
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA fluorescent methylation-switchable probe for highly sensitive analysis of FTO N6-methyladenosine demethylase activity in cells†
Adeline
Cheong
a,
Joanne J. A.
Low
a,
Andrea
Lim
ab,
Paul M.
Yen
b and
Esther C. Y.
Woon
 *a
*a
aDepartment of Pharmacy, National University of Singapore, 18 Science Drive 4, 117543, Singapore. E-mail: esther.woon@nus.edu.sg; Fax: +65 6779 1554; Tel: +65 6516 2932
bProgram of Cardiovascular and Metabolic Disorders, Duke-NUS Medical School, 8 College Road, 169857, Singapore
First published on 7th August 2018
Abstract
N 6-Methyladenosine (m6A) is one of the most abundant epigenetic modifications on mRNA. It is dynamically regulated by the m6A demethylases FTO and ALKBH5, which are currently attracting intense medical interest because of their strong association with several human diseases. Despite their clinical significance, the molecular mechanisms of m6A demethylases remain unclear, hence there is tremendous interest in developing analytical tools to facilitate their functional studies, with a longer term view of validating their therapeutic potentials. To date, no method exists which permits the analysis of m6A-demethylase activity in cells. To overcome this challenge, herein, we describe the first example of a fluorescent m6A-switchable oligonucleotide probe, which enables the direct detection of FTO demethylase activity both in vitro and in living cells. The m6A probe provides a simple, yet powerful visual tool for highly sensitive detection of demethylase activity. Through the use of m6A-probe, we were able to achieve real-time imaging and single-cell flow cytometry analyses of FTO activity in HepG2 cells. We also successfully applied the probe to monitor dynamic changes in FTO activity and m6A methylation levels during 3T3-L1 pre-adipocyte differentiation. The strategy outlined here is highly versatile and may, in principle, be adapted to the study of a range of RNA demethylases and, more widely, other RNA modifying enzymes. To the best of our knowledge, the present study represents not only the first assay for monitoring FTO activity in living cells, but also a new strategy for sensing m6A methylation dynamics.
Introduction
N 6-Methyladenosine (m6A) is an important post-transcriptional modification that is present in more than half of all mammalian messenger RNAs (mRNAs).1–3 It is currently of intense chemical and biological interest because of its critical roles in various key cellular processes, such as the regulation of mRNA stability, translation efficiency and alternative splicing.4–10 In recent years, it has become increasingly clear that the m6A methylation level in mRNA is dynamically regulated by a complex interplay between the m6A methyltransferases (MTases), such as the METTL3-METTL14-WTAP complex,11–13 which catalyse the installation of m6A modification on mRNA and the m6A demethylases, such as FTO (fat mass and obesity-associated protein)14 and ALKBH5 (AlkB homologue 5),15 which directly remove the m6A mark via an oxidative-demethylation mechanism. Both FTO and ALKBH5 belong to the broader family of iron- and 2-oxoglutarate (2OG)-dependent AlkB dioxygenases which also includes Escherichia coli AlkB and eight human homologues, ALKBH1-8.16–18 There is emerging evidence that the aberrant expression of these m6A demethylases may underlie the pathogenesis of a number of human diseases. FTO, in particular, is strongly associated with a range of metabolic and neurodegenerative disorders, including obesity,19 diabetes,20 non-alcoholic steatohepatitis (NASH)21 and Alzheimer's diseases,22 whilst ALKBH5 is likely involved in spermatogenetic defect and male infertility.15,23 Despite the strong physiological links, the exact molecular mechanisms for the m6A demethylases remain unclear. Hence there is currently tremendous interest in developing analytical tools to facilitate the functional and mechanistic studies of these enzymes, with a longer term view of validating their therapeutic potentials.However, the m6A demethylase activity of FTO and ALKBH5 has proven challenging to assay, especially in cells, in part due to the small size and chemical inertness of the N6-methyl group they remove. Consequently, at present, only few methods have been reported for their analysis. This includes the detection of demethylated DNA/RNA product using HPLC or MALDI-TOF mass spectrometry,14,15,24–26 gel electrophoresis of enzyme-digested substrates,26 fluorescence analysis of fluorescein-labelled ssDNA substrate,26 immuno-detection with m6A-specific antibodies14,15,24–26 and, more recently, RNA aptamer-based and MazF endonuclease-based fluorescence assays.27,28 Whilst these methods are useful, many of them are limited by poor sensitivity, low throughput and incompatibility with live-cell applications. Moreover, to date, no method exists which permits the real-time analysis of m6A-demethylase activities in living cells; this severely hinders the study of these clinically-important enzymes.
We,29 and others,30–33 have previously showed that although m6A is able to participate in Watson–Crick base pairing in DNA/RNA duplexes, this is achieved by restricting the N6-methylamino group in an energetically unfavourable anti-conformation (with respect to N1 in the purine ring). Thus the presence of m6A modification has a general destabilising effect on DNA/RNA duplexes and could, in fact, trigger a major overall conformational change in certain sequence contexts. Indeed, m6A has been shown to induce a dramatic duplex–hairpin conversion in some RNA sequences.29,31 The structure-modifying influence of m6A has also been observed in cellular RNAs, such as human MALAT1 (metastasis associated lung adenocarcinoma transcript 1), where m6A apparently modulates RNA–protein interactions by altering the local RNA structure.34,35 Inspired by these fascinating observations, we envisaged that m6A-induced conformational change could provide a basis for sensing m6A (de)methylation in cells.
Herein, we describe the first example of a fluorescent m6A-switchable probe, which enables the highly sensitive and selective detection of FTO demethylase activity both in vitro and in living cells. The analytical principle is illustrated in Fig. 1a. The m6A-probe is a structurally dynamic oligonucleotide probe which, by design, has the capacity to switch between different conformations according to its m6A methylation status. When the probe is methylated, it preferentially adopts a B-like hairpin structure. Removal of the m6A methyl group by FTO, however, triggers a rapid and spontaneous transformation to an A-duplex. To visualise this conformational change, we employed the pyrene-labelled uridine 2′-O-(1-pyrenylmethyl)uridine (UP; Fig. 1b) as the fluorescent reporter. UP is particularly suited for our probe design because its fluorescence emission is highly sensitive to its local structural environment. Previous photophysical studies showed that UP is quenched when stacked with other nucleobases in a single-stranded or hairpin loop environment due to photo-induced charge transfer from the pyrene to neighbouring nucleobases.36–39 However it emits brightly when in a compact A-duplex environment due to disposition of pyrene to the non-quenching minor groove.36–39 Although UP and other pyrene-modified nucleotides have been widely used to probe DNA/RNA structures, base-flipping and protein–nucleic acid interactions,36–39 to date, UP has not been applied to the study of m6A methylation dynamics. There is also no report of pyrene-based probe that switches fluorescence in response to methylation status.
 | ||
| Fig. 1 The fluorescence m6A-switchable probe strategy. (a) The m6A-probe is conformationally-responsive to its m6A methylation status. Removal of m6A methylation by FTO triggers a dramatic switch from hairpin to duplex structure, with concomitant activation of the fluorescent nucleotide 2′-O-(1-pyrenylmethyl)uridine (UP). (b) Structures of Up and m6A. (c) Proposed mechanism for the demethylation of m6A-probe by FTO. The N6-methyl group is first oxidised to a hydroxymethyl group, which then fragments spontaneously to give formaldehyde and the demethylated base. ALKBH5 could not demethylate the m6A-probe. (d) Schematic representation of the m6A-probe and its demethylated counterpart. The probe design is highly versatile and could be adapted to the construction of fluorescence-switchable probes containing other nucleic acid base modifications, including N1-methyladenosine (m1A), N1-methylguanosine (m1G) and N3-methylcytosine (m3dC). | ||
As we shall demonstrate, the fluorescent m6A-probe provides a simple, yet powerful visual tool that permits the highly sensitive detection of FTO demethylase activity. Through the use of m6A-probe, we were able to achieve real-time imaging and single-cell flow cytometry analysis of FTO activity in living cells. We also successfully applied the probe to monitor dynamic changes in FTO activity and m6A methylation levels during 3T3-L1 pre-adipocyte differentiation. The strategy outlined here is simple, inexpensive and amenable to high-throughput format. It is also highly versatile and may, in principle, be applied to the study of a range of RNA demethylases and, more widely, other RNA modifying enzymes. Prior to this study, there is no report of assay method that enables the direct monitoring of FTO m6A-demethylase activity in living cells. We are also not aware of any cell-based assay that specifically target FTO over ALKBH5 (the only other human enzyme currently known to demethylate m6A in RNAs). Hopefully the development of this highly selective approach will advance our understanding of the roles of FTO in m6A-mediated epigenetic processes and its mechanistic connection to human diseases.
