
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
[P4H]+[Al(OTeF5)4]−: protonation of white phosphorus with the Brønsted superacid H[Al(OTeF5)4](solv)†‡
Anja
Wiesner
 ,
Simon
Steinhauer
,
Helmut
Beckers
,
Christian
Müller
* and
Sebastian
Riedel
*
,
Simon
Steinhauer
,
Helmut
Beckers
,
Christian
Müller
* and
Sebastian
Riedel
*
Institut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstr. 34/36, 14195 Berlin, Germany. E-mail: c.mueller@fu-berlin.de; s.riedel@fu-berlin.de
First published on 23rd August 2018
Abstract
A sustainable transformation of white phosphorus (P4) into chemicals of higher value is one of the key aspects in modern phosphorus research. Even though the chemistry of P4 has been investigated for many decades, its chemical reactivity towards the simplest electrophile, the proton, is still virtually unknown. Based on quantum-chemical predictions, we report for the first time the successful protonation of P4 by the Brønsted acid H[Al(OTeF5)4](solv). Our spectroscopic results are in agreement with acid-mediated activation of P4 under protonation of an edge of the P4-tetrahedron and formation of a three-center two-electron P–H–P bond. These investigations are of fundamental interest as they permit the activation of P4 with the simplest electrophile as a new prototype reaction for this molecule.
Introduction
White phosphorus (P4), discovered by Henning Brand in 1669 while searching for the philosopher's stone, is the thermodynamically least stable and most reactive form of phosphorus at room temperature and consists of tetrahedral P4 molecules. Despite its spontaneous flammability and severe toxicity, P4 is the easiest form to produce on an industrial scale and is therefore the commercially most important allotrope.1 Especially its conversion to PCl3 is of high interest, as it is a base chemical for the production of many organophosphorus compounds.From a historical point of view, two important chemical reactions of P4 are described in every good textbook of inorganic chemistry:2 (a) the slow oxidation of P4 vapor to P4O10 under emission of light. This chemoluminescence has coined the name phosphorus, which is derived from the greek mythology (“light-bearer”). (b) the activation and disproportionation of P4 by aqueous solutions of alkali metal hydroxides. In this way, the industrially relevant phosphine gas (PH3) is obtained in high purity next to the alkali metal salt of hypophosphorous acid (NaH2PO2). More recent studies deal with the degradation of white phosphorus in the presence of other strong nucleophiles, such as organolithium and organomagnesium compounds, carbenes or silylenes, under the topic “P4-activation and functionalization”.3–6 From a mechanistic point of view, a charged nucleophile (Nu−) interacts with one of the three energetically degenerate LUMOs of the P4 molecule (Fig. 1a) under opening of the P4 tetrahedron to yield a substituted butterfly-like bicyclo[1,1,0]tetraphospha-butane anion (Fig. 1b, I).7
 | ||
| Fig. 1 Molecular orbital scheme of P4 (a)8 and examples of nucleophilic and electrophilic attacks at P4 and the assumed structures for protonated P4 (b). | ||
Electrophiles, on the other hand, should react at an edge of the tetrahedron, as the two energetically degenerate highest occupied molecular orbitals (HOMO and HOMO−1) have large coefficients at two adjacent phosphorus atoms (Fig. 1a). The situation is, however, much more complicated and the formation of various products is usually observed in this seemingly simple reaction. In the case of Ph2P+ and NO+, the insertion of these small molecules into one of the P–P bonds is indeed observed, as also theoretically predicted for NO+ (Fig. 1b, II).9–11 In the case of Ag+ as an example of an electrophilic transition metal center, a weak coordination of Ag+ to the edge of the P4 tetrahedron occurs (Fig. 1b, III).12 However, depending on the steric demand of the metal fragment, the coordination of P4via the apex can be enforced, even though the interaction of the energetically low-lying HOMO−5 with the electrophile is necessary to achieve this coordination mode (Fig. 1b, IV).13
