,15 free radical thiol–ene coupling16 or Ferrier reaction17 has also been described. While these methods have provided robust access to S-linked glycopeptides, in most cases the sugar moiety is attached to the peptide through cysteine or sometimes homocysteine residues. Strikingly, to the best of our knowledge there is no report concerning S-linked glycopeptides analogues of O-glycosylated tyrosines,18 despite the relevance of such compounds for glycobiological investigations. Tyrosine O-glycosylation,19 is indeed a rather underexplored PTM, and O-GalNAc Tyr residues were identified only recently in human20 and in murine proteins.21 Although this modification is rare, it was found associated with pathogenic conditions for the amyloid precursor protein (APP) in Alzheimer's disease. Characterizing the O-glycosylation landscape of human plasma, platelets and endothelial cells also showed the presence of such O-GalNAc Tyr linkages in several proteins involved in hemostasis.22 Very recently, pathogenic bacteria toxins able to inactivate eukaryotic Rho GTPases by catalyzing the O-GlcNAcylation of a Tyr residue have also been characterized.23 These recent findings prompted us to develop synthetic routes to O-glycotyrosine thioanalogues, by either preparing glycosylaminoacid building blocks suitable for peptide synthesis, or using a post-synthetic protocol based on the glycoconjugation of unprotected peptide under aqueous conditions. Both approaches are based on the reaction of para-iodophenylalanine (p-IPhe) residues with glycosyl thiols using highly chemoselective Pd-catalysis conditions.
In our efforts to functionalize sugars under transition-metal catalysis,24 we recently reported an efficient protocol for the palladium-catalyzed coupling of aryl halides with various α- and β-glycosyl thiols.25 The C–S bond-forming reaction was achieved within minutes at room temperature by using G3-XantPhos palladacycle pre-catalyst26 (1 mol%), in the presence of Et3N in THF. In order to expand the synthetic utility of this transformation onto more complex systems such as peptides and proteins, we herein extend the scope of this reaction for (i) the preparation of glycoaminoacid building blocks (ii) the late stage functionalization of unprotected peptides under aqueous conditions and (iii) application to a wide variety of protected and unprotected glycosyl thiols. In particular we demonstrate for the first time the application of this chemistry to the reputedly challenging 2-deoxy-2-N-acetamido sugar α-thioGalNAc.
This work thus opens unprecedented opportunities for chemical glycobiology studies of tyrosine glycosylation through precise oxygen-to-sulfur substitution. Besides such possible application, we also showed the utility of this straightforward methodology for the synthesis of various complex neoglycoconjugates, a growing area of research where the saccharide moiety can be exploited to modulate the bioactivity or physicochemical properties of a given peptide or protein.27
To establish the appropriate conditions for the coupling reaction, tetra-O-acetylated 1-thio-β-D-glucopyranose 1a and N-Boc-DL-4-iodophenylalanine 2a were initially selected as model coupling substrates (for the optimization of the reaction conditions see ESI†). A full conversion and 96% yield of isolated product 3a was obtained using 3 mol% of G3-XantPhos catalyst.28 Taking into account that suitable conditions for peptides and proteins functionalization include aqueous media and ambient temperature, we performed the coupling of 1a with 2a in different aqueous media (see ESI† for details). Thus, conducting the coupling reaction in THF/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) afforded 3a in an excellent yield of 92% (see ESI†). Motivated by this water-tolerant coupling procedure, we next expanded the scope of this Pd-catalyzed coupling of structurally diverse α- and β-thiol derivatives of mono-, di- and tri-saccharides with various amino acids 2a–f (Table 1). All the coupling reactions proceeded in good yields and without epimerization at anomeric position.29 In addition to 1a, tetra-O-acetylated 1-thio-β-D-galactopyranose 1b, tri-O-acetylated N-Ac-1-thio-β-D-glucosaminopyranose 1c, tetra-O-benzoylated 1-thio-β-D-glucopyranose 1d and tetra-O-acetylated 1-thio-β-D-mannopyranose 1e were efficiently coupled with both N-Boc-DL-p-iodophenylalanine 2a and its methyl ester 2b to give the corresponding thioglycoconjugates 3a–p in good yields. A slightly lower yield was observed with 1c as nucleophilic partner to give thioglycosylated amino acids 3e and 3f in 72% and 59% yields, respectively. Moreover, the reaction is not limited to monosaccharides, but can be applied to more complex di- and trisaccharide derivatives. Thus, hepta-O-acetylated 1-thio-β-D-cellobiose 1f and deca-O-acetylated 1-thio-β-D-maltotriose 1g were reacted with 2a or 2b to give corresponding glycoconjugates 3j–l in good yields. Noteworthy the reaction works also well with unprotected sugars. As an example, the reaction of 2b with 1-thio-β-D-glucose 1h afforded product 3m in a satisfactory 63% yield and with a total retention of the β-anomeric configuration as confirmed from the distinct J1,2 coupling of 9.7 Hz. 1a and 1h were readily coupled with the L-enantiomer of Fmoc-p-iodo-phenylalanine 2d to furnish 3o and 3p in good yields. Interestingly, the latter compounds are two building blocks suitably protected for their use in Fmoc-based SPPS. In addition to para-iodophenylalanine derivatives, N-Boc-3-iodo-D-tyrosine 2e as well as N-Boc p-iodobenzyl L-cysteine 2f were used successfully in this coupling to furnish thioglycoconjugates 3q, r in 82% and 75% yields, respectively.