Results and discussion
Design strategy for m6A-switchable probes
We begin by identifying oligonucleotide sequences that are structurally-responsive to m6A methylation changes. To engender probes that are sufficiently stable for in vitro and live-cell applications, we considered a series of 2′-O-methyl RNAs of varying length, each containing a single m6A modification (Table 1; for details of chemical synthesis, see ESI†). The probes are modelled after the general palindromic sequence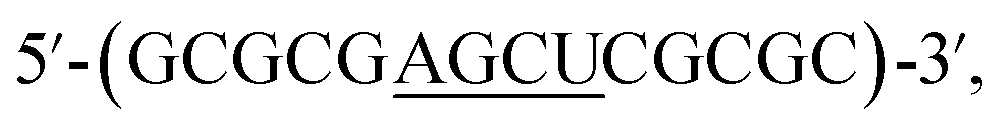 which consists of a middle ‘AGCU segment’ (underlined), flanked on both side by two ‘CG segments’. Because of their self-complementary nature, they can inherently form two main conformations in solution, namely (1) duplex structure, by engaging in intermolecular base pairing, and (2) hairpin structure, by folding back on themselves.40 We hypothesised that, in certain sequence contexts, the introduction of an m6A modification in the ‘middle segment’ could significantly disrupt duplex base-pairing, thereby forcing the probe to adopt a hairpin conformation (where m6A could be stably localised within the hairpin loop). Removal of the m6A modification, however, is expected to restore full base pairing ability and thus favours the alternative duplex structure.
which consists of a middle ‘AGCU segment’ (underlined), flanked on both side by two ‘CG segments’. Because of their self-complementary nature, they can inherently form two main conformations in solution, namely (1) duplex structure, by engaging in intermolecular base pairing, and (2) hairpin structure, by folding back on themselves.40 We hypothesised that, in certain sequence contexts, the introduction of an m6A modification in the ‘middle segment’ could significantly disrupt duplex base-pairing, thereby forcing the probe to adopt a hairpin conformation (where m6A could be stably localised within the hairpin loop). Removal of the m6A modification, however, is expected to restore full base pairing ability and thus favours the alternative duplex structure.
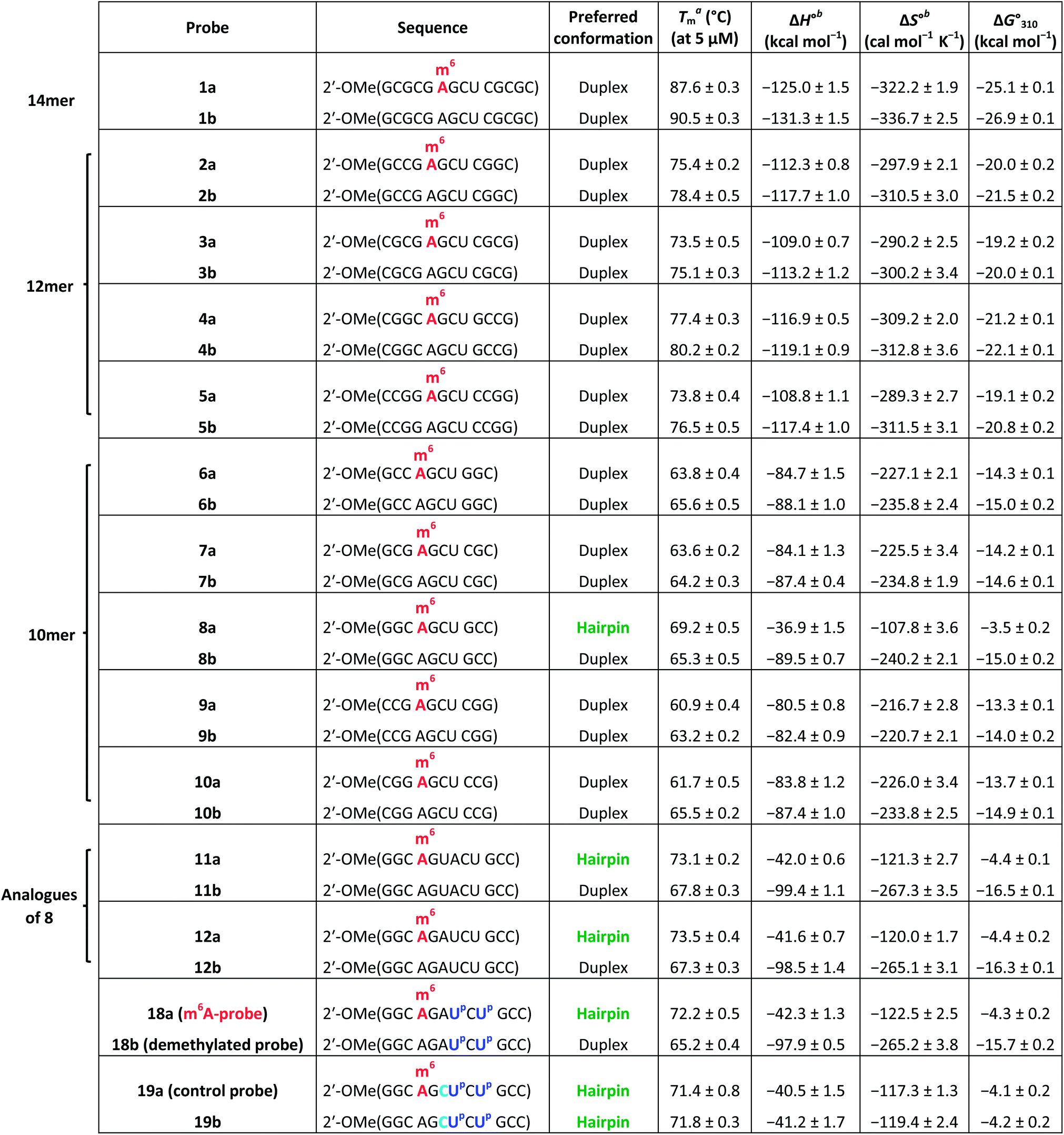
| a Melting temperature (Tm) was determined by UV melting analysis at 5 μM total strand concentration (in 10 mM sodium phosphate buffer containing 150 mM NaCl, pH 7.4). b ΔH° and ΔS° values for duplexes were determined from 1/Tmversus ln(strand concentration) plot, whilst that of hairpins were obtained from α (the fraction of strands remaining hybridised) versus temperature plot by curve fitting to a two-state transition model. |
|---|
|
The m6A-probe switches between hairpin and duplex conformations according to its methylation status
To test our hypothesis, we first compared the UV-melting profile of each of the methylated probes (1a–12a) with that of their non-methylated counterparts (1b–12b) to obtain a qualitative picture of the influence of m6A methylation on their secondary structures (Fig. 2 and S1–S6†). The assignment of hairpin and duplex conformations was initially made based on the hyperchromicity of the melting curves and the dependence of melting temperatures (Tms) on strand concentration; melting transitions that exhibit low UV hyperchromicity (∼10%) and concentration-independent Tms are suggestive of a monomolecular hairpin structure, whereas those that show strong hyperchromicity (∼20%) and concentration-dependent Tms are characteristic of a bimolecular duplex structure. The results are summarised in Table 1. Amongst the sequences investigated, probes 8, 11 and 12 were found to be structurally-responsive towards m6A methylation status. Consistent with our design strategy, they preferentially adopt a hairpin structure when methylated, but convert to a duplex structure upon removal of the m6A modification. The transformation of the probe from hairpin to duplex structure appears to be primarily an enthalpy-driven process, as evidenced by the highly favourable enthalpy change (e.g. ΔΔH° (12a → 12b) = −56.9 kcal mol−1) which counteracts the large entropy cost (ΔΔS° (12a → 12b) = −145.1 cal mol−1 K−1) (Table 1, Fig. S4 and S6†). Moreover, the magnitude of destabilisation caused by a single m6A base on 2′-O-methyl RNA duplexes is relatively small, ranging from 0.4–1.8 kcal mol−1, which is similar to values reported for RNA duplexes (0.35–1.7 kcal mol−1),29–33 suggesting that m6A is generally well-accommodated within 2′-O-methyl RNA duplexes.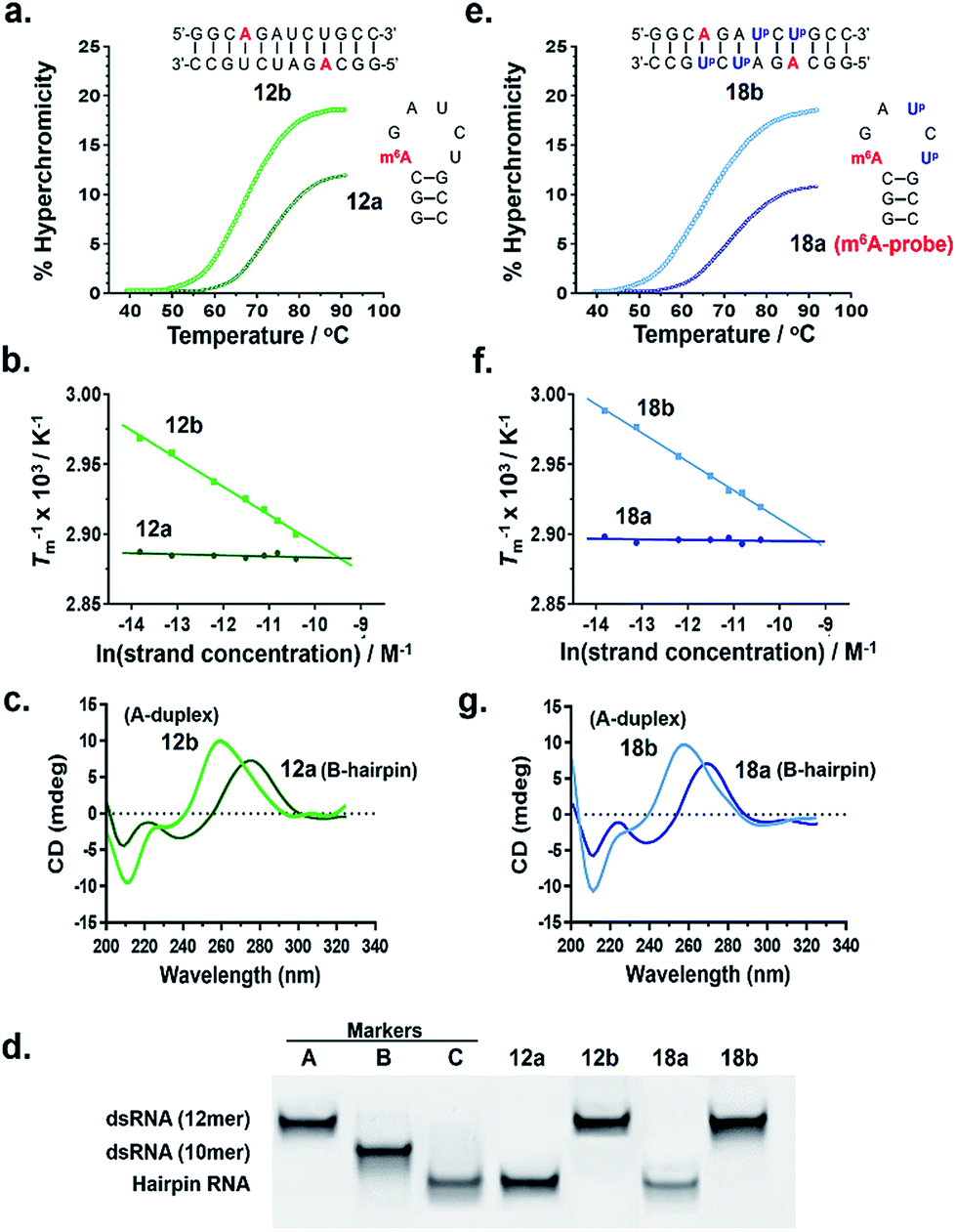 | ||
| Fig. 2 Verification of the m6A-probe design. Both probe 12a and its Up-labelled analogue 18a (m6A-probe) are able to spontaneously adopt two distinct conformations (hairpin or duplex) in response to their m6A methylation status, as demonstrated by their (a, e) UV melting profiles, (b, f) van't Hoff plots, (c, g) CD spectra, and (d) native polyacrylamide gel. All UV and CD experiments were performed at 5 μM strand concentration in 10 mM sodium phosphate buffer containing 150 mM NaCl, pH 7.4. Gel marker A: 12mer dsRNA r(CGCGCGCGCGCG);2 marker B: 10mer dsRNA r(CGCGCGCGCG);2 marker C: 12mer hairpin RNA (CGCGAAUUCGCG). For the conformational analyses of probes 8a and 11a, see Fig. S8.† | ||
To confirm the observed conformational change, we performed circular dichroism (CD) analysis of probes 8, 11 and 12, which clearly revealed a B-like hairpin structure for the methylated probes and an A-form duplex for the corresponding non-methylated equivalents (Fig. 2c and S8†). Native polyacrylamide gel electrophoresis further revealed only one discrete band for each strand, indicating that they exist exclusively as hairpin or duplex structure, rather than a mixture of conformations (Fig. 2d and S8d†). Thus these probes likely undergo complete hairpin–duplex conversion in response to methylation changes. We appreciate that for more vigorous proof of secondary structure, other analytical methods such as 2D NMR studies are required. Nevertheless, data from the present study clearly demonstrated that probes 8, 11 and 12 have the capacity to spontaneously adopt two distinct conformations according to their methylation status, thus they could potentially function as m6A-switchable probes for sensing m6A demethylase activity.