In contrast to the experimental observations made for the reaction of P4 with nucleophiles as well as coordinatively and electronically unsaturated transition metal complexes, experimental proof for the structure of the elusive [P4H]+ cation in solution is still missing in the literature. In fact, weak acids do not react with P4 due to the rather poor nucleophilicity and weak basicity of white phosphorus. Common strong acids, such as sulfuric acid (H2SO4) and nitric acid (HNO3) cannot be used for the generation of [P4H]+ as they directly oxidize P4 to either phosphorous acid (H3PO3) and sulfur dioxide (SO2), or to phosphoric acid (H3PO4), nitrogen oxide (NO2) and water (H2O), respectively. Hydrogen chloride (HCl) can react with P4 to form phosphine gas (PH3) and phosphorus trichloride (PCl3). Based on ab initio calculations, Fluck et al.14 predicted in 1979 that the weakly bound proton in [P4H]+ is located at the apex of the tetrahedron (Fig. 1b, V), while protonation at the edge was predicted to be energetically less favored (Fig. 1b, VI). The authors exclude protonation at the P3-face. More recent ab initio molecular orbital calculations at the MP2/6-31G(d,p) level of theory in 1996 by Abboud, Yáñez and co-workers reveal, however, that the thermodynamically most favourable process is the protonation at the edge under formation of a three-center two-electron (3c-2e) P–H–P bond (Fig. 1b, VI).15 The same group determined the gas-phase basicity of P4 by means of Fourier transform ion cyclotron resonance mass spectrometry. In 2000, Ponec and co-workers provided an additional theoretical support for the existence of a non-classical 3c-2e P–H–P bond in [P4H]+ using the generalized population analysis.16 More recently, Lobayan and Bochicchio used a topological analysis of the electron density to describe the 3c-2e P–H–P bond in [P4H]+.17
Results
Taking the above-mentioned considerations into account, we anticipated that strong acids of conjugated weakly coordinating and non-reactive anions should be excellent reagents for the protonation of P4. Reed and Nixon, for instance, have shown that phosphabenzenes can be protonated by the in situ generated Brønsted superacid H(CHB11Me5Br6).18 Also these phosphorus heterocycles are known for their extremely weak basicity. As one of us19 has recently reported a novel aluminum-based superacidic system containing the weakly coordinating anion [Al(OTeF5)4]−, we report here now the synthesis and the first spectroscopic proof on the structure of [P4H]+ in solution.According to quantum-chemical calculations at the B3LYP/def2-TZVPP level, the protonation of P4 can be achieved by a medium consisting of the Brønsted superacid H[Al(OTeF5)4](solv) and ortho-difluorobenzene (o-DFB), see eqn (1).19 This is due to a slightly lower proton affinity of o-DFB (741.6 kJ mol−1) compared to P4 (748.4 kJ mol−1), as computed at CCSD(T)/aug-cc-pVTZ level of theory.
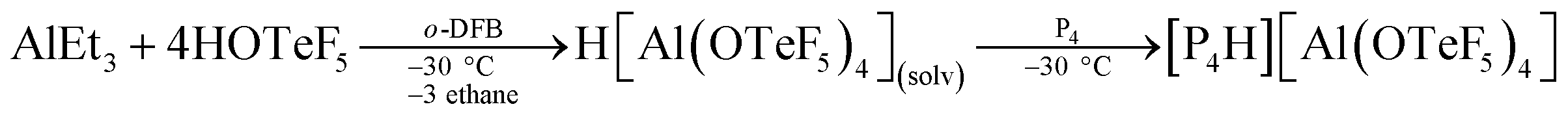 | (1) |
The reaction product of P4 and the Brønsted superacid was obtained as a temperature-, moisture- and oxygen-sensitive salt. It shows a clean low-temperature proton-coupled 31P NMR spectrum with two equally intense signals at δ = −481.7 and δ = −405.8 ppm with a weak roof effect (Fig. 2a). No other signals were observed in the 31P NMR spectrum in the region between δ = 400 ppm and δ = −800 ppm.