:2) afforded 3a in an excellent yield of 92% (see ESI†). Motivated by this water-tolerant coupling procedure, we next expanded the scope of this Pd-catalyzed coupling of structurally diverse α- and β-thiol derivatives of mono-, di- and tri-saccharides with various amino acids 2a–f (Table 1). All the coupling reactions proceeded in good yields and without epimerization at anomeric position.29 In addition to 1a, tetra-O-acetylated 1-thio-β-D-galactopyranose 1b, tri-O-acetylated N-Ac-1-thio-β-D-glucosaminopyranose 1c, tetra-O-benzoylated 1-thio-β-D-glucopyranose 1d and tetra-O-acetylated 1-thio-β-D-mannopyranose 1e were efficiently coupled with both N-Boc-DL-p-iodophenylalanine 2a and its methyl ester 2b to give the corresponding thioglycoconjugates 3a–p in good yields. A slightly lower yield was observed with 1c as nucleophilic partner to give thioglycosylated amino acids 3e and 3f in 72% and 59% yields, respectively. Moreover, the reaction is not limited to monosaccharides, but can be applied to more complex di- and trisaccharide derivatives. Thus, hepta-O-acetylated 1-thio-β-D-cellobiose 1f and deca-O-acetylated 1-thio-β-D-maltotriose 1g were reacted with 2a or 2b to give corresponding glycoconjugates 3j–l in good yields. Noteworthy the reaction works also well with unprotected sugars. As an example, the reaction of 2b with 1-thio-β-D-glucose 1h afforded product 3m in a satisfactory 63% yield and with a total retention of the β-anomeric configuration as confirmed from the distinct J1,2 coupling of 9.7 Hz. 1a and 1h were readily coupled with the L-enantiomer of Fmoc-p-iodo-phenylalanine 2d to furnish 3o and 3p in good yields. Interestingly, the latter compounds are two building blocks suitably protected for their use in Fmoc-based SPPS. In addition to para-iodophenylalanine derivatives, N-Boc-3-iodo-D-tyrosine 2e as well as N-Boc p-iodobenzyl L-cysteine 2f were used successfully in this coupling to furnish thioglycoconjugates 3q, r in 82% and 75% yields, respectively.

Having demonstrated the excellent catalytic activity of the G3-XantPhos pre-catalyst with aminoacids, we next turned our attention to the generalization of the method with respect to IPhe-containing N-Boc protected di- and tripeptides (Table 2). In all cases studied, the coupling reactions proceeded selectively and cleanly in high yields (glycoconjugates 5a–f). The established C–S bond formation protocol is compatible with the indole NH of tryptophan (5b), phenol of tyrosine (5c and 5f) and carboxylic acids (5e), indicating a large tolerance towards functional groups.

To open a complementary synthetic pathway towards larger thioglycopeptides, we next assessed the suitability of the synthesized building blocks in standard Fmoc-based SPPS protocols. For a proof-of-concept, we selected lipo-triazolopeptide 6a, a synthetic derivative of kisspeptin-10 (KP10), an endogenous neuropeptide playing a pivotal role in the central control of the reproductive system in mammals and non-bird vertebrates. KP10 acts through the activation of its cognate G-protein-coupled receptor, KiSS1R. KP10 has a very short half-life in blood (<1 min) and 6a was rationally designed with the aim to overcome this limitation, by including both a 1,4-disubstituted 1,2,3-triazole as a protease-resistant amide bond surrogate30 to improve metabolic stability and an N-palmitoylated isoglutamate albumin-binding motif to decrease renal excretion.316a shows a dramatically enhanced in vivo activity as compared with KP10, while also benefiting from an increased in vitro potency towards KiSS1R. We chose the Tyr1 residue (KP10 numbering), that as has been shown by Ala-scanning to be tolerant to modifications, to incorporate a model β-thioglucosyl analogue.32,33 Note that, as many lipopeptides, 6a shows very limited solubility under aqueous conditions, and is prone to hydrogel formation such as the parent KP10.34 Our choice of this peptide as a model compound was in part driven by our curiosity to see if glycoconjugation could improve its physicochemical properties.
The use of the building block 3p, instead of a standard SPPS Fmoc-Tyr(tBu)-OH furnished the expected thioglyco-lipo-triazolopeptide 6b in similar yields as for 6a (Fig. 1). This demonstrates the compatibility of our thioglycoaminoacid building block with standard Fmoc-SPPS protocols, including couplings, piperidine-mediated Fmoc deprotections and TFA-based cleavage. 6b was tested for its agonist activity towards KiSSR, and showed similar activity (EC50 = 0.03–0.3 nM) as compared with 6a (EC50 0.02–0.2 nM, mean and 95% confidence interval). Physicochemical properties of 6b were also briefly evaluated. However, no marked differences were observed as compared to 6a: the glycopeptide is not soluble in water at a concentration of 0.1 mM, and slowly forms a gel after solubilization in DMSO followed by dilution with water (0.1 mM final concentration, 95:5 water/DMSO).
