DOI:
10.1039/C8SC02920B
(Edge Article)
Chem. Sci., 2019,
10, 293-309
Luminescent ruffled iridium(III) porphyrin complexes containing N-heterocyclic carbene ligands: structures, spectroscopies and potent antitumor activities under dark and light irradiation conditions†
Received
3rd July 2018
, Accepted 27th September 2018
First published on 10th October 2018
Abstract
A panel of iridium(III) porphyrin complexes containing axial N-heterocyclic carbene (NHC) ligand(s) were synthesized and characterized. X-ray crystal structures of the bis-NHC complexes [IrIII(ttp)(IMe)2]+ (2a), [IrIII(oep)(BIMe)2]+ (2d), [IrIII(oep)(IiPr)2]+ (2e) and [IrIII(F20tpp)(IMe)2]+ (2f) display ruffled porphyrin rings with mesocarbon displacements of 0.483–0.594 Å and long Ir–CNHC bonds of 2.100–2.152 Å. Variable-temperature 1H NMR analysis of 2a reveals that the macrocycle porphyrin ring inversion takes place in solution with an activation barrier of 40 ± 1 kJ mol−1. The UV-vis absorption spectra of IrIII(por)–NHC complexes display split Soret bands. TD-DFT calculations and resonance Raman experiments show that the higher-energy Soret band is derived from the 1MLCT dπ(Ir) → π*(por) transition. The near-infrared phosphorescence of IrIII(por)–NHC complexes from the porphyrin-based 3(π, π*) state features broad emission bands at 701–754 nm with low emission quantum yields and short lifetimes (Φem < 0.01; τ < 4 μs). [IrIII(por)(IMe)2]+ complexes (por = ttp and oep) are efficient photosensitizers for 1O2 generation (Φso = 0.64 and 0.88) and are catalytically active in the light-induced aerobic oxidation of secondary amines and arylboronic acid. The bis-NHC complexes exhibit potent dark cytotoxicity towards a panel of cancer cells with IC50 values at submicromolar levels. The cytotoxicity of these complexes could be further enhanced upon light irradiation with IC50 values as low as nanomolar levels in association with the light-induced generation of reactive oxygen species (ROS). Bioimaging of [IrIII(oep)(IMe)2]+ (2c) treated cells indicates that this Ir complex mainly targets the endoplasmic reticulum. [IrIII(oep)(IMe)2]+ catalyzes the photoinduced generation of singlet oxygen and triggers protein oxidation, cell cycle arrest, apoptosis and the inhibition of angiogenesis. It also causes pronounced photoinduced inhibition of tumor growth in a mouse model of human cancer.
Introduction
The recent years have witnessed a bloom in the applications of metal–N-heterocyclic carbene (NHC) complexes in catalysis,1 materials science2 and medicine.3 These burgeoning research activities have benefited greatly from improved knowledge regarding the coordination chemistry of metal–NHC complexes.4 Nonetheless, there are only a handful of examples of metalloporphyrins bearing NHC ligand(s) thus far. In this regard, the groups of Albrecht,5 Woo6 and Che7 have contributed to the synthesis and investigation of the spectroscopic properties and/or catalytic activities of d6 metalloporphyrin complexes of CoIII, RhIII, IrIII and RuII ions bearing axial NHC ligand(s). Several characteristics of these metalloporphyrins have been noted, including (1) gentle dearomatization of the porphyrin ligand; (2) elongation of the axial metal–ligand bond trans to the NHC ligand; and (3) pronounced ruffling of the porphyrin ligand scaffold.5,6 The last feature, namely, NHC-induced ruffling deformation, is particularly intriguing because the out-of-plane deformation of metalloporphyrin is recognized to have a profound impact on its photophysical (e.g., electronic absorption, emission, excited state dynamics) and physiochemical properties (e.g., axial ligand affinity, electron transfer rate), both of which have important implications in biological processes involving the ubiquitous tetrapyrrole compounds.8 While many studies on nonplanar metalloporphyrins have focused on metal ions of relatively small size, such as NiII8a,9 and ZnII,10 and those with MnIII11 and FeIII,12 in which nonplanarity is primarily induced by the peripheral substituents of the porphyrin, fewer studies have been performed on axial ligand-induced porphyrin ring deformation in d6 metalloporphyrins.5,6,13