 | ||
| Fig. 2 (a) Low-temperature (T = −40 °C) experimental 31P NMR spectra of [P4H][Al(OTeF5)4] in o-DFB (external lock: [D6]acetone). The atom labelling is indicated in accordance with the AX2Y2 spin system. (b) 31P NMR spectrum (162 MHz, top) and simulated PX and PY signals (bottom) of the species [P4H]+(edge). (c) Experimental and simulated 1H NMR spectrum (401 MHz). The full spectra are provided in Fig. S1 and S2.‡ (d) Simulated 31P NMR PX and PY signals of the species [P4H]+(apex). | ||
This spectrum, which also reveals an additional splitting owing to the higher order of the system, is in accordance with an AX2Y2 spin system (1J(31PX, 31PY) = 233.95 Hz, 1J(1HA, 31PY) = 36.70 Hz, 2J(31PX, 1HA) = 4.91 Hz). This can only result from the protonation of the P4 molecule at the P–P-edge. Furthermore, a triplet of triplets at δ = −5.35 ppm appears in the 1H NMR spectrum (Fig. 2c) showing the corresponding couplings of the proton to PX and PY, respectively. Interestingly, both the chemical shifts and coupling constants are in excellent agreement with the simulated spectra of [P4H]+(edge), obtained by quantum-chemical calculations (Fig. 2b and c and S3, Table S1‡). For comparison reasons, Fig. 2d shows the simulated 31P NMR spectrum of the species [P4H]+(apex), which clearly differs from the experimental results. The NMR studies clearly prove the presence of [P4H][Al(OTeF5)4] and that P4 is protonated at an edge of the tetrahedron as predicted by quantum-chemical calculations.15–17
We further started to investigate the dynamics of the cation in solution. Interestingly, variable temperature NMR spectroscopy indicates a coalescence of the signals at T = −10 °C (Fig. 3).
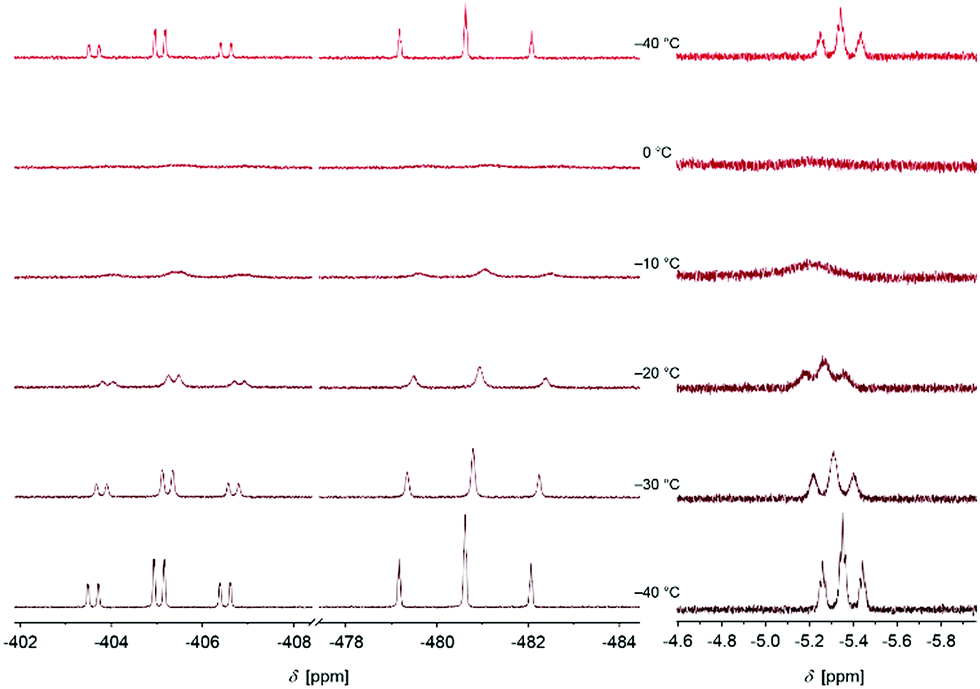 | ||
| Fig. 3 Excerpt of the 31P and 1H NMR spectra of [P4H][Al(OTeF5)4] in o-DFB (external lock: [D6]acetone) at various temperatures. The sample was first measured at T = −40 °C, annealed stepwise to T = 0 °C and cooled down again to T = −40 °C afterwards. | ||