Direct visualisation of methylation changes using pyrene-labelled uridine as the fluorescent reporter
We next investigated whether it is possible to monitor the conformational dynamics of the probe using 2′-O-(1-pyrenylmethyl)uridine (UP) as the fluorescent sensor (Fig. 1b). To identify suitable positions for fluorophore placement, each uridine residue in probes 8, 11 and 12 was systematically replaced with a UP residue. Fluorescence analysis (λex 340 nm) of the resulting probes 13–17 (Table S2†) indicates that UP emission is most effectively quenched (via PET) when incorporated at the U7 or U9 positions of probe 12 (Fig. 3, S9 and S10†). This gives starting methylated probes with very low fluorescence quantum yields (ΦF) of 0.04 (16a) and 0.06 (17a), respectively. Their non-methylated counterparts, 16b and 17b, however, are brightly fluorescent (ΦF 0.24 and 0.25) and exhibit at least 4-fold greater fluorescence intensity.Due to the modular nature of our probe design, it is possible to further amplify the fluorescence signal of the probe through judicious placement of several UP monomers, as exemplified by 18a where the concurrent incorporation of two UP residues led to a dramatic increase in fluorescence light-up response (ΔΦF ∼ 12-fold; ε340 = 9.4 × 104 M−1 cm−1), with concomitant appearance of a broad excimer emission peak at ∼475 nm (Fig. 3b). We appreciate that the presence of bulky UP residue might alter the hairpin conformation of 18a, nevertheless, our results from UV-melting, CD and native PAGE analyses revealed little or no change in its overall secondary structure compared with parent probe 12a (Fig. 2 and S7†). The non-methylated equivalent 18b also retained the ability to dimerise into stable A-duplex structure (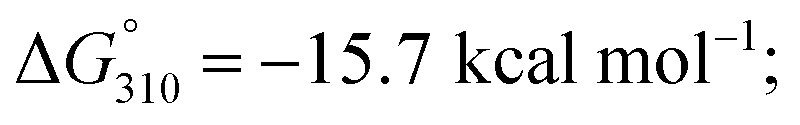 Table 1).
Table 1).
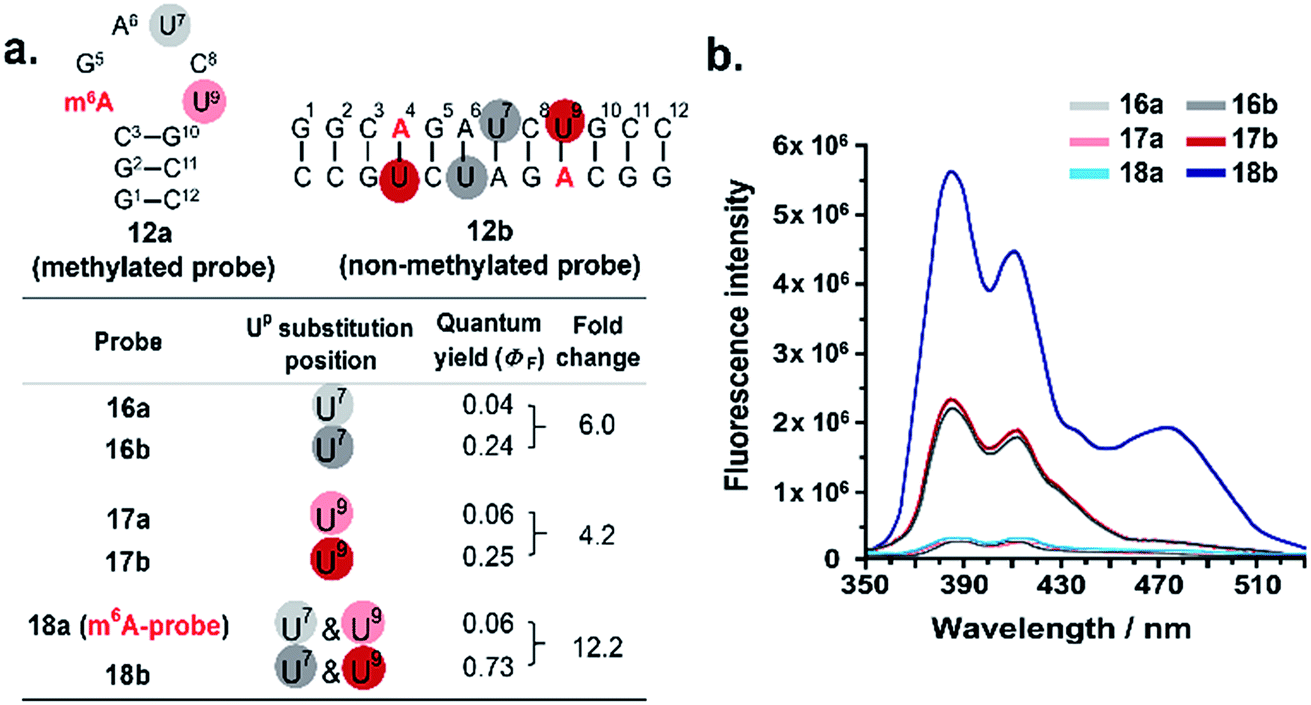 | ||
| Fig. 3 Effects of Up substitution position on fluorescence light-up response. (a) Uridine residues at U7 and U9 positions of probes 12a and 12b (colour coded) were systematically replaced with the fluorescence nucleotide Up to generate probes 16–18. (b) The fluorescence emission spectra (λex 340 nm) of the probes were recorded at 5 μM strand concentration under physiologically-relevant conditions (10 mM sodium phosphate buffer containing 150 mM NaCl, pH 7.4, 37 °C). Two pyrene emission peaks at λem ∼385 nm and ∼410 nm were observed. The concurrent placement of two UP residues at U7 and U9 positions i.e.18a (m6A-probe) led to a dramatic ∼12-fold enhancement in fluorescence light-up response, with concomitant appearance of a broad excimer emission peak at ∼475 nm. The fluorescence quantum yields were determined with ±10% accuracy. For the effects of Up substitution on probes 8 and 11, see Fig. S9 and S10.† | ||
Taken together, these results demonstrated that it is possible to visualise methylation changes in the probe using UP as the fluorescent reporter. Because 18a gave the greatest fluorescence switch-on response and signal-to-noise ratio, it was selected as our representative m6A-probe for further investigation.