The Soret and Q bands in the electronic absorption spectra of metalloporphyrins of d6 RuII, OsII, RhIII and IrIII generally display marked hypsochromic shifts with respect to their free base porphyrins due to dπ(M)–π*(por) interaction.14 Axial ligand(s) can also perturb the absorption spectra by modulating the electronic properties of the metal ion and hence the metal–porphyrin bonding interaction (cis-effect).14 The literature reports that d6 metalloporphyrins display red to infrared phosphorescence derived from the 3(π, π*) IL state of the porphyrin ligand or the 3(dπ, π*) metal-to-ligand charge transfer (MLCT) state upon photoexcitation.15 Nevertheless, only IrIII–porphyrin complexes emit strong red to near-infrared (NIR) phosphorescence in solution at room temperature, with emission quantum yields up to 0.30 and emission lifetimes up to 97 μs.16a–c Due to these advantageous luminescence properties, the use of cationic IrIII-oep-derived bioconjugates for cellular imaging and as phosphorescent probes for intracellular oxygen sensing has been reported by Papkovsky and co-workers.16b On the other hand, two near-IR-absorbing BODIPY assemblies of IrIII–porphyrin, recently reported by Shen, Chan, Mack and co-workers,16d have been shown to display excellent photostability and efficiency in photosensitized singlet oxygen generation with a quantum yield of up to 0.85.16d These encouraging findings have shed light on the potential photobiological uses of IrIII–porphyrin complexes, which remain unexplored.
Here, we describe the synthesis and characterization of a panel of luminescent ruffled iridium(III) porphyrin complexes containing mono-NHC and bis-NHC ligand(s), namely, [IrIII(por)(NHC)Cl] (1a–1d) and [IrIII(por)(NHC)2]+ (2a–2f), respectively. Steady-state and time-resolved spectroscopic studies and theoretical calculations in conjunction with resonance Raman spectroscopy were undertaken to examine the effects of NHC ligation on the photophysical properties and electronic structures of the IrIII–porphyrin complexes. In addition, the photochemical reactivity via the light-induced generation of singlet oxygen as well as the cytotoxicity and phototoxicity of these bis-NHC iridium(III) porphyrin complexes were investigated both in vitro and in vivo.
Results
Synthesis
The reported synthetic approaches to metalloporphyrin-NHC complexes include the thermal decarboxylation of imidazolium carboxylate,5 deprotonation of the imidazolium salt with a strong base7 and incorporation of the free imidazoylidene.6 In this work, the coordination of NHC ligands to iridium(III) porphyrin was accomplished by reacting bis(NHC)silver(I) complex17 with [IrIII(por)Cl(CO)] via transmetalation. This synthetic protocol offers the advantage that the reaction can be conducted under mild, aerobic conditions. Mono-NHC complexes [IrIII(por)(NHC)Cl] (1a–1d) were prepared by stirring a mixture of [IrIII(por)Cl(CO)] with half equivalents of the respective bis(NHC)silver(I) complexes in CH2Cl2 at room temperature for 12 h (Scheme 1). After the removal of AgCl salt by filtration through a short plug of Celite (when por = tmp, where H2tmp = meso-tetramesitylporphyrin) or a silica gel column (when por = F20tpp, where H2F20tpp = meso-tetrakis(pentafluorophenyl)porphyrin), the desired complexes were recrystallized from CH2Cl2/hexane and obtained in 60–74% isolated yields. Bis-NHC complexes 2a–2f were similarly synthesized by reacting stoichiometric amounts of bis(NHC)silver(I) complexes with [IrIII(por)Cl(CO)] in CH2Cl2 at room temperature or at 40 °C for 12 h to 4 days (Scheme 2). The complexes were then purified by flash column chromatography on a silica gel column using CH2Cl2/EtOAc as the eluent and were obtained in 70–96% isolated yields. [IrIII(oep)(CNPhOMe)2]+ (3) and [IrIII(oep)(py)2]+ (4) were also prepared for comparative study (see (ESI†)).