The triplet of triplets observed at T = −40 °C in the 1H NMR spectrum broadens with increasing temperature resulting in a broad singlet (approx. FWHM = 75 Hz) at the coalescence temperature. The chemical shift slightly changes from δ = −5.35 ppm at T = −40 °C to δ = −5.19 ppm at T = 0 °C. In the 31P NMR spectrum, a similar process is observed. The two signals are broadened at the coalescence temperature and shifted to a higher field by 0.5 ppm for PX and PY at T = 0 °C. This process is reversible by re-cooling the sample to T = −40 °C again. No signal for P4 is detected during this process. Based on these observations, we anticipate a dynamic intramolecular migration of the proton on the P4 surface. From the experimental dynamic-NMR data, the corresponding barrier can be estimated to ΔG‡ = 54.2 kJ mol−1.
We noticed, that if an excess of P4 is present in the reaction mixture, the previously described [P9]+ cation20 is formed next to [P4H]+. Upon warming a sample containing a mixture of [P4H]+ and P4 from T = −40 °C to T = −10 °C, a fast and full conversion to [P9]+ is observed, as detected by NMR spectroscopy. This observation indicates that activation of the P4 molecule by protonation already occurs at low temperature, while broad band UV/Vis irradiation is necessary to form [P9]+ from a P4/[P4NO]+ mixture, as reported in the literature before.10
The [P4H]+ cation was further analyzed by means of mass spectrometry. In the mass spectrum (positive mode), a signal allocated to [P4H]+ appears at m/z = 124.9. In addition, signals due to [P4]+ (m/z = 123.8) as well as of [nTe]+ and [nTeH]+ in a natural isotope distribution (n = 122, 124–126) arise with less intensity. Furthermore, the cations [P3]+, [o-DFB]+, [o-DFB–H]+ and [P5]+ were found. The mass spectrum recorded in the negative mode shows only signals of the four anions [AlF3(OTeF5)]−, [AlF2(OTeF5)2]−, [AlF(OTeF5)3]− and [Al(OTeF5)4]−, see Fig. S7–S9.‡
Finally, we investigated [P4H][Al(OTeF5)4] by means of Raman spectroscopy both in an o-DFB solution at T = −30 °C and as a neat powder at T = −78 °C in the solid state (Fig. 4). In both spectra, two prominent bands can be observed. The band around ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 1615 cm−1 corresponds to the symmetrical P–H–P stretching mode and the band at = 598 cm−1 occurs slightly shifted with respect to the breathing mode of neat P4 and is assigned to the corresponding mode of [P4H]+.
= 1615 cm−1 corresponds to the symmetrical P–H–P stretching mode and the band at = 598 cm−1 occurs slightly shifted with respect to the breathing mode of neat P4 and is assigned to the corresponding mode of [P4H]+.