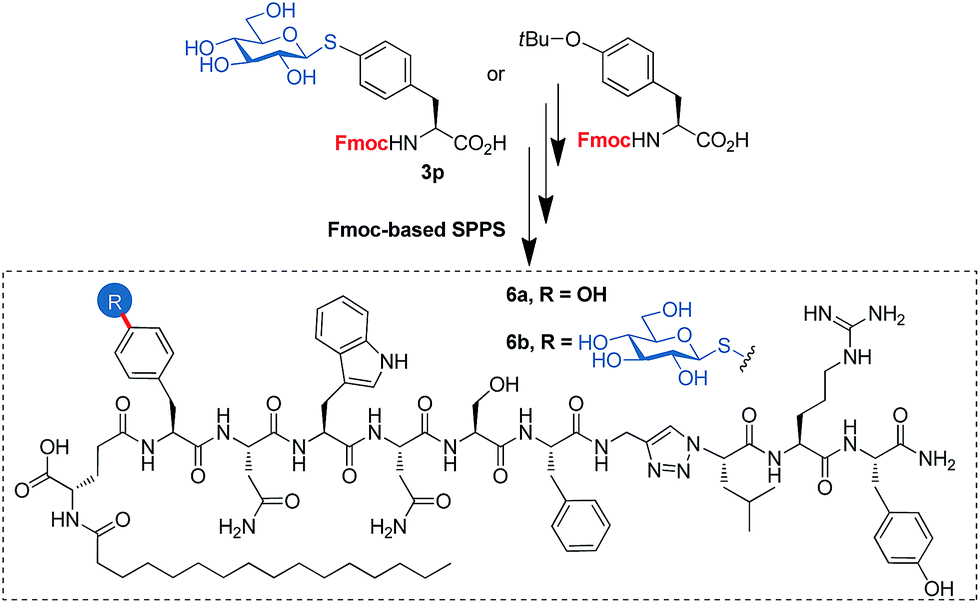 | ||
| Fig. 1 Lipo-triazolopeptide analogue 6a of the neuropeptide KP10, and its thioglucosylated derivative 6b. | ||
To assess the utility of our palladium-catalyzed procedure in the synthesis of very long thioglycopeptides through post-SPPS thioglycosylation, we next investigated the synthesis of analogues of the human mucin MUC1. MUC1 is a densely O-glycosylated glycoprotein produced as a membrane-anchored as well as a soluble form, both expressed by many epithelial cells. Their highly enhanced expression by many tumor cells types is accompanied by their aberrant glycosylation giving rise to largely reduced O-glycan chain complexity and density. Therefore, tumor-associated carbohydrate antigens such as Tn and its sialylated form (STn), are frequently found on epithelial tumor cells and are considered as clinically relevant tumor markers.35 The extracellular and soluble domains of MUC1 proteins contain a variable number of tandem repeat sequences of 20 amino acids (APDTRPAPGSTAPPAHGVTS),36 each presenting 5 potential O-glycosylation sites.37 Thus, Tn antigens on MUC1 act as a signature associated with cancer38 and represent promising targets for the development of synthetic antitumor vaccines.39 However, many attempts to produce MUC1-based anticancer-vaccines illustrated the very low immunogenicity of the natural MUC1 glycoprotein. It is thus necessary to generate analogues of MUC1 to improve the immune response against the native oncogenic glycoprotein. As an example, unnatural derivatives were recently synthesized by incorporation of a linker placed between the peptide and the α-GalNAc moiety, in order to favor the presentation of the sugar for molecular recognition events.40 The crystal structure of major histocompatibility complex-I (MHC-I) with such a linker-containing neoglycopeptide indeed showed that while the aglycone part of the antigen binds to the MHC, the carbohydrate moiety can facilitate the recognition of T-cell receptors (TCR) and therefore stimulate immune response.41 Here, we propose to produce new MUC1 mimics in which the GalNAc moieties would also be distant from the peptide chain and could thus present novel properties interesting for the production of cancer vaccines.
We first examined the coupling reactions between tri-O-acetylated β-thio-GlcNAc 1c, its unprotected congener 1i and α-thioGalNAc 1j with two fully-unprotected modified 60-amino-acid sequences of MUC1 (e.g. 3 tandem repeats) in which threonine residues of the immunogenic APDTR sequence were replaced by p-iodo-L-phenylalanine in position 24 (mono-iodo peptide 7a, Fig. 2) or in positions 4, 24 and 44 (tri-iodo peptide 7b).