The m6A-probe enables highly sensitive detection of FTO demethylase activity
We first examined whether the m6A-probe could serve as fluorogenic substrate for FTO. In our in vitro m6A-probe assay, 10 μM of the probe was incubated with FTO (0.5 μM) in the presence of Fe(II) (cofactor; 100 μM), 2-oxoglutarate (2OG; cosubstrate; 100 μM) and L-ascorbate (2 mM) under physiologically-relevant conditions (50 mM HEPES buffer, pH 7.4, 37 °C). The fluorescence of the reaction (λex 340 nm) was monitored over time and the emergence of fluorescent demethylated probe 18b was indicated by an increase in emission at 410 nm. As shown in Fig. 4a, strong fluorescence could be detected within 30 min of incubation, suggesting that the probe was readily demethylated by FTO. Maximum signal was reached in approximately 2 h, giving a ∼10-fold increase in fluorescence intensity. Such a large fluorescence light-up response is remarkable considering only a single methyl group change between the probe and the demethylated product. Thus the use of m6A-probe enables highly sensitive detection of FTO demethylase activity.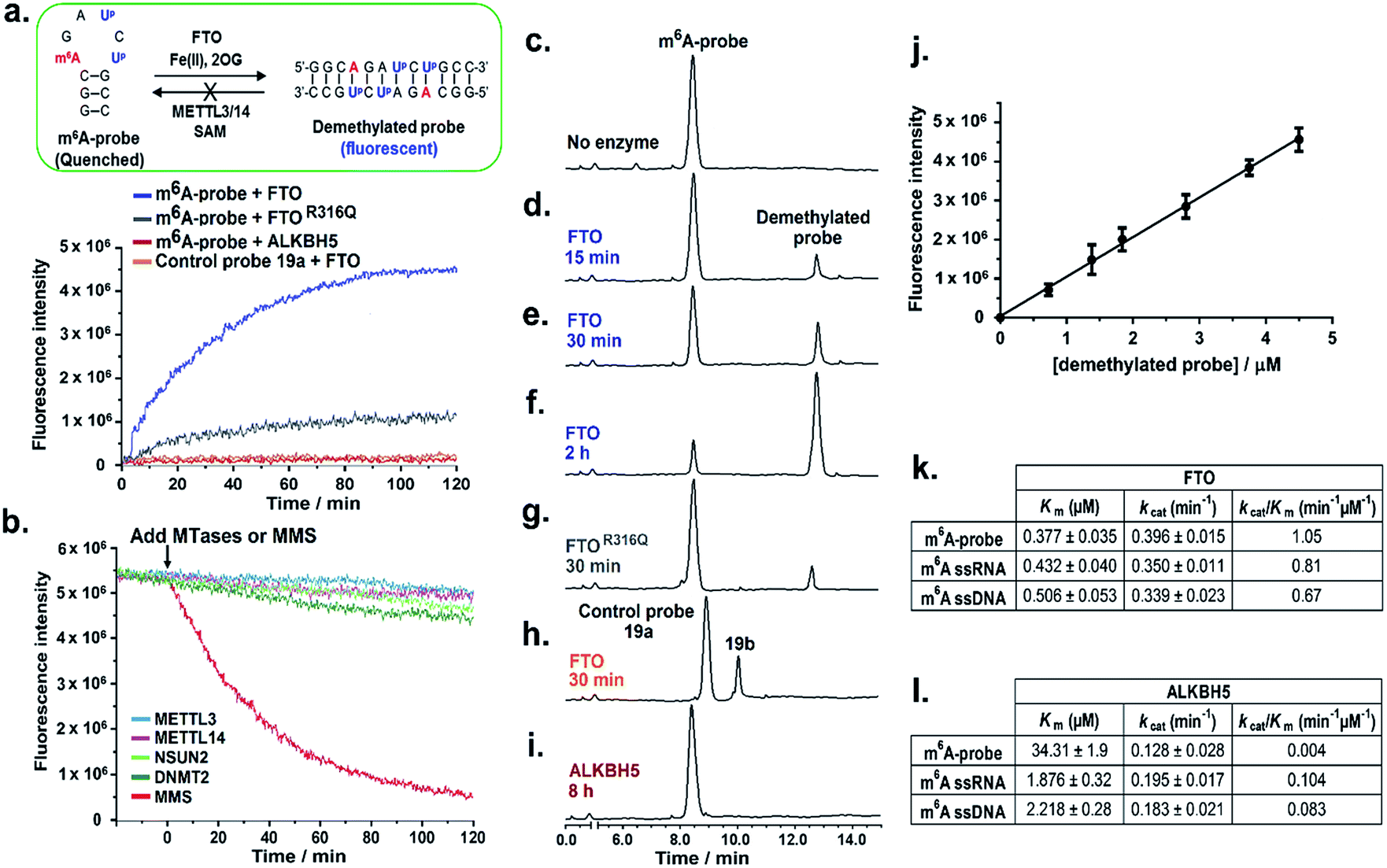 | ||
| Fig. 4 The m6A-probe is remarkably selective for FTO over ALKBH5. (a) Time-course fluorescence analyses (λex 340 nm; λem 410 nm) of the m6A-probe (10 μM) in the presence of FTO (0.5 μM), FTOR316Q (0.5 μM), and ALKBH5 (2 μM) showed that probe fluorescence is activated solely by FTO and not by ALKBH5. (b) There was no appreciable change in the fluorescence intensity (λem 410 nm) of the demethylated probe when exposed to m6A MTases (METTL3 and METTL14) and m5C MTases (NSUN2 and DNMT2), implying no re-methylation of the demethylated probe by these MTases. Treatment with methyl methanesulfonate (MMS) however caused a significant decline in fluorescence. HPLC analysis of m6A-probe (c) in the absence of enzyme (control), and after treatment with FTO for (d) 15 min, (e) 30 min, and (f) 2 h. There was a time-dependent increase in formation of demethylated product with a concomitant decrease in m6A-probe substrate. Treatment of m6A-probe with (g) FTOR316Q, (h) control probe 19a, and (i) ALKBH5. The assignment of HPLC peaks was made by comparison with known standards. (j) There is an excellent linear relationship between the fluorescence intensity of the demethylated probe and its concentration. Data are expressed as mean ± SD of three replicates. Kinetic parameters for the demethylation of m6A-probe, m6A-ssRNA (5′-GCGG-m6A-CUCCAGAUG-3′) and m6A-ssDNA (5′-GCGG-m6A-CTCCAGATG-3′) by (k) FTO and (l) ALKBH5. | ||
Consistent with fluorescence analysis, HPLC analysis of the reaction mixture at various time points showed a time-dependent increase in formation of demethylated product with concomitant decrease in m6A-probe level (Fig. 4c–f). Moreover, the product concentration correlates linearly with the fluorescence intensity (Fig. 4j), hence the fluorescence signal provides a direct indication of FTO demethylase activity.
To confirm that the observed fluorescence increase was specifically triggered by FTO demethylase activity and not due to auto-fluorescence or other non-specific mechanisms, we repeated the m6A-probe assay using an FTO missense mutant, FTOR316Q, which has significantly reduced (80% less) demethylase activity.14 As anticipated, FTOR316Q gave a greatly diminished fluorescence response compared with wild-type FTO, clearly demonstrating that fluorescence activation was indeed induced by FTO-dependent demethylase activity (Fig. 4a and g). Consistent with this finding, control experiments lacking FTO or any of the key assay components i.e. 2OG (cosubstrate) or Fe(II) (cofactor) did not result in any fluorescence increase (Fig. S12†). Subsequent kinetic analysis using a HPLC-based assay revealed that the m6A-probe is a reasonably good substrate for FTO (kcat/Km = 1.05 min−1 μM−1; Fig. 4k and S11†), and could, in fact, be demethylated with similar efficiency as ssRNA and ssDNA substrates containing the GG(m6A)CU consensus motif. This result is interesting because it suggests that, in addition to DNA and RNA substrates, FTO is also able to demethylate m6A modification residing on 2′-O-methyl RNAs, consistent with recent report that FTO demethylates N6,2′-O-dimethyladenosine (m6Am) modification in the 5′ cap of mRNA.46
To further explore whether the hairpin–duplex structural change is truly essential for probe fluorescence activation, we prepared control probe 19a-an analogue of m6A-probe wherein the adenine base at position 6 was replaced with cytosine (Fig. 1d). This single base substitution renders the control probe non-palindromic, thus unable to undergo hairpin–duplex conversion on demethylation, as verified by UV-melting analysis (Table 1 and Fig. S7†). Treatment of the control probe with FTO failed to produce any fluorescence increase (Fig. 4a) despite clear evidence of demethylated product formation by HPLC analysis (Fig. 4h). Thus, fluorescence turn-on is strictly dependent on a switch in probe conformation from hairpin to duplex structure.
The m6A-probe is remarkably selective for FTO over ALKBH5 and other subfamily members
We next evaluated the specificity of the m6A-probe by testing it against several FTO subfamily members, namely ALKBH2,41–43 ALKBH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 43,44 and ALKBH5.15,45 These enzymes are of particular interest because they not only share high levels of sequence and structural homology with FTO, but also possess direct nucleic acid demethylase activities.25 The results from HPLC-based and fluorescence assays showed negligible demethylated product formation and fluorescence activation for all enzymes investigated, suggesting that the m6A-probe is highly selective for FTO over its subfamily members (Fig. S12†). The apparent lack of fluorescence response towards ALKBH5 is notable because ALKBH5 is also known to demethylate m6A on mRNAs (Fig. 4a and l). Such exceptional selectively renders the probe potentially useful for studying the respective roles of FTO and ALKBH5 in m6A-mediated epigenetic processes. The mechanism by which the m6A-probe discriminates against ALKBH5 is unclear at present, however, in light that ALKBH5 could effectively demethylate the RNA equivalent of the m6A-probe, selectivity is likely attributed to the 2′-O-methyl backbone.