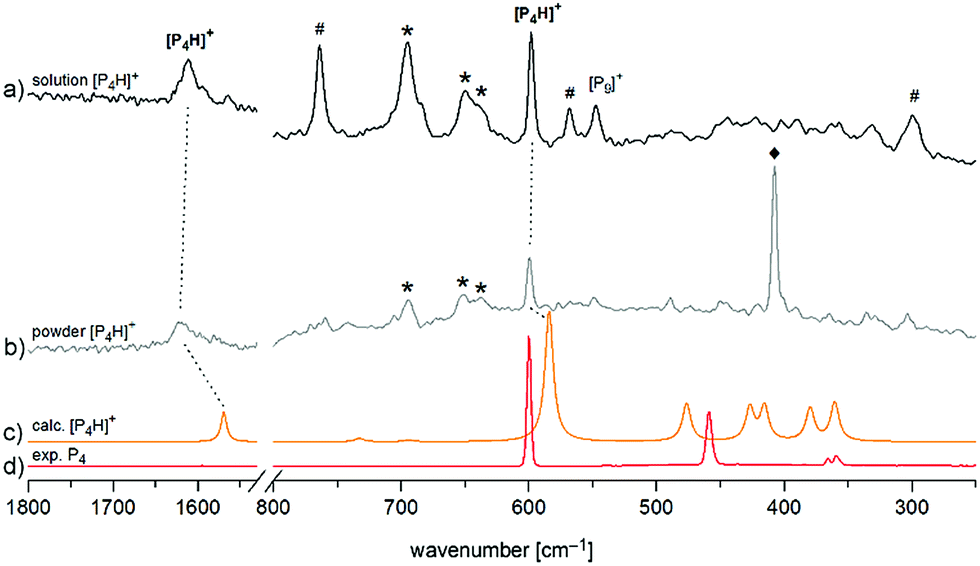 | ||
| Fig. 4 Enlarged Raman spectrum of (a) [P4H][Al(OTeF5)4] in o-DFB at T = −30 °C and (b) [P4H][Al(OTeF5)4] washed with n-pentane at T = −78 °C, (c) calculated spectrum of [P4H]+ at the B3LYP/def2-TZVPP level of theory and (d) experimental spectrum of solid P4 at T = −196 °C. Bands of the anion [Al(OTeF5)4]− at = 695, 650 and 637 cm−1 are marked by an asterisk (*). Bands of the solvent (o-DFB: #, n-pentane: ♦) are indicated as well. Full spectra are provided in Fig. S5 and S6.‡ | ||
Both the experimental band positions agree well with the computed wavenumbers at the B3LYP/def2-TZVPP level of theory at = 1569 cm−1 (A1) and = 584 cm−1 (A1), respectively. Further Raman bands are predicted between = 360 cm−1 and = 476 cm−1 but they are difficult to assign in the experimental spectrum due to their rather low intensities and their partial interference with the bands of [P9]+ impurities. It should be pointed out that special care must be taken by isolating [P4H][Al(OTeF5)4] as a solid, as one sample exploded during the Raman measurement at dry ice temperature after approx. 300 scans at 75 mW, Fig. 4b. Attempts to record low-temperature IR spectra of the [P4H]+ cation were unsuccessful, as the strongest IR band of [P4H]+ is hidden by very prominent bands of the anion at = 713 cm−1 and = 695 cm−1. Also, the strongest band of o-DFB occurs at = 750 cm−1. Nevertheless, low temperature (T = −30 °C) IR spectra of the liquid phase of [P4H][Al(OTeF5)4] have been recorded using a glass fiber ATR head, which are provided in Fig. S10 and S11.‡
Our quantum-chemical calculations at the coupled-cluster CCSD(T)/aug-cc-pVTZ level agree very well with the experimental results of the protonation of a P4 edge. This position is also expected from the MO diagram, where the HOMO orbital is located along the P4 edge (Fig. 1a), leading to a three-center two-electron P–H–P bond. This gives rise to a C2v symmetric structure with an elongation of the PY⋯PY bond of 20.4 pm compared to the bond length of 221.8 pm in the P4 tetrahedral structure. The bond distances PY–PX and PX–PX are less affected by 1.4 and 6.3 pm, respectively (Fig. 5). The computed minimum structure for protonation at the apex of the P4 molecule is 61.4 kJ mol−1 higher in energy than the global minimum structure. An apex protonation would also lead to a computed P–H stretching mode at = 2502 cm−1, which