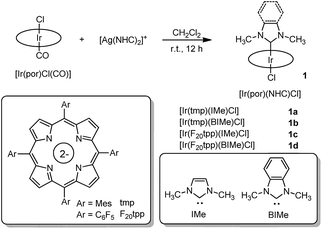 |
| | Scheme 1 Synthesis of [IrIII(por)(NHC)Cl] complexes 1a–1d. | |
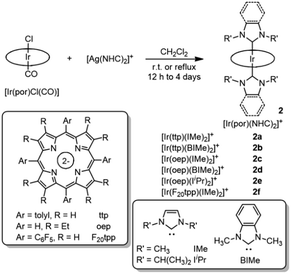 |
| | Scheme 2 Synthesis of [IrIII(por)(NHC)2]+ complexes 2a–2f. | |
The number of coordinated NHC ligands was revealed by the 1H NMR and FAB-MS analyses of the complexes and is dependent on the steric bulk of the porphyrin ligand. Specifically, whereas a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 bis(NHC)silver(I) complex with [IrIII(por)Cl(CO)] (por = ttp or oep) recipe gave solely [IrIII(por)(NHC)2]+ complexes, the same stoichiometry furnished only [IrIII(tmp)(NHC)Cl] and trace [IrIII(tmp)(NHC)]+ complexes in the case of [IrIII(tmp)Cl(CO)]. The reaction of bis(NHC)silver(I) complex with [IrIII(F20tpp)Cl(CO)] to give [IrIII(F20tpp)(IMe)2]+ (2f) was completed at elevated temperature with extended reaction time (refluxing CH2Cl2 for 4 days). The FAB-MS spectra of complexes 1a–1d all show peaks attributed to [M − Cl]+ fragments, suggesting that the Ir–Cl bond is weakened by the trans-axial NHC ligand. The IR oxidation-state marker bands of all complexes 1a–1d and 2a–2f fall into the range of 1018–1022 cm−1, which is comparable to that of their parental complexes [IrIII(por)Cl(CO)] and consistent with the iridium(III) oxidation state.18 Mono-NHC complexes 1a, 1b and 1d are stable in the solid state and in CDCl3 solution for 1 week with the exception of 1c, which decomposes within 1 day under ambient conditions. Bis-NHC complexes 2a–2e are remarkably air-stable in the solid state and in CDCl3 solution in the dark for 1 month, as revealed by 1H NMR spectroscopy and UV-vis absorption spectroscopy, except for 2f, which gradually deteriorates in CDCl3 in 3 days.
:1 bis(NHC)silver(I) complex with [IrIII(por)Cl(CO)] (por = ttp or oep) recipe gave solely [IrIII(por)(NHC)2]+ complexes, the same stoichiometry furnished only [IrIII(tmp)(NHC)Cl] and trace [IrIII(tmp)(NHC)]+ complexes in the case of [IrIII(tmp)Cl(CO)]. The reaction of bis(NHC)silver(I) complex with [IrIII(F20tpp)Cl(CO)] to give [IrIII(F20tpp)(IMe)2]+ (2f) was completed at elevated temperature with extended reaction time (refluxing CH2Cl2 for 4 days). The FAB-MS spectra of complexes 1a–1d all show peaks attributed to [M − Cl]+ fragments, suggesting that the Ir–Cl bond is weakened by the trans-axial NHC ligand. The IR oxidation-state marker bands of all complexes 1a–1d and 2a–2f fall into the range of 1018–1022 cm−1, which is comparable to that of their parental complexes [IrIII(por)Cl(CO)] and consistent with the iridium(III) oxidation state.18 Mono-NHC complexes 1a, 1b and 1d are stable in the solid state and in CDCl3 solution for 1 week with the exception of 1c, which decomposes within 1 day under ambient conditions. Bis-NHC complexes 2a–2e are remarkably air-stable in the solid state and in CDCl3 solution in the dark for 1 month, as revealed by 1H NMR spectroscopy and UV-vis absorption spectroscopy, except for 2f, which gradually deteriorates in CDCl3 in 3 days.