43,44 and ALKBH5.15,45 These enzymes are of particular interest because they not only share high levels of sequence and structural homology with FTO, but also possess direct nucleic acid demethylase activities.25 The results from HPLC-based and fluorescence assays showed negligible demethylated product formation and fluorescence activation for all enzymes investigated, suggesting that the m6A-probe is highly selective for FTO over its subfamily members (Fig. S12†). The apparent lack of fluorescence response towards ALKBH5 is notable because ALKBH5 is also known to demethylate m6A on mRNAs (Fig. 4a and l). Such exceptional selectively renders the probe potentially useful for studying the respective roles of FTO and ALKBH5 in m6A-mediated epigenetic processes. The mechanism by which the m6A-probe discriminates against ALKBH5 is unclear at present, however, in light that ALKBH5 could effectively demethylate the RNA equivalent of the m6A-probe, selectivity is likely attributed to the 2′-O-methyl backbone.
Besides selectivity, the m6A-probe also exhibits excellent sensitivity and dynamic range. Titration of the probe (10 μM) with decreasing concentrations of FTO indicated that m6A demethylase activity could be detected at FTO concentration as low as 10 nM (Fig. S13†). This is at least a 100-fold more sensitive than conventional assay performed under similar conditions, where micromolar enzyme concentrations are typically required.
The m6A-probe is not methylated by m6A MTases and is highly stable in cell lysate
One potential concern with the use of m6A-probe in cells is that the demethylated probe could, in principle, be re-methylated back to the hairpin structure by cellular m6A methyltransferases (MTases), leading to a reversal of fluorescence activation. To investigate this possibility, we assayed the demethylated probe against METTL3 and METTL14,11–13 which are the only two m6A MTases identified to date. We also tested it against m5C RNA MTases, NSUN247,48 and DNMT2,49 to examine for possible methylation on the ‘CG segment’ of the probe. Interestingly, in all cases, there was no methylation of the demethylated probe, even after prolonged incubation (8 h), as determined by MALDI-TOF MS analysis (Fig. S14†); there was also no appreciable change in the fluorescence intensity of the probe (Fig. 4b). In sharp contrast, control experiment using the methylating agent methylmethane sulfonate (MMS) resulted in a rapid decline in fluorescence signal, suggesting that probe fluorescence could indeed be reversed by methylation (Fig. 4b). Recent studies suggested that many of the MTases exhibit strict sequence and/or structural requirements for substrate recognition. The m6A MTases for instance specifically recognise and bind to the GG(m6A)CU consensus motif.11–13 This may explain, at least in part, the apparent lack of methylation of the demethylated probes by these enzymes.
To investigate the potential loss of probe fluorescence due to other nucleic acid modifying enzymes or cellular nucleases, we further performed time-course fluorescence analysis of the m6A-probe in HepG2 cell lysate, where FTO is abundantly expressed.21 As shown in Fig. S15,† the demethylated probe product formed rapidly in cell lysate where it remained strongly emissive for at least 24 h with no obvious decline in fluorescence. This suggests that the probe has good photostability and is fairly resistant to nuclease degradation. Our combined results, therefore, established m6A-probe as a highly sensitive and selective probe suitable for visualising FTO demethylase activity in cells.
The m6A-probe assay enables high-throughput screening of FTO inhibitors
On the basis of these promising data, we next investigated whether the developed m6A-probe assay could be applied to the high-throughput screening of FTO inhibitor. To assess the performance of our assay, a set of structurally-diverse small molecules (Fig. 5a) consisting of known FTO inhibitors (24PDCA,50 LipotF,25 Rhein,24 IOX350) and non-inhibitor isonicotinic acid40 were individually incubated at various concentrations with FTO (50 nM) and m6A-probe (10 μM) in a 384-well plate, and their fluorescence spectra simultaneously recorded on a fluorescence microplate reader. The inhibition curves are shown in Fig. 5b. With the exception of isonicotinic acid, all compounds investigated induced a clear dose-dependent decrease in fluorescence intensity. The determined IC50s are also comparable with previously reported values (Fig. 5a), thus the m6A-probe assay is able to successfully identify FTO inhibitors across diverse chemical scaffolds. To further evaluate the quality and reproducibility of our m6A-probe assay for screening in a 384-well format, we determined the Z′ factor using the method described by Zhang et al.51 (for experimental details, see ESI†). The average Z′ factor for our assay was found to be 0.68, which indicates excellent statistical reliability (Fig. 5c). Taken together, the results validate our m6A-probe assay as a robust method for high-throughput screening of FTO inhibitors.
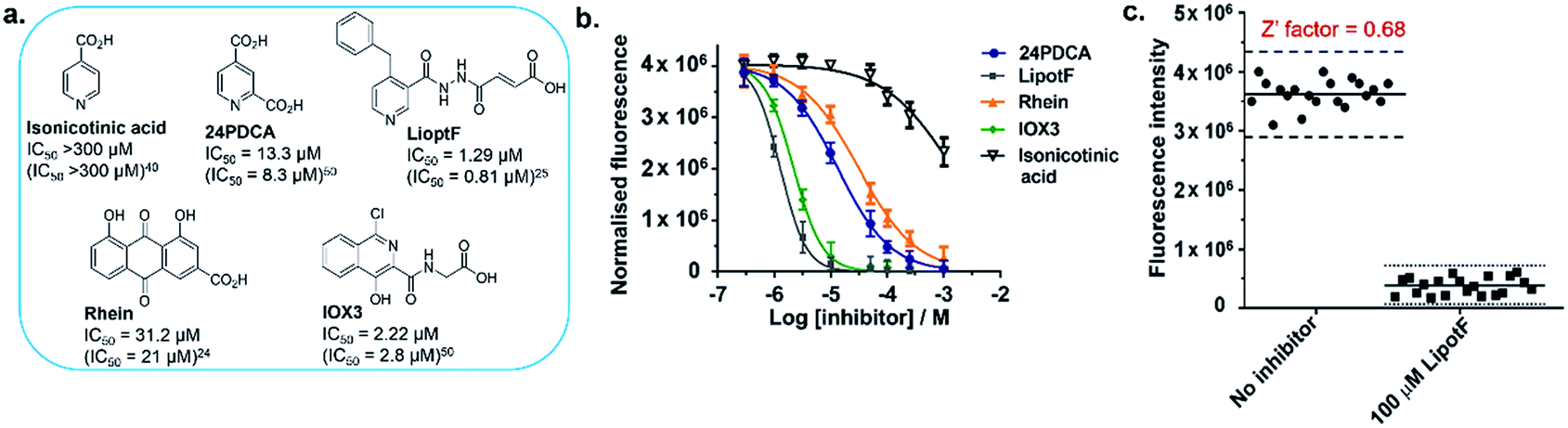 | ||
| Fig. 5 The m6A-probe assay provides a robust method for the high-throughput screening of FTO inhibitors. (a) Structures of reported FTO inhibitors. The determined IC50s were comparable with previously reported values (in parentheses). (b) Enzyme inhibition curves for the FTO inhibitors, as determined using the m6A-probe assay. Percent enzyme activity was calculated by normalising to the fluorescence signal without inhibitor (100%) and without m6A-probe (0%). Data are expressed as mean ± SD of three replicates. (c) The Z′ factor for the m6A-probe assay was determined in a 384-well format, using the method described by Zhang et al.51 A total of 40 independent assays were run with either LipotF (positive control) or DMSO (negative control). | ||
The m6A-probe enables real-time monitoring of FTO activity in living cells
We next proceeded to explore the utility of the m6A-probe for imaging FTO activity in live-cells. To this end, 10 μM of the m6A-probe or control probe 19a was delivered into HepG2 cells via streptolysin-O (SLO) reversible permeabilisation method52 and then analysed using fluorescence microscopy; it has been suggested that this method is more rapid and efficient compared with conventional transfection methods.52 Preliminary experiments in our lab further showed that SLO-permeabilisation is an effective method for m6A-probe delivery and that SLO treatment does not significantly affect FTO expression in HepG2 cells (Fig. S16†). As shown in Fig. 6a and b, bright blue fluorescence could be observed within the cells after 1 h incubation. This represents a 7-fold increase in fluorescence relative to the control probe (Fig. 6c), which is fairly substantial considering suboptimal filter settings (λex 340–380 nm; λem 435–485 nm). Notably, the fluorescence appears to accumulate in the cytoplasm, with little or no detectable signal in the nucleus (Fig. S17†). In light of emerging evidence suggesting that FTO shutters between the nucleus and the cytoplasm where it likely serves different cellular functions,53–55 the selective distribution of m6A-probe in the cytoplasm may allow us to specifically study FTO activity on cytoplasmic mRNAs. Despite toxicity concerns associated with the use of pyrene-based fluorophore in general, MTT cytotoxicity assays showed that the m6A-probe is reasonably well-tolerated by HepG2 and 3T3-L1 cells, with >80% of the cells remaining viable after treatment with 50 μM probe for 24 h (Fig. S18†). The probe also did not significantly affect FTO expression in cells (Fig. S16†).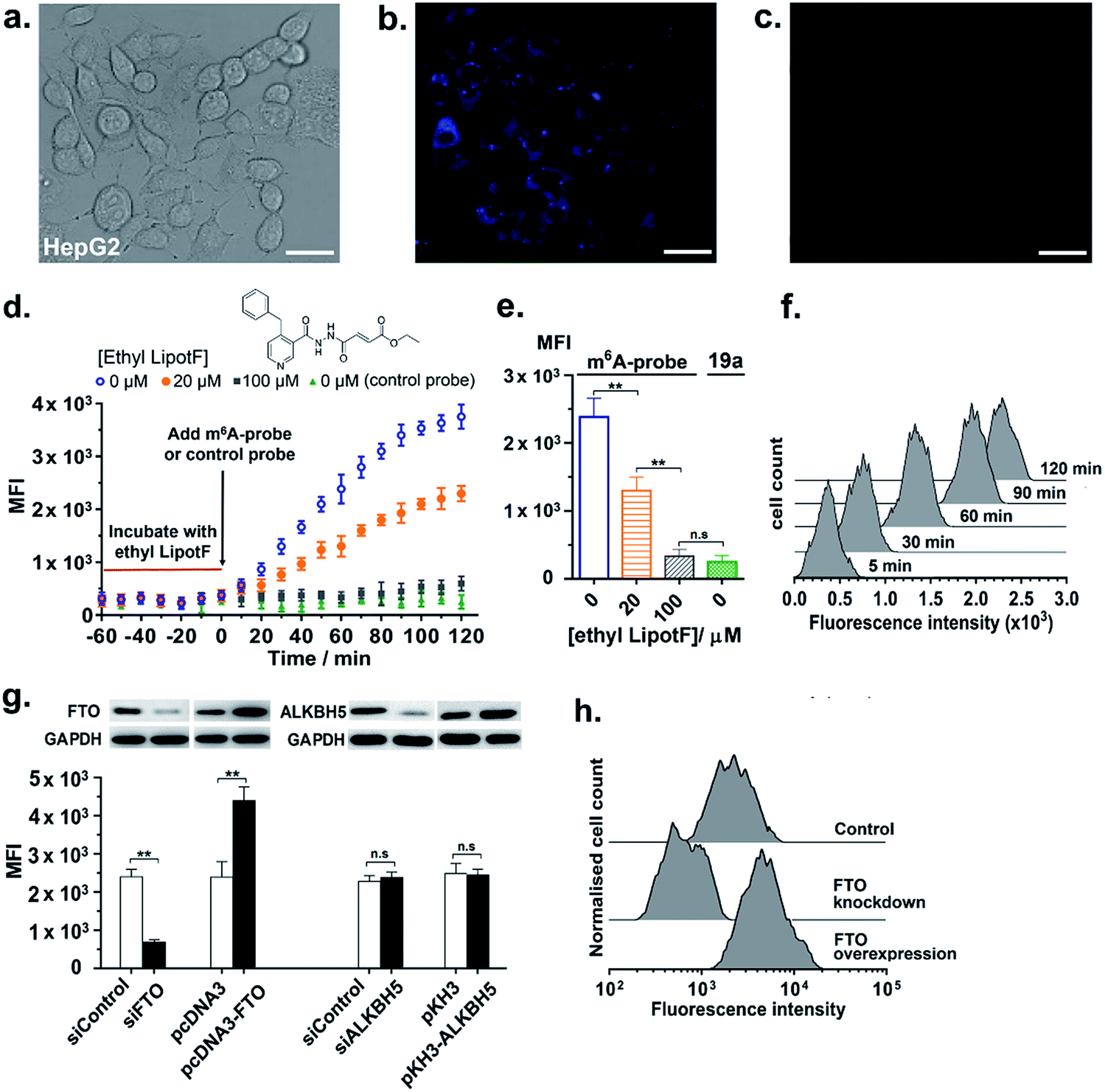 | ||