is more than = 900 cm−1 above the experimentally observed band at = 620 cm−1. Surprisingly, the protonation and simultaneous opening of the tetrahedral structure lead to a P4-butterfly type minimum structure (Fig. 5), while protonation of the triangle surface shows a higher order saddle point. Both structures will be higher in energy by 74.3 and 88.5 kJ mol−1 compared to the global minimum structure of [P4H]+, see Fig. 5. For details of the computed structural parameters see Tables S2 and S3.‡
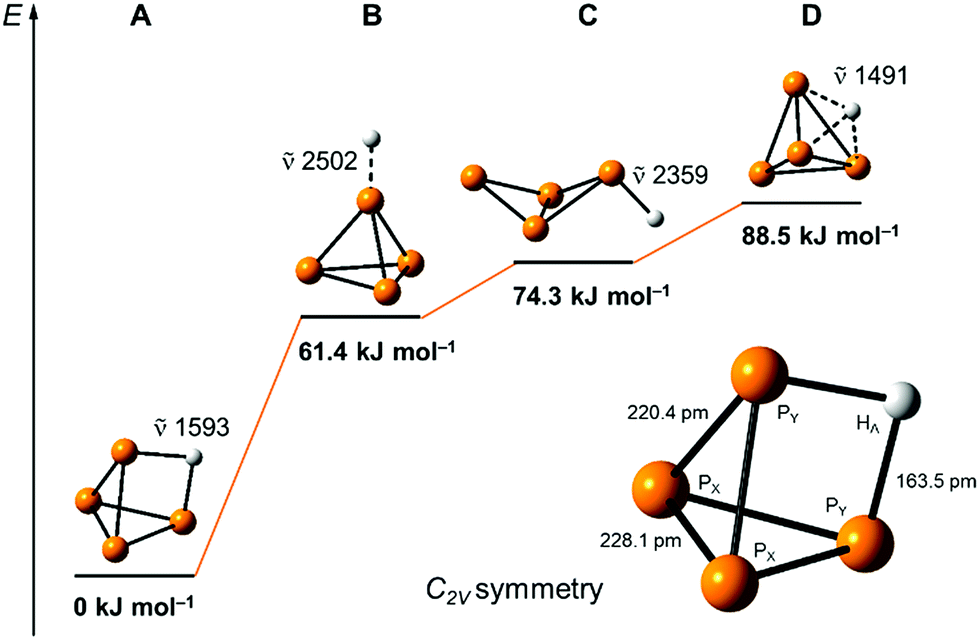 | ||
| Fig. 5 Computed relative energies and P–H vibrations of optimized [P4H]+ structures at the CCSD(T)/aug-cc-pVTZ level of theory. | ||
Conclusions
Based on these results, we could reveal for the first time the structure of protonated white phosphorus in solution. Both experimental results and quantum-chemical calculations provide evidence for a protonation at the edge of the P4 molecule. The opening of the P4-tetrahedron via the simplest electrophile (H+) under formation of a three-center two-electron P–H–P bond is of fundamental interest for understanding the reactivity of this intriguing phosphorus allotrope. It is expected that this groundbreaking result is important for the development of chemical processes related to the activation and further functionalization of elemental phosphorus by electrophiles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Freie Universität Berlin and Deutsche Forschungsgemeinschaft (GRK 1582, Fluorine as a Key Element). We thank Dr Andreas Springer from the Core Facility BioSupraMol at the FU Berlin for measuring the mass spectra. Computing time was made available by High-Performance Computing at ZEDAT/Freie Universität Berlin.Notes and references
- F. Krafft, Angew. Chem., Int. Ed. Engl., 1969, 8, 660 CrossRef PubMed.
- R. Steudel, Chemie der Nichtmetalle, Synthesen - Strukturen - Bindung - Verwendung de Gruyter, Berlin, edn 4, 2014 Search PubMed.
- M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236 CrossRef PubMed.
- (a) J. E. Borger, A. W. Ehlers, M. Lutz, J. Chris Slootweg and K. Lammertsma, Angew. Chem., Int. Ed., 2014, 53, 12836 CrossRef PubMed; (b) L. Xu, Y. Chi, S. Du, W.-X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2016, 55, 9187 CrossRef PubMed; (c) M. Arrowsmith, M. S. Hill, A. L. Johnson, G. Kociok-Köhn and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 54, 7882 CrossRef PubMed.