X-ray crystal structures
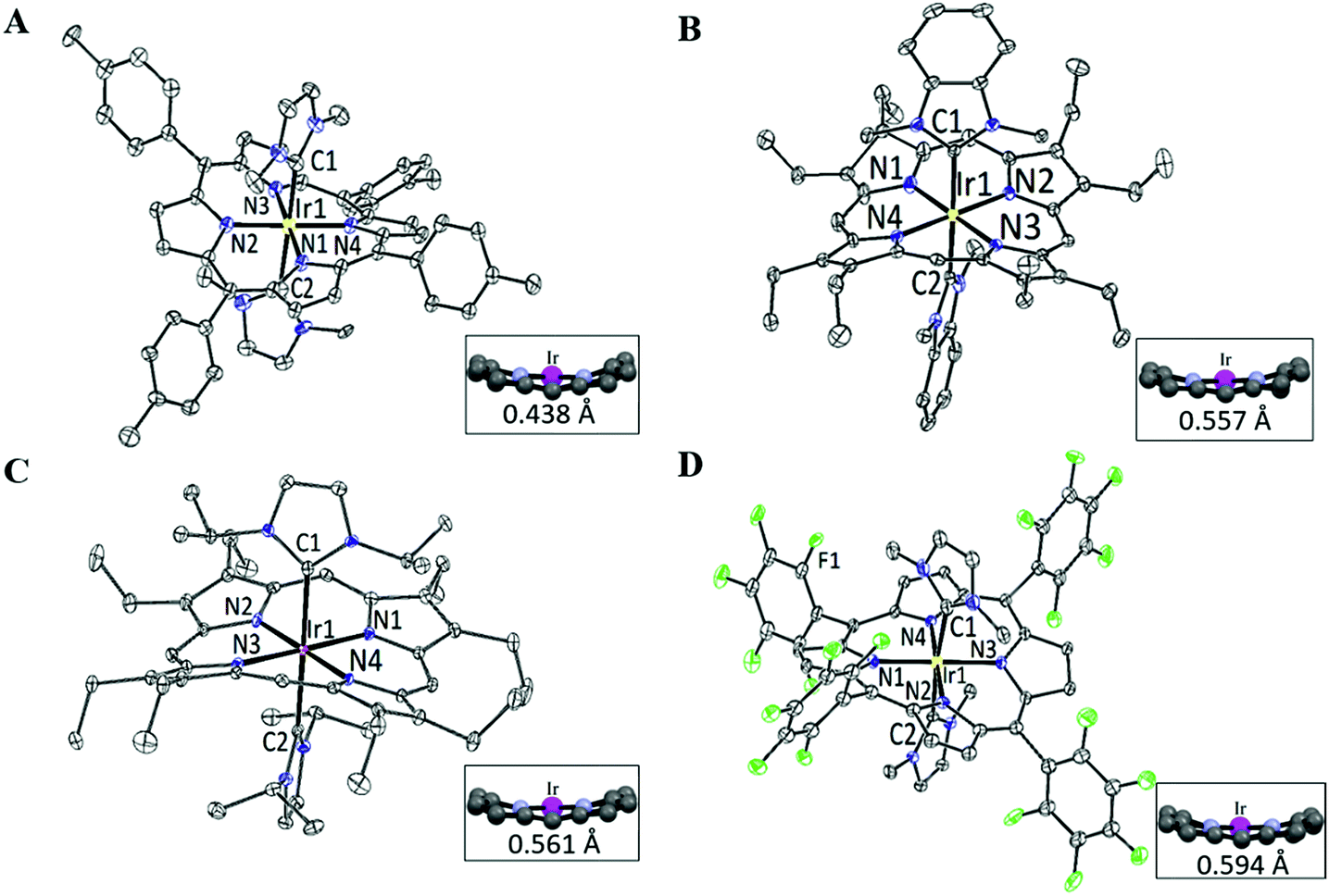
The structures of 2a, 2d, 2e and 2f were determined by X-ray diffraction analysis, and their perspective views are depicted in Fig. 1. Selected bond distances and angles are summarized in Table 1. All complexes adopt a slightly distorted octahedral geometry, with the porphyrin ring exhibiting a significant ruffling deformation. The mean deviations of the mesocarbon atom from the least square plane of the porphyrin ring in 2a, 2d, 2e and 2f are 0.483, 0.557, 0.561 and 0.594 Å, respectively. The planes of the NHC ligands are slightly to moderately tilted with respect to the Ir–CNHC axis. The mean pitch angles of the NHC ligands (measurement of out-of-plane tilting) of 2a, 2d, 2e and 2f are 7.5°, 4.6°, 1.4° and 5.1° respectively. The pitch angle of 2a is slightly larger than that of [IrIII(ttp)(IBuMe)Me] (6.4°).6 The ruffling deformation of the porphyrin ring combined with the tilting of NHC ligands is considered to be a means to relieve the unfavorable steric repulsion caused by the close proximity of the NHC ligands and porphyrin ring. Ruffling deformation of porphyrin rings has also been observed in other NHC-bearing metalloporphyrin complexes.5,6 In all the X-ray