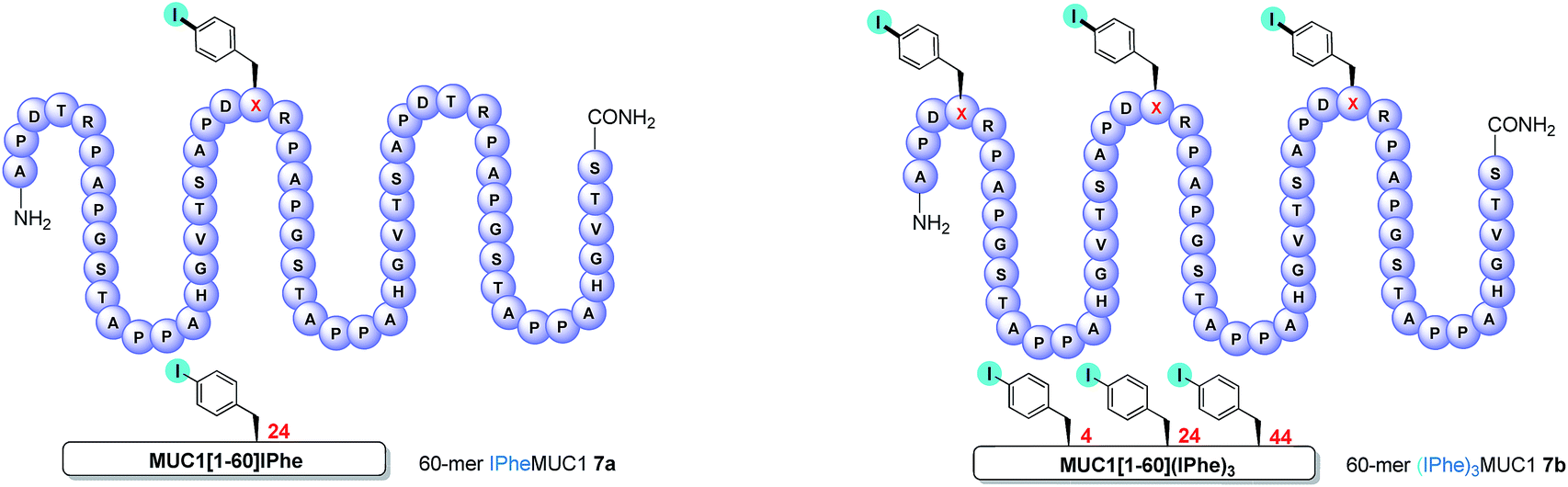 | ||
| Fig. 2 Schematic representations of MUC1-derived mono-iodo peptide 7a and tri-iodo peptide 7b. X = p-iodophenylalanine. | ||
First attempts were initiated to find optimal conditions for Pd-catalyzed coupling with mono-iodo peptide MUC1 7a (1 mM). O-Acetylated β-thioGlcNAc 1c was used as the model thiosugar. It was found that the quantity of G3-XantPhos reagent is crucial as the use of almost 5 equivalents was required to obtain a complete conversion of the starting material over 1 h. Under these conditions thioglycoconjugate 8a was isolated in an excellent 80% yield (Fig. 3). Notably, coupling of unprotected β-thioGlcNAc 1i with 7a under these conditions furnished exclusively 8b without any detectable peptide side products (Fig. 3). Finally, peptide 8c bearing α-thioGalNAc was obtained by coupling of the challenging and unstable α-GalNAc thiol 1j with 7a in a good yield. These selective thioglycosylations of MUC1-derived mono-iodopeptide 7a represent an important preliminary result in view of modifying more complex peptides and proteins. These promising results encouraged us to investigate this coupling reaction with the tri-iodo peptide 7b (Fig. 2). This coupling was effective in all cases to produce conjugates 8d–f in good yields.
 | ||
| Fig. 3 Pd-catalyzed thioglycoconjugation of MUC1-derived mono-iodo peptide 7a and tri-iodo peptide 7b with 1c, 1i and 1j. aIsolated yield. | ||
In order to characterize, from a biochemical point of view, the obtained thioglycopeptides we next verified if the thioGalNAc-containing 8c and 8f could be recognized by an O-GalNAc-specific lectin. For that purpose, we conjugated a biotin moiety at the N-terminus of each thioglycopeptide 8c and 8f as well as on iodopeptides 7a and 7b used as negative controls (see ESI† for details).42 The biotinylated MUC1 analogues were efficiently immobilized on NeutrAvidin-coated plates in a quantitative and saturable manner, as judged through recognition by specific anti MUC1 antibodies (see ESI† for details).
We then checked if the Vicia villosa plant lectin (VVL) specific for α- or β-linked terminal O-GalNAc could still recognize the GalNAc-thioglycopeptides. The results of our ELISA assays, presented in Fig. 4, show efficient lectin binding since the apparent Kd was 26.0 ± 7.3 nM for the mono-GalNAc thioglycopeptide (biotinylated 8c) and 27.2 ± 12.1 nM for the tri-GalNAc thioglycopeptide (biotinylated 8f). Such values are in very good agreement with those recently obtained on O-GalNAc-Tyr containing glycopeptides43 with apparent Kd values ranging from 1.4 ± 0.1 to 243 ± 113 nM for neoglycoproteins showing different GalNAc densities. In our work the two thioglycopeptides tested gave roughly similar apparent Kd values indicating no effect on the lectin binding of the GalNAc density (1 vs. 3 GalNAc per peptide). These VVL binding results suggest the usefulness of our simple synthetic strategy in order to produce thioglycopeptides which could be used as mimics of Tyr-GalNAc containing peptides.