| Fig. 6 The m6A-probe enables real-time monitoring of FTO demethylase activity in living cells. Correlated (a) bright field and (b) fluorescent images of HepG2 cells after 1 h treatment with m6A-probe (10 μM; λex 340–380 nm; λem 435–485 nm). (c) Negligible fluorescence was observed for cells treated with control probe 19a (10 μM). The image in (c) is from a different experiment than the paired images in (a) and (b). Scale bar, 50 μm. (d) Time-course flow cytometry analysis of FTO inhibition in HepG2 cells. Cells were incubated with various concentrations of ethyl LipotF (0, 20 and 100 μM; colour coded) for 1 h before treatment with m6A-probe. MFI is the mean fluorescence intensity of at least 20000 live cells (λex 355 nm; λem 379 nm). (e) A plot of MFI at 60 min showed a clear dose-dependent decrease in fluorescence intensity. (f) A set of representative flow cytometry profiles for cells treated with 20 μM ethyl LipotF. (g, h) Fluorescence responses of m6A-probe to different FTO and ALKBH5 expression levels in HepG2 cells. The probe responds only to changes in the levels of FTO and not ALKBH5. GAPDH was used as loading control. Data are expressed as mean ± SD of three biological replicates. *P < 0.05; **P < 0.01; n.s = not significant. | ||
In addition to live-cell imaging, we further demonstrated that the m6A-probe is compatible with flow cytometry analysis (λex 355 nm; λem 379 nm) and could provide real-time monitoring of FTO inhibition in cells. As shown in the time-course flow cytometry profiles (Fig. 6d–f), incubation of HepG2 cells with various concentrations of ethyl LipotF (a cell-permeable FTO inhibitor developed in our lab; PubChem ID: 126970770)25 for 1 hour prior to the addition of m6A-probe led to a dose-dependent reduction in fluorescence signal. The control probe 19a, however, did not produce any fluorescence change, as anticipated.
Flow cytometry experiment further demonstrated that the m6A-probe is highly sensitive towards changes in cellular FTO expression levels. In particular, stable overexpression of FTO in HepG2 cells caused a profound increase in probe fluorescence, whilst FTO knockdown via transient transfection with FTO-specific siRNA led to a significant reduction in emission (Fig. 6g, h and S19†). The fluorescence intensity was unchanged with ALKBH5 overexpression or knockdown, thus, consistent with our in vitro data, the m6A-probe is exclusively activated by FTO and not by ALKBH5. The m6A-probe therefore provides a single assay platform that permits applications both in vitro and in living cells.
The m6A-probe provides a powerful tool for following dynamic changes in endogenous FTO level during cellular differentiation
It is increasingly clear that both FTO expression level and m6A methylation pattern are dynamically regulated during cellular differentiation7,56 and pathological processes, such as tumourigenesis,57 DNA damage,58 heat shock59 and stress responses.60 Recent study by Zhao et al. in particular showed that FTO expression levels decreased during 3T3-L1 pre-adipocyte FTO FTO expression levels decreased during 3T3-L1 pre-adipocyte differentiation and this correlated inversely with global m6A levels.7 We were therefore interested in exploring whether the m6A-probe is able to report dynamic changes in endogenous FTO activity during cellular differentiation. To this end, 3T3-L1 pre-adipocytes were treated with the probe at different time points of adipogenesis i.e. Day 0 (prior to treatment with mitotic inducing agent), 3, 6, and 9 and then analysed by fluorescence microscopy and flow cytometry. Consistent with the above-mentioned report,7 cells on Day 3, 6, and 9 of differentiation exhibited a 1.4-, 2.0- and 5.4-fold decrease in fluorescence intensity, respectively, relative to Day 0 (Fig. 7a and b), suggesting a time-dependent reduction in FTO level and/or activity during adipogenesis. Remarkably, the observed fluorescence decrease also corresponds to a 1.3-, 1.6-, and 2.1-fold increase in m6A levels in mRNAs, respectively, as determined by dot-blot analysis (Fig. 7c and d). Thus the fluorescence intensity provides a direct read-out of cellular FTO activity. Taken together, the results showed that our m6A-probe can provide a simple, yet highly sensitive tool for following dynamic changes in cellular FTO activity. | ||
| Fig. 7 The m6A-probe provides a powerful tool for following dynamic changes in endogenous FTO activity during cellular differentiation. Differentiation of 3T3-L1 pre-adipocyte was induced by incubation with the differentiation cocktail (IBMX/DEX/Insulin) on Day 0. Cells at different time points of adipogenesis (Day 0, 3, 6, 9) were treated with m6A-probe (10 μM), then analysed with fluorescence microscopy and flow cytometry. (a) Correlated bright field (top panel) and fluorescence (bottom panel) images of 3T3-L1 cells during different phases of adipogenesis. (b) Flow cytometry analysis showed a clear time-dependent decrease in probe fluorescence, suggesting a reduction in FTO activity during adipogenesis. MFI is the mean fluorescence intensity of at least 20000 live cells (λex 355 nm; λem 379 nm). Data are expressed as mean ± SD of three biological replicates. *P < 0.05; **P < 0.01. (c) The level of m6A in mRNA during different stages of adipogenesis was determined by dot-blot analysis with m6A antibody and quantified by Grayscale analysis with ImageJ software. (d) The observed decreased in fluorescence was accompanied with an increase in m6A levels in mRNA, thus fluorescence signal provides a direct read-out of cellular FTO activity. | ||
The m6A-probe strategy could be adapted to the study of a range of nucleic acid modifications
Because our m6A-probe design exploits the universal principle of Watson–Crick base pairing, it may conceptually be applied to the construction of fluorescent switchable-probes for other nucleic acid modifications. To explore the general utility of our probe strategy, we further prepared analogues of m6A-probe containing other physiologically-relevant base modifications, namely N1-methyladenosine (m1A; an epigenetic modification in mRNAs),61N1-methylguanosine (m1G; a post-transcriptional modification on bacterial rRNA and tRNA)62 and N3-methylcytosine (m3dC; a DNA base lesion)63 (Fig. 1d). Remarkably, the removal of these modified bases triggered a similar hairpin–duplex conformational change, as demonstrated by CD analysis (Fig. S20†). The observed conformational change was again accompanied with significant fluorescence light-up responses (ΔΦF > 9; Fig. S20d†). Thus our m6A-probe strategy is highly versatile and could potentially be adapted to the study of a range of nucleic acid-modifying enzymes.Conclusions
Overall, we have provided proof of principle that a novel strategy employing fluorescent m6A-switchable probe could enable highly sensitive detection of FTO m6A-demethylase activity both in vitro and in living cells. The developed m6A-probe assay is simple, inexpensive and permits the direct analysis of FTO activity without recourse to radioactive substrates or cumbersome techniques. This assay also requires only small amount of protein (low nanomolar concentrations), thus avoiding the drawbacks of high protein consumption and limited dynamic range associated with current assay methods. Through the use of the m6A-probe, the removal of a single methyl group is dramatically amplified (∼10-fold). This enables highly sensitive detection of FTO demethylase activity.Importantly, the m6A-probe provides a powerful visual tool for direct imaging and single-cell flow cytometry analysis of FTO activity. In this study, we have successfully applied the probe to the real-time monitoring of FTO inhibition in HepG2 cells. We also demonstrated that the m6A-probe is highly sensitive towards cellular FTO expression levels and could report on subtle changes in FTO activity during 3T3-L1 pre-adipocyte differentiation. Thus the m6A-probe may provide insights into the dynamic regulation of cellular processes by FTO. We further showed that the m6A-probe strategy can be applied to the screening of FTO inhibitors across diverse chemical scaffolds. Given the very high level of current interest in FTO as an m6A ‘eraser’ and as disease target, the development of a single assay platform that allows both the functional study of FTO and the high-throughput discovery of FTO therapeutic leads shall be of significant biological and medical interest.