crystal structures, the planes of the NHC ligands are approximately orthogonal to each other, with an interplanar angle of 84.8° for 2a, 69.2° for 2d, 87.7° for 2e and 80.9° for 2f. The orientation of NHC ligands with respect to Ir–Npor, described by the torsional angle of NNHC–CNHC–Ir–Npor, has an average value of 40.3° for 2a, 35.0° for 2d, 43.5° for 2e and 37.0° for 2f. The near-staggered orientation of the NHC ligands with respect to Ir–Npor suggests minimal Ir–CNHC π bonding interaction.5a Evidently, the arrangement of NHC ligands is related to the structural requirement for ruffling deformation of porphyrin ligands.
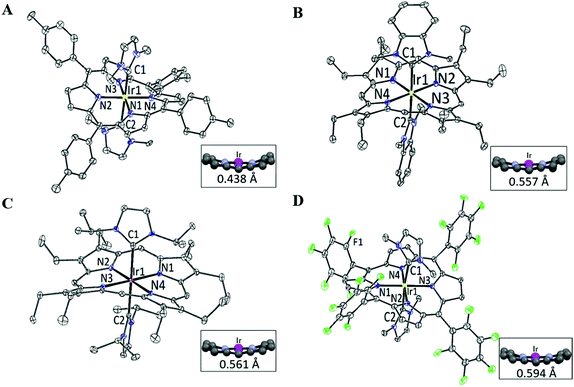 |
| | Fig. 1 Perspective views of (A) [IrIII(ttp)(IMe)2]+2a, (B) [IrIII(oep)(BIMe)2]+2d, (C) [IrIII(oep)(IiPr)2]+2e and (D) [IrIII(F20tpp)(IMe)2]+2f at 30% probability of the thermal ellipsoid. Hydrogen atoms, cocrystallized solvent molecules and counteranions are omitted for clarity. Insets show the side views of the porphyrin cores and the average mesocarbon displacements (Å) from the mean porphyrin planes. | |
Table 1 Selected bond lengths (Å) and bond angles (°) of 2a, 2d, 2e and 2f
|
|
2a
|
2d
|
2e
|
2f
|
|
Ir–Nmean is the average bond distance of four Ir–Npor bonds.