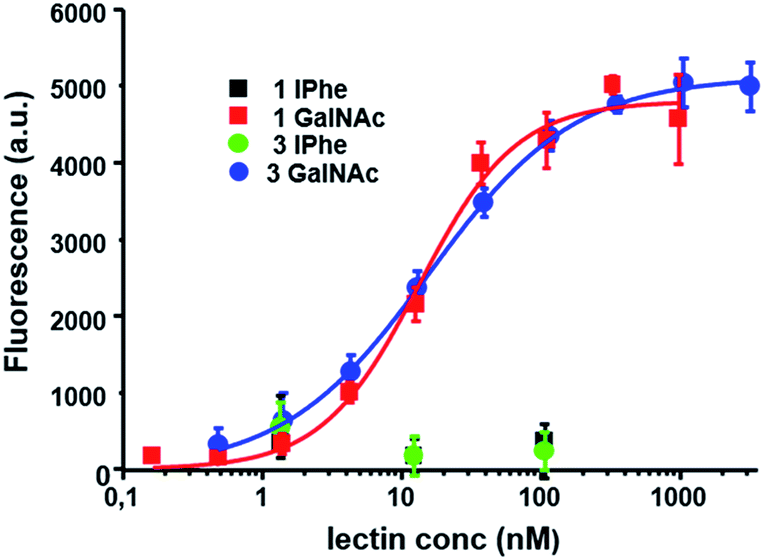 | ||
| Fig. 4 ELISA-based assays showing FITC-labelled VVL binding of the different peptides and glycopeptides tested. The fluorescence values are the means of three independent experiments realized at the same peptide concentration and error bars indicate the standard deviation. Apparent Kd values were determined from assays at different peptide concentrations (see ESI† for details). | ||
Conclusions
In conclusion, we have developed a highly chemoselective and efficient method to access a wide range of complex thioglycopeptides using the powerful palladacycle pre-catalyst G3-XantPhos. This protocol provides a straightforward method to access thioglyco-aminoacid building blocks and for the late-stage thioglyco functionalization of unprotected peptides. An iodophenyl “tag” positioned using the non-natural amino acid p-IPhe allows post-synthetic site-selective thioglycoconjugation of a MUC1-derived peptide using β-S-GlcNAc and α-S-GalNAc thiosugars. The reaction is compatible with nearly all natural amino acids, which are inert under these conditions (at the exclusion of free thiol cysteine). Since several methods have been developed for the incorporation of p-iodophenylalanine residues in recombinant proteins,44 these results also pave the way to more complex and larger thioglycoproteins, through semi-synthesis approaches. We expect this simple and general protocol to be of broad utility for the development of new tools to explore and modulate biological systems, in particular for the study of tyrosine O-glycosylation, a recently emerging field of investigation. Interestingly, while we were preparing this manuscript, a very complementary methodology was reported for chemoselective tyrosine O-glycosylation through Ca(OH)2 mediated coupling of unprotected peptides with glycosyls fluorides.45Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge support of this project by ANR (ANR-15-CE29-0002 CarNuCat, ANR-15-CE07-0022 Easyminiprot and ANR-15-CE20-0015 KISS projects), Région Centre-Val-de-Loire (Capriss projet), CNRS, University Paris Sud and by la Ligue Contre le Cancer through an Equipe Labellisée 2014 grant. BioCIS laboratory is a member of the Laboratory of Excellence LERMIT supported by a grant (ANR-10-LABX-33).Notes and references
- For reviews on thioglycosides, see: (a) H. Driguez, Top. Curr. Chem., 1997, 187, 85–116 CrossRef CAS; (b) H. Driguez, ChemBioChem, 2001, 2, 311–318 CrossRef CAS PubMed; (c) K. Pachamuthu and R. R. Schmidt, Chem. Rev., 2006, 106, 160–187 CrossRef CAS PubMed; (d) G. Lian, X. Zhang and B. Yu, Carbohydr. Res., 2015, 403, 13–22 CrossRef CAS PubMed.
- For representative examples, see: (a) B. Aguilera and A. Fernández-Mayoralas, J. Org. Chem., 1998, 63, 2719–2723 CrossRef CAS PubMed; (b) M. Sandgren, G. I. Berglund, A. Shaw, J. Stahlberg, L. Kenne, T. Desmet and C. Mitchinson, J. Mol. Biol., 2004, 342, 1505–1517 CrossRef CAS PubMed; (c) J. Buckingham, J. A. Brazier, J. Fisher and R. Cosstick, Carbohydr. Res., 2007, 342, 16–22 CrossRef CAS PubMed; (d) K. Young-Wan, A. L. Lovering, C. Hongming, T. Kantner, L. P. McIntosh, N. C. J. Strynadka and S. G. Withers, J. Am. Chem. Soc., 2006, 128, 2202–2203 CrossRef PubMed , as well as ref. 1c.
- For representative examples, see: (a) R. T. Dere and X. Zhu, Org. Lett., 2008, 10, 4641–4644 CrossRef CAS PubMed; (b) C. O'Reilly and P. V. Murphy, Org. Lett., 2011, 13, 5168–5171 CrossRef PubMed; (c) X. Zeng, R. Smith and X. Zhu, J. Org. Chem., 2013, 78, 4165–4170 CrossRef CAS PubMed.
- C. A. De Leon, P. M. Levine, T. W. Craven and M. R. Pratt, Biochemistry, 2017, 56, 3507–3517 CrossRef CAS PubMed.