One limitation of this approach is that, as oppose to genetically-encoded sensors, it is necessary to deliver the m6A-probe into cells exogenously, and this introduces additional handling steps. The probe delivery efficiency also likely varies between different cell types. Moreover, this method is not suitable for base modifications that do not significantly impact duplex stability, such as 5-methylcytosine (m5C) and N7-methylguanosine (m7G). Nevertheless, the strategy outlined here should be applicable to the wide number of other base modifications that are known to disrupt duplex base pairing. Indeed, we have demonstrated that probes containing modifications, such as m1A, m1G and m3dC also undergo spontaneous hairpin–duplex transformation, with concomitant fluorescence activation. Hence the developed assay could, in principle, be adapted to the study of a range of nucleic acid-modifying enzymes.
Prior to this study, there is no report of assay method that enables the direct analysis of FTO m6A-demethylase activity in living cells. We are also not aware of any cell-based assay that selectively target FTO over ALKBH5 (the only other human enzyme currently known to demethylate m6A in RNAs). The development of this highly selective approach may prove valuable in dissecting the respective roles of FTO and ALKBH5 in m6A-mediated epigenetic processes, which hopefully will advance our understanding of mechanistic links between FTO and human diseases.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Singapore Ministry of Health's National Medical Research Council (NMRC/BNIG/2008/2013) and the Singapore Ministry of Education (AcRF Tier 1 Grant R148-000-231-114 and R148-000-238 114).Notes and references
- D. Dominissini, S. Moshitch-Moshkovitz, S. Schwartz, M. Salmon-Divon, L. Ungar, S. Osenberg, K. Cesarkas, J. Jacob-Hirsch, N. Amariglio, M. Kupiec, R. Sorek and G. Rechavi, Nature, 2012, 485, 201–206 CrossRef PubMed.
- K. D. Meyer, Y. Saletore, P. Zumbo, O. Elemento, C. E. Mason and S. R. Jaffrey, Cell, 2012, 149, 1635–1646 CrossRef PubMed.
- B. Linder, A. V. Grozhik, A. O. Olarerin-George, C. Meydan, C. E. Mason and S. R. Jaffrey, Nat. Methods, 2015, 12, 767–772 CrossRef PubMed.
- Y. Fu, D. Dominissini, G. Rechavi and C. He, Nat. Rev. Genet., 2014, 15, 293–306 CrossRef PubMed.
- K. D. Meyer and S. R. Jaffrey, Nat. Rev. Mol. Cell Biol., 2014, 15, 313–326 CrossRef PubMed.
- X. Wang, Z. Lu, A. Gomez, G. C. Hon, Y. Yue, D. Han, Y. Fu, M. Parisien, Q. Dai, G. Jia, B. Ren, T. Pan and C. He, Nature, 2014, 505, 117–120 CrossRef PubMed.
- X. Zhao, Y. Yang, B.-F. Sun, Y. Shi, X. Yang, W. Xiao, Y.-J. Hao, X.-L. Ping, Y.-S. Chen, W.-J. Wang, K.-X. Jin, X. Wang, C.-M. Huang, Y. Fu, X.-M. Ge, S.-H. Song, H. S. Jeong, H. Yanagisawa, Y. Niu, G.-F. Jia, W. Wu, W.-M. Tong, A. Okamoto, C. He, J. M. R. Danielsen, X.-J. Wang and Y.-G. Yang, Cell Res., 2014, 24, 1403–1419 CrossRef PubMed.
- Y. Wang, Y. Li, J. I. Toth, M. D. Petroski, Z. Zhang and J. C. Zhao, Nat. Cell Biol., 2014, 16, 191–198 CrossRef PubMed.
- X. Wang, B. S. Zhao, I. A. Roundtree, Z. Lu, D. Han, H. Ma, X. Weng, K. Chen, H. Shi and C. He, Cell, 2015, 161, 1388–1399 CrossRef PubMed.
- M. Bartosovic, H. C. Molares, P. Gregorova, D. Hrossova, G. Kudla and S. Vanacova, Nucleic Acids Res., 2017, 45, 11356–11370 CrossRef PubMed.
- J. Liu, Y. Yue, D. Han, X. Wang, Y. Fu, L. Zhang, G. Jia, M. Yu, Z. Lu, X. Deng, Q. Dai, W. Chen and C. He, Nat. Chem. Biol., 2014, 10, 93–95 CrossRef PubMed.
- X. Wang, J. Feng, Y. Xue, Z. Guan, D. Zhang, Z. Liu, Z. Gong, Q. Wang, J. Huang, C. Tang, T. Zou and P. Yin, Nature, 2016, 534, 575–578 CrossRef PubMed.
- P. Wang, K. A. Doxtader and Y. Nam, Mol. Cell, 2016, 63, 306–317 CrossRef PubMed.
- G. Jia, Y. Fu, X. Zhao, Q. Dai, G. Zheng, Y. Yang, C. Yi, T. Lindahl, T. Pan, Y.-G. Yang and C. He, Nat. Chem. Biol., 2011, 7, 885–887 CrossRef PubMed.
- G. Zheng, J. A. Dahl, Y. Niu, P. Fedorcsak, C.-M. Huang, C. J. Li, C. B. Vågbø, Y. Shi, W.-L. Wang, S.-H. Song, Z. Lu, R. P. G. Bosmans, Q. Dai, Y.-J. Hao, X. Yang, W.-M. Zhao, W.-M. Tong, X.-J. Wang, F. Bogdan, K. Furu, Y. Fu, G. Jia, X. Zhao, J. Liu, H. E. Krokan, A. Klungland, Y.-G. Yang and C. He, Mol. Cell, 2013, 49, 18–29 CrossRef PubMed.
- T. Gerken, C. A. Girard, Y.-C. L. Tung, C. J. Webby, V. Saudek, K. S. Hewitson, G. S. H. Yeo, M. A. McDonough, S. Cunliffe, L. A. McNeill, J. Galvanovskis, P. Rorsman, P. Robins, X. Prieur, A. P. Coll, M. Ma, Z. Jovanovic, I. S. Farooqi, B. Sedgwick, I. Barroso, T. Lindahl, C. P. Ponting, F. M. Ashcroft, S. O'Rahilly and C. J. Schofield, Science, 2007, 318, 1469–1472 CrossRef PubMed.
- B. I. Fedeles, V. Singh, J. C. Delaney, D. Li and J. M. Essigmann, J. Biol. Chem., 2015, 290, 20734–20742 CrossRef PubMed.
- C. Schofield and R. Hausinger, 2-Oxoglutarate-Dependent Oxygenases, Royal Society of Chemistry, Cambridge, 2015 Search PubMed.
- T. M. Frayling, N. J. Timpson, M. N. Weedon, E. Zeggini, R. M. Freathy, C. M. Lindgren, J. R. B. Perry, K. S. Elliott, H. Lango, N. W. Rayner, B. Shields, L. W. Harries, J. C. Barrett, S. Ellard, C. J. Groves, B. Knight, A.-M. Patch, A. R. Ness, S. Ebrahim, D. A. Lawlor, S. M. Ring, Y. Ben-Shlomo, M.-R. Jarvelin, U. Sovio, A. J. Bennett, D. Melzer, L. Ferrucci, R. J. F. Loos, I. Barroso, N. J. Wareham, F. Karpe, K. R. Owen, L. R. Cardon, M. Walker, G. A. Hitman, C. N. A. Palmer, A. S. F. Doney, A. D. Morris, G. D. Smith, A. T. Hattersley and M. I. McCarthy, Science, 2007, 316, 889–894 CrossRef PubMed.
- R. M. Freathy, N. J. Timpson, D. A. Lawlor, A. Pouta, Y. Ben-Shlomo, A. Ruokonen, S. Ebrahim, B. Shields, E. Zeggini, M. N. Weedon, C. M. Lindgren, H. Lango, D. Melzer, L. Ferrucci, G. Paolisso, M. J. Neville, F. Karpe, C. N. A. Palmer, A. D. Morris, P. Elliott, M.-R. Jarvelin, G. D. Smith, M. I. McCarthy, A. T. Hattersley and T. M. Frayling, Diabetes, 2008, 57, 1419–1426 CrossRef PubMed.
- A. Lim, J. Zhou, R. A. Sinha, B. K. Singh, S. Ghosh, K.-H. Lim, P. K.-H. Chow, E. C. Y. Woon and P. M. Yen, Biochem. Biophys. Res. Commun., 2016, 479, 476–481 CrossRef PubMed.
- C. Graff, L. Keller, W. Xu, H.-X. Wang, B. Winblad and L. Fratiglioni, Alzheimer's Dementia, 2010, 6, S111 CrossRef.
- C. Tang, R. Klukovich, H. Peng, Z. Wang, T. Yu, Y. Zhang, H. Zheng, A. Klungland and W. Yan, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E325–E333 CrossRef PubMed.