- (a) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 7052 CrossRef PubMed; (b) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2007, 129, 14180 CrossRef PubMed; (c) O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530 CrossRef PubMed; (d) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389 RSC; (e) C. L. Dorsey, B. M. Squires and T. W. Hudnell, Angew. Chem., Int. Ed., 2013, 52, 4462 CrossRef PubMed; (f) C. D. Martin, C. M. Weinstein, C. E. Moore, A. L. Rheingold and G. Bertrand, Chem. Commun., 2013, 49, 4486 RSC; (g) M. Cicač-Hudi, J. Bendi, S. H. Schlindwein, M. Bispringhoff, M. Nieger, H. Grützmacher and D. Gudat, Eur. J. Inorg. Chem., 2016, 5, 649 CrossRef.
- (a) Y. Xiong, S. Yao, M. Brym and M. Driess, Angew. Chem., 2007, 119, 4595 CrossRef; (b) S. Khan, R. Michel, S. S. Sen, H. W. Roesky and D. Stalke, Angew. Chem., Int. Ed., 2011, 50, 11786 CrossRef PubMed.
- R. Riedel, R. H.-D. Hausen and E. Fluck, Angew. Chem., 1985, 97, 1050 CrossRef.
- B. M. Cossairt and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 15501 CrossRef PubMed.
- J. J. Weigand, M. Holthausen and R. Fröhlich, Angew. Chem., 2009, 121, 301 CrossRef.
- T. Köchner, S. Riedel, A. J. Lehner, H. Scherer, I. Raabe, T. A. Engesser, F. W. Scholz, U. Gellrich, P. Eiden, R. A. Paz Schmidt, D. A. Plattner and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 8139 CrossRef PubMed.
- K. F. Hoffmann, A. Wiesner, N. Subat, S. Steinhauer, S. Riedel and Z. Anorg, Z. Anorg. Allg. Chem., 2018 DOI:10.1002/zaac.201800174.
- I. Krossing and L. van Wüllen, Chem. - Eur. J., 2002, 8, 700 CrossRef.
- (a) P. Dapporto, S. Midollini and L. Sacconi, Angew. Chem., Int. Ed., 1979, 18, 469 CrossRef; (b) M. Peruzzini, S. Mañas, A. Romerosa and A. Vacca, Mendeleev Commun., 2000, 10, 134 CrossRef; (c) T. Gröer, G. Baum and M. Scheer, Organometallics, 1998, 17, 5916 CrossRef.
- E. Fluck, C. M. E. Pavlidou and R. Janoschek, Phosphorus Sulfur Relat. Elem., 1979, 6, 469 CrossRef.
- J.-L. M. Abboud, M. Herreros, R. Notario, M. Esseffar, O. Mó and M. Yáñez, J. Am. Chem. Soc., 1996, 118, 1126 CrossRef.
- R. Bochicchio, L. Lain, A. Torre and R. Ponec, Croat. Chem. Acta, 2000, 73, 1039 Search PubMed.
- R. M. Lobayan and R. Bochicchio, J. Phys. Chem. A, 2015, 119, 7000 CrossRef PubMed.
- Y. Zhang, F. S. Tham, J. F. Nixon, C. Taylor, J. C. Green and C. A. Reed, Angew. Chem., Int. Ed., 2008, 47, 3801 CrossRef PubMed.
- A. Wiesner, T. W. Gries, S. Steinhauer, H. Beckers and S. Riedel, Angew. Chem., Int. Ed., 2017, 56, 8263 CrossRef PubMed.
- T. Köchner, T. A. Engesser, H. Scherer, D. A. Plattner, A. Steffani and I. Krossing, Angew. Chem., Int. Ed., 2012, 51, 6529 CrossRef PubMed.
Footnotes |
| † Dedicated to Prof. Dr Peter Jutzi on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03023e |
| This journal is © The Royal Society of Chemistry 2018 |