- N. Martínez-Sáez, J. Castro-López, J. Valero-González, D. Madariaga, I. Compañón, V. J. Somovilla, M. Salvadó, J. L. Asensio, J. Jiménez-Barbero, A. Avenoza, J. H. Busto, G. J. L. Bernardes, J. M. Peregrina, R. Hurtado-Guerrero and F. Corzana, Angew. Chem., Int. Ed., 2015, 54, 9830–9834 CrossRef PubMed.
- M. K. Tarrant, H.-S. Rho, Z. Xie, Y. L. Jiang, C. Gross, J. C. Culhane, G. Yan, J. Qian, Y. Ichikawa, T. Matsuoka, N. Zachara, F. A. Etzkorn, G. W. Hart, J. S. Jeong, S. Blackshaw, H. Zhu and P. A. Cole, Nat. Chem. Biol., 2012, 8, 262–269 CrossRef CAS PubMed.
- Z. Amso, S. W. Bisset, S.-H. Yang, P. W. R. Harris, T. H. Wright, C. D. Navo, M. L. Patchett, G. E. Norris and M. A. Brimble, Chem. Sci., 2018, 9, 1686–1691 RSC.
- G. E. Norris and M. L. Patchett, Curr. Opin. Struct. Biol., 2016, 40, 112–119 CrossRef CAS PubMed.
- J. C. Maynard, A. L. Burlingame and K. F. Medzihradszky, Mol. Cell. Proteomics, 2016, 15, 3405–3411 CrossRef CAS PubMed.
- (a) M. R. Levengood and W. A. van der Donk, Nat. Protoc., 2006, 1, 3001–3010 CrossRef CAS PubMed; (b) D. P. Galonić, W. A. van der Donk and D. Y. Gin, Chem.–Eur. J., 2003, 9, 5997–6006 CrossRef PubMed.
- (a) D. A. Thayer, H. N. Yu, M. C. Galan and C. H. Wong, Angew. Chem., Int. Ed., 2005, 44, 4596–4599 CrossRef CAS PubMed; (b) X. M. Zhu and R. R. Schmidt, Chem.–Eur. J., 2004, 10, 875–887 CrossRef CAS PubMed; (c) S. Knapp and D. S. Myers, J. Org. Chem., 2002, 67, 2995–2999 CrossRef CAS PubMed; (d) D. P. Galonić, W. A. van der Donk and D. Y. Gin, J. Am. Chem. Soc., 2004, 126, 12712–12713 CrossRef PubMed.
- S. B. Cohen and R. L. Halcomb, Org. Lett., 2001, 3, 405–407 CrossRef CAS PubMed.
- G. J. L. Bernardes, E. J. Grayson, S. Thompson, J. M. Chalker, J. C. Errey, F. El Oualid, T. D. W. Claridge and B. G. Davis, Angew. Chem., Int. Ed., 2008, 47, 2244–2247 CrossRef CAS PubMed.
- D. Crich and F. Yang, J. Org. Chem., 2008, 73, 7017–7027 CrossRef CAS PubMed.
- E. Calce, G. Digilio, V. Menchise, M. Saviano and S. De Luca, Chem.–Eur. J., 2018, 24, 6231–6238 CrossRef CAS PubMed.
- A. Dondoni, A. Massi, P. Nanni and A. Roda, Chem.–Eur. J., 2009, 15, 11444–11449 CrossRef CAS PubMed.
- (a) D. Ellis, S. E. Norman and H. M. I. Osborn, Tetrahedron, 2008, 64, 2832–2854 CrossRef CAS; (b) L. Lázár, M. Csávás, M. Herczeg, P. Herczegh and A. Borbás, Org. Lett., 2012, 14, 4650–4653 CrossRef PubMed.
- Besides thioanalogues of glycosylated tyrosines, only one example of peptide arylthioglycoside has been reported to our knowledge: A. Novoa, S. Barluenga, C. Serba and N. Winssinger, Chem. Commun., 2013, 49, 7608–7610 RSC.
- P. Lafite and R. Daniellou, Nat. Prod. Rep., 2012, 29, 729–738 RSC.
- (a) A. Halim, G. Brinkmalm, U. Rüetschi, A. Westman-Brinkmalm, E. Portelius, H. Zetterberg, K. Blennow, G. Larson and J. Nilsson, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11848–11853 CrossRef CAS PubMed; (b) S. Y. Vakhrushev, C. Steentoft, M. B. Vester-Christensen, E. P. Bennett, H. Clausen and S. B. Levery, Mol. Cell. Proteomics, 2013, 12, 932–944 CrossRef CAS PubMed.
- J. C. Trinidad, R. Schoepfer, A. L. Burlingame and K. F. Medzihradszky, Mol. Cell. Proteomics, 2013, 12, 3474–3488 CrossRef CAS PubMed.
- S. L. King, H. J. Joshi, K. T. Schjoldager, A. Halim, T. D. Madsen, M. H. Dziegiel, A. Woetmann, S. Y. Vakhrushev and H. H. Wandall, Blood Adv., 2017, 1, 429–442 CrossRef CAS PubMed.