- B. Chen, F. Ye, L. Yu, G. Jia, X. Huang, X. Zhang, S. Peng, K. Chen, M. Wang, S. Gong, R. Zhang, J. Yin, H. Li, Y. Yang, H. Liu, J. Zhang, H. Zhang, A. Zhang, H. Jiang, C. Luo and C.-G. Yang, J. Am. Chem. Soc., 2012, 134, 17963–17971 CrossRef PubMed.
- J. D. W. Toh, L. Sun, L. Z. M. Lau, J. Tan, J. J. A. Low, C. W. Q. Tang, E. J. Y. Cheong, M. J. H. Tan, Y. Chen, W. Hong, Y.-G. Gao and E. C. Y. Woon, Chem. Sci., 2015, 6, 112–122 RSC.
- Y. Huang, J. Yan, Q. Li, J. Li, S. Gong, H. Zhou, J. Gan, H. Jiang, G.-F. Jia, C. Luo and C.-G. Yang, Nucleic Acids Res., 2015, 43, 373–384 CrossRef PubMed.
- N. Svensen and S. R. Jaffrey, Cell Chem. Biol., 2016, 23, 415–425 CrossRef PubMed.
- M. Imanishi, S. Tsuji, A. Suda and S. Futaki, Chem. Commun., 2017, 53, 12930–12933 RSC.
- S. Zou, J. D. W. Toh, K. H. Q. Wong, Y.-G. Gao, W. Hong and E. C. Y. Woon, Sci. Rep., 2016, 6, 25677 CrossRef PubMed.
- J. D. Engel and P. H. J. von Hippel, Biol. Chem., 1978, 253, 927–934 Search PubMed.
- R. Micura, W. Pils, C. Höbartner, K. Grubmayr, M. O. Ebert and B. Jaun, Nucleic Acids Res., 2001, 29, 3997–4005 CrossRef PubMed.
- E. Kierzek and R. Kierzek, Nucleic Acids Res., 2003, 31, 4472–4480 CrossRef PubMed.
- C. Roost, S. R. Lynch, P. J. Batista, K. Qu, H. Y. Chang and E. T. Kool, J. Am. Chem. Soc., 2015, 137, 2107–2115 CrossRef PubMed.
- N. Liu, Q. Dai, G. Zheng, C. He, M. Parisien and T. Pan, Nature, 2015, 518, 560–564 CrossRef PubMed.
- K. I. Zhou, M. Parisien, Q. Dai, N. Liu, L. Diatchenko, J. R. Sachleben and T. Pan, J. Mol. Biol., 2016, 428, 822–833 CrossRef PubMed.
- K. Yamana, R. Iwase, S. Furutani, H. Tsuchida, H. Zako, T. Yamaoka and A. Murakami, Nucleic Acids Res., 1999, 27, 2387–2392 CrossRef PubMed.
- M. Nakamura, Y. Fukunaga, K. Sasa, Y. Ohtoshi, K. Kanaori, H. Hayashi, H. Nakano and K. Yamana, Nucleic Acids Res., 2005, 33, 5887–5895 CrossRef PubMed.
- M. E. Østergaard, P. Cheguru, M. R. Papasani, R. A. Hill and P. J. Hrdlicka, J. Am. Chem. Soc., 2010, 132, 14221–14228 CrossRef PubMed.
- Y. Park, D. Nim-anussornkul, T. Vilaivan, T. Morii and B. H. Kim, Bioorg. Med. Chem. Lett., 2018, 28, 77–80 CrossRef PubMed.
- T. Yang, A. Cheong, X. Mai, S. Zou and E. C. Y. Woon, Chem. Commun., 2016, 52, 6181–6184 RSC.
- C.-G. Yang, C. Yi, E. M. Duguid, C. T. Sullivan, X. Jian, P. A. Rice and C. He, Nature, 2008, 452, 961–965 CrossRef PubMed.
- T. Duncan, S. C. Trewick, P. Koivisto, P. A. Bates, T. Lindahl and B. Sedgwick, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16660–16665 CrossRef PubMed.
- P. A. Aas, M. Otterlei, P. Ø. Falnes, C. B. Vågbø, F. Skorpen, M. Akbari, O. Sundheim, M. Bjørås, G. Slupphaug, E. Seeberg and H. E. Krokan, Nature, 2003, 421, 859–863 CrossRef PubMed.
- O. Sundheim, C. B. Vågbø, M. Bjørås, M. M. L. Sousa, V. Talstad, P. A. Aas, F. Drabløs, H. E. Krokan, J. A. Tainer and G. Slupphaug, EMBO J., 2006, 25, 3389–3397 CrossRef PubMed.
- W. Aik, J. S. Scotti, H. Choi, L. Gong, M. Demetriades, C. J. Schofield and M. A. McDonough, Nucleic Acids Res., 2014, 42, 4741–4754 CrossRef PubMed.
- J. Mauer, X. Luo, A. Blanjoie, X. Jiao, A. V. Grozhik, D. P. Patil, B. Linder, B. F. Pickering, J.-J. Vasseur, Q. Chen, S. S. Gross, O. Elemento, F. Debart, M. Kiledjian and S. R. Jaffrey, Nature, 2017, 541, 371–375 CrossRef PubMed.
- S. Hussain, A. A. Sajini, S. Blanco, S. Dietmann, P. Lombard, Y. Sugimoto, M. Paramor, J. G. Gleeson, D. T. Odom, J. Ule and M. Frye, Cell Rep., 2013, 4, 255–261 CrossRef PubMed.
- V. Khoddami and B. R. Cairns, Nat. Biotechnol., 2013, 31, 458–464 CrossRef PubMed.
- A. Jeltsch, A. Ehrenhofer-Murray, T. P. Jurkowski, F. Lyko, G. Reuter, S. Ankri, W. Nellen, M. Schaefer and M. Helm, RNA Biol., 2017, 14, 1108–1123 CrossRef PubMed.
- M. Das, T. Yang, J. Dong, F. Prasetya, Y. Xie, K. Wong, A. Cheong and E. C. Y. Woon, Chem.–Asian J., 2018 DOI:10.1002/asia.201800729.
- J.-H. Zhang, T. D. Chung and K. R. Oldenburg, J. Biomol. Screening, 1999, 4, 67–73 CrossRef PubMed.
- I. Walev, S. C. Bhakdi, F. Hofmann, N. Djonder, A. Valeva, K. Aktories and S. Bhakdi, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3185–3190 CrossRef PubMed.
- P. Gulati, E. Avezov, M. Ma, R. Antrobus, P. Lehner, S. O'Rahilly and G. S. H. Yeo, Biosci. Rep., 2014, 34, 621–628 CrossRef PubMed.
- P. Gulati, M. K. Cheung, R. Antrobus, C. D. Church, H. P. Harding, Y.-C. L. Tung, D. Rimmington, M. Ma, D. Ron, P. J. Lehner, F. M. Ashcroft, R. D. Cox, A. P. Coll, S. O'Rahilly and G. S. H. Yeo, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2557–2562 CrossRef PubMed.
- A. Aas, P. Isakson, C. Bindesbøll, E. A. Alemu, A. Klungland and A. Simonsen, PLoS One, 2017, 12, e0168182 CrossRef PubMed.
- A. Klungland, J. A. Dahl, G. Greggains, P. Fedorcsak and A. Filipczyk, Nat. Methods, 2017, 14, 18–22 CrossRef PubMed.
- J. Zhou, J. Wan, X. Gao, X. Zhang, S. R. Jaffrey and S.-B. Qian, Nature, 2015, 526, 591–594 CrossRef PubMed.
- Y. Xiang, B. Laurent, C.-H. Hsu, S. Nachtergaele, Z. Lu, W. Sheng, C. Xu, H. Chen, J. Ouyang, S. Wang, D. Ling, P.-H. Hsu, L. Zou, A. Jambhekar, C. He and Y. Shi, Nature, 2017, 543, 573–576 CrossRef PubMed.
- Q. Cui, H. Shi, P. Ye, L. Li, Q. Qu, G. Sun, G. Sun, Z. Lu, Y. Huang, C.-G. Yang, A. D. Riggs, C. He and Y. Shi, Cell Rep., 2017, 18, 2622–2634 CrossRef PubMed.
- M. Engel, S. Roeh, C. Eggert, P. M. Kaplick, L. Tietze, J. Arloth, P. Weber, M. Rex-Haffner, M. Jakovcevski, M. Uhr, M. Eder, C. T. Wotjak, M. V. Schmidt, J. M. Deussing, E. B. Binder and A. Chen, bioRxiv, 2017, 200402.
- D. Dominissini, S. Nachtergaele, S. Moshitch-Moshkovitz, E. Peer, N. Kol, M. S. Ben-Haim, Q. Dai, A. Di Segni, M. Salmon-Divon, W. C. Clark, G. Zheng, T. Pan, O. Solomon, E. Eyal, V. Hershkovitz, D. Han, L. C. Doré, N. Amariglio, G. Rechavi and C. He, Nature, 2016, 530, 441–446 CrossRef PubMed.
- H. Zhou, I. J. Kimsey, E. N. Nikolova, B. Sathyamoorthy, G. Grazioli, J. McSally, T. Bai, C. H. Wunderlich, C. Kreutz, I. Andricioaei and H. M. Al-Hashimi, Nat. Struct. Mol. Biol., 2016, 23, 803–810 CrossRef PubMed.
- J. C. Delaney and J. M. Essigmann, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14051–14056 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, including full synthesis procedure, protein purification methods, fluorescence and spectroscopic studies, thermodynamic analyses, biochemical and cell-based experimental conditions. See DOI: 10.1039/c8sc02163e |
| This journal is © The Royal Society of Chemistry 2018 |