- (a) T. Jank, X. Bogdanović, C. Wirth, E. Haaf, M. Spoerner, K. E. Böhmer, M. Steinemann, J. H. Orth, H. R. Kalbitzer, B. Warscheid, C. Hunte and K. Aktories, Nat. Struct. Mol. Biol., 2013, 20, 1273–1280 CrossRef CAS PubMed; (b) T. Jank, S. Eckerle, M. Steinemann, C. Trillhaase, M. Schimpl, S. Wiese, D. M. van Aalten, W. Driever and K. Aktories, Nat. Commun., 2015, 6, 7807 CrossRef CAS PubMed.
- (a) A. Bruneau, J.-D. Brion, M. Alami and S. Messaoudi, Chem. Commun., 2013, 49, 8359–8361 RSC; (b) E. Brachet, J.-D. Brion, S. Messaoudi and M. Alami, Adv. Synth. Catal., 2013, 355, 477–490 CrossRef CAS; (c) E. Brachet, J.-D. Brion, M. Alami and S. Messaoudi, Adv. Synth. Catal., 2013, 355, 2627–2636 CrossRef CAS; (d) E. Brachet, J.-D. Brion, M. Alami and S. Messaoudi, Chem.–Eur. J., 2013, 19, 15276–15280 CrossRef CAS PubMed; (e) R. A. A. Al-Shuaeeb, G. Galvani, G. Bernadat, J.-D. Brion, M. Alami and S. Messaoudi, Org. Biomol. Chem., 2015, 13, 10904–10916 RSC; (f) A. Chabrier, A. Bruneau, S. Benmahdjoub, B. Benmerad, S. Belaid, J.-D. Brion, M. Alami and S. Messaoudi, Chem.–Eur. J., 2016, 22, 15006–15010 CrossRef CAS PubMed; (g) T. T. H. Luong, J.-D. Brion, E. Lescop, M. Alami and S. Messaoudi, Org. Lett., 2016, 18, 2126–2129 CrossRef CAS PubMed; (h) R. A. A. AL-Shuaeeb, D. Montoir, M. Alami and S. Messaoudi, J. Org. Chem., 2017, 82, 6720–6728 CrossRef CAS PubMed.
- A. Bruneau, M. Roche, A. Hamze, J.-D. Brion, M. Alami and S. Messaoudi, Chem.–Eur. J., 2015, 21, 8375–8379 CrossRef CAS PubMed.
- Fort the preparation of the precatalyst, see: (a) N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916–920 RSC, for a review related to the use of Pd-G3 precatalysts, see: (b) A. Bruneau, M. Roche, M. Alami and S. Messaoudi, ACS Catal., 2015, 5, 1386–1396 CrossRef CAS.
- S. V. Moradi, W. M. Hussein, P. Varamini, P. Simerska and I. Toth, Chem. Sci., 2016, 7, 2492–2500 RSC.
- The coupling of 1a and 2a was also performed in the presence of different other Pd-based catalysts such as Pd(OAc)/SPhos, Pd(OAc)/XPhos and Pd(OAc)2/disodium 2-aminopyrimidine-4,6-diol. However, none of the catalysts did produce the desired product 3a even at high loading (see ESI,† P5).
- In all cases herein reported, when the peak corresponding to the anomeric proton could be clearly identified on the 1H NMR spectrum, it showed characteristic J1,2 coupling constants indicating the retention of the expected anomeric configuration in the glycosylated products.
- (a) I. Valverde, F. Lecaille, G. Lalmanach, V. Aucagne and A. F. Delmas, Angew. Chem., Int. Ed., 2012, 51, 718–722 CrossRef CAS PubMed; (b) V. Aucagne, I. E. Valverde, P. Marceau, N. Dendane, M. Galibert and A. F. Delmas, Angew. Chem., Int. Ed., 2012, 51, 11320–11324 CrossRef CAS PubMed; (c) M. Beltramo, V. Robert, M. Galibert, J.-B. Madinier, P. Marceau, H. Dardente, N. De Roux, D. Lomet, A. F. Delmas, A. Caraty and V. Aucagne, J. Med. Chem., 2015, 58, 3459–3470 CrossRef CAS PubMed; (d) M. Galibert, V. Piller, F. Piller, V. Aucagne and A. F. Delmas, Chem. Sci., 2015, 6, 3617–3623 RSC; (e) M. Galibert, M. Wartenberg, F. Lecaille, A. Saidi, S. Mavel, A. Joulin-Giet, B. Korkmaz, D. Brömme, V. Aucagne, A. F. Delmas and G. Lalmanach, Eur. J. Med. Chem., 2018, 143, 201–210 CrossRef PubMed.
- C. Decourt, V. Robert, K. Anger, M. Galibert, J.-B. Madinier, H. Dardente, D. Lomet, A. F. Delmas, A. Caraty, A. Herbison, X. Liu, G. Anderson, V. Aucagne and M. Beltramo, Sci. Rep., 2016, 6, 26908 CrossRef CAS PubMed.
- (a) M. J. Orsini, M. A. Klein, M. P. Beavers, P. J. Connolly, S. A. Middleton and K. H. Mayo, J. Med. Chem., 2007, 50, 462–471 CrossRef CAS PubMed; (b) A. E. Curtis, J. H. Cooke, J. E. Baxter, J. R. C. Parkinson, A. Bataveljic, M. A. Ghatei, S. R. Bloom and K. G. Murphy, Am. J. Physiol.: Endocrinol. Metab., 2010, 298, E296–E303 CrossRef CAS PubMed.
- Proteomic studies showed that the precursor of KP10, KISS1, can be post-translationally modified by phosphorylation at this Tyr residue. See: L. M. Brill, A. R. Salomon, S. B. Ficarro, M. Mukherji, M. Stettler-Gill and E. C. Peters, Anal. Chem., 2004, 76, 2763–2772 CrossRef CAS PubMed.
- N. Nishizawa, Y. Takatsu, K. Sumano, A. Kiba, J. Ban, S. Tsutsumi, H. Matsui, S. I. Matsumoto, M. Yamaguchi, Y. Ikeda, M. Kusaka, T. Ohtaki, F. Itoh and T. Asami, J. Med. Chem., 2016, 59, 8804–8811 CrossRef CAS PubMed.
- G. F. Springer, J. Mol. Med., 1997, 75, 594–602 CrossRef CAS PubMed.
- S. J. Gendler, C. A. Lancaster, J. Taylor-Papadimitriou, T. Duhig, N. Peat, J. Burchell, L. Pemberton, E. N. Lalani and D. Wilson, J. Biol. Chem., 1990, 265, 15286–15293 CAS.
- C. Sihlbom, I. van Dijk Härd, M. E. Lidell, T. Noll, G. C. Hansson and M. Bäckström, Glycobiology, 2009, 19, 375–381 CrossRef CAS PubMed.
- (a) V. Lakshminarayanan, P. Thompson, M. A. Wolfert, T. Buskas, J. M. Bradley, L. B. Pathangey, C. S. Madsen, P. A. Cohen, S. J. Gendler and G.-J. Boons, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 261–266 CrossRef CAS PubMed; (b) M. K. Hossain and K. A. Wall, Vaccines, 2016, 4, 25 CrossRef PubMed.
- (a) V. Lakshminarayanan, P. Thompson, M. A. Wolfert, T. Buskas, J. M. Bradley, L. B. Pathangey, C. S. Madsen, P. A. Cohen, S. J. Gendler and G. J. Boons, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 261–266 CrossRef CAS PubMed; (b) K. Yamamoto, T. Ueno, T. Kawaoka, S. Hazama, M. Fukui, Y. Suehiro, Y. Hamanaka, Y. Ikematsu, K. Imai, M. Oka and Y. Hinoda, Anticancer Res., 2005, 25, 3575–3579 CAS.
- V. Rojas-Ocáriz, I. Compañón, C. Aydillo, J. Castro-Lopez, J. Jiménez-Barbero, R. Hurtado-Guerrero, A. Avenoza, M. M. Zurbano, J. M. Peregrina, J. H. Busto and F. Corzana, J. Org. Chem., 2016, 81, 5929–5941 CrossRef PubMed.
- J. A. Speir, U. M. Abdel-Motal, M. Jondal and I. A. Wilson, Immunity, 1999, 10, 51–61 CrossRef CAS PubMed.
- N-Terminal biotinylation of 7a by reaction with biotin sulfoNHS ester under standard conditions led to a mixture of the expected mono-biotinylated together with non- and multi-biotinylated species, despite the absence of Lys residues in the sequence. We attribute these results to the formation of Ser/Thr esters, and optimized conditions for 7a, 7b, 8c and 8f to avoid such over-biotinylation, see ESI† for details.
- R. Gibadulli, D. W. Farnswort, J. J. Barchi and J. C. Gildersleeve, ACS Chem. Biol., 2017, 12, 2172–2182 CrossRef PubMed.
- (a) M. Ibba, P. Kast and H. Hennecke, Biochemistry, 1994, 33, 7107–7112 CrossRef CAS PubMed; (b) P. Kast and H. Hennecke, J. Mol. Biol., 1991, 222, 99–124 CrossRef CAS PubMed; (c) D. Kiga, K. Sakamoto, K. Kodama, T. Kigawa, T. Matsuda, T. Yabuki, M. Shirouzu, Y. Harada, H. Nakayama, K. Takio, Y. Hasegawa, Y. Endo, I. Hirao and S. Yokoyama, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9715–9723 CrossRef CAS PubMed; (d) K. Kirshenbaum, I. S. Carrico and D. A. Tirrell, ChemBioChem, 2002, 3, 235–237 CrossRef PubMed; (e) J. W. Chin, T. A. Cropp, J. C. Anderson, M. Mukherji, Z. Zhang and P. G. Schultz, Science, 2003, 301, 964–967 CrossRef CAS PubMed; (f) L. Wang, J. Xie, A. A. Deniz and P. G. Schultz, J. Org. Chem., 2003, 68, 174–176 CrossRef CAS PubMed; (g) K. Kodama, S. Fukuzawa, H. Nakayama, T. Kigawa, K. Sakamoto, T. Yabuki, N. Matsuda, M. Shirouzu, K. Takio, K. Tachibana and S. Yokoyama, ChemBioChem, 2006, 7, 134–139 CrossRef CAS PubMed.
- T. J. Wadzinski, A. Steinauer, L. Hie, G. Pelletier, A. Schepartz and S. J. Miller, Nat. Chem., 2018, 10, 644–652 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General, experimental procedures, and characterization of all new compounds. See DOI: 10.1039/c8sc02370k |
| This journal is © The Royal Society of Chemistry 2018 |