|
| Ir–CNHC1 |
2.140(17) |
2.128(3) |
2.151(5) |
2.080(3) |
| Ir–CNHC2 |
2.164(18) |
2.128(3) |
2.143(5) |
2.120(3) |
| Ir–Nmeana |
2.027(11) |
2.023(3) |
2.147(16) |
1.985(17) |
| CNHC1–Ir–CNHC2 |
177.9(16) |
177.62(15) |
179.4(16) |
174.5(10) |
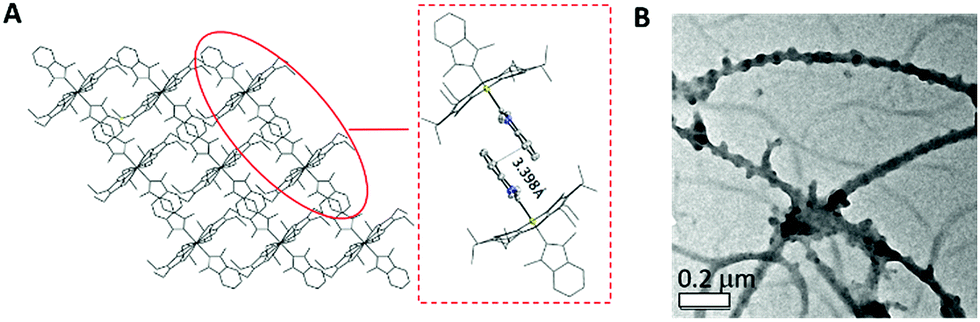
The average Ir–N distances of 2a, 2d, 2e and 2f are 2.027, 2.023, 2.017 and 1.985 Å, respectively, which are comparable to those of other organoiridium(III) porphyrin complexes.16,19 The mean Ir–CNHC distances of 2a, 2d, 2e and 2f are 2.152, 2.128, 2.147 and 2.100 Å, respectively, which are longer than those of non-porphyrin-type Ir–NHC complexes reported in the literature.20 The elongated Ir–CNHC bond in the [IrIII(por)(NHC)2]+ complexes is a consequence of the combination of (1) the strong trans influence of the NHC ligands and (2) the unfavorable steric interactions between the N-alkyl substituents on the NHC ligands and the porphyrin ring. While the influence of the N-alkyl groups of the NHC ligand on the Ir–CNHC distance is marginal (cf.2dvs.2e), the change of the porphyrin from tpp to F20tpp ligand reduces the Ir–CNHC distance by 0.052 Å (cf.2avs.2f). In comparison, the Ir–CNHC distances in [IrIII(ttp)(IMe)2]+ (2a) are discernibly shorter than that reported in [IrIII(ttp)(IBuMe)Me] (2.194(4)Å).6 This finding indicates that the methyl ligand has a stronger trans influence than NHC. In addition, the extensive intermolecular π–π stacking interactions between the molecules of 2d to form 1D polymeric chains are worth noting (Fig. 2A). The neighboring BIMe ligands are parallel to one another with an interplanar angle of 0° and a short interplanar distance of 3.398 Å. Intriguingly, the intermolecular interactions have presumably led to the self-assembled fibrous structure of 2d observed by TEM upon precipitating the complex in a THF/H2O (1:90, v/v) mixture (Fig. 2B).
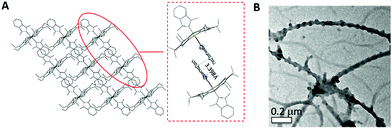 |
| | Fig. 2 (A) The crystal packing diagram of 2b viewed along the b-axis and an enlarged image of a pair of 2b molecules with an interplanar distance of 3.398 Å. (B) TEM image obtained from THF/H2O (1:90, v/v) mixture. | |
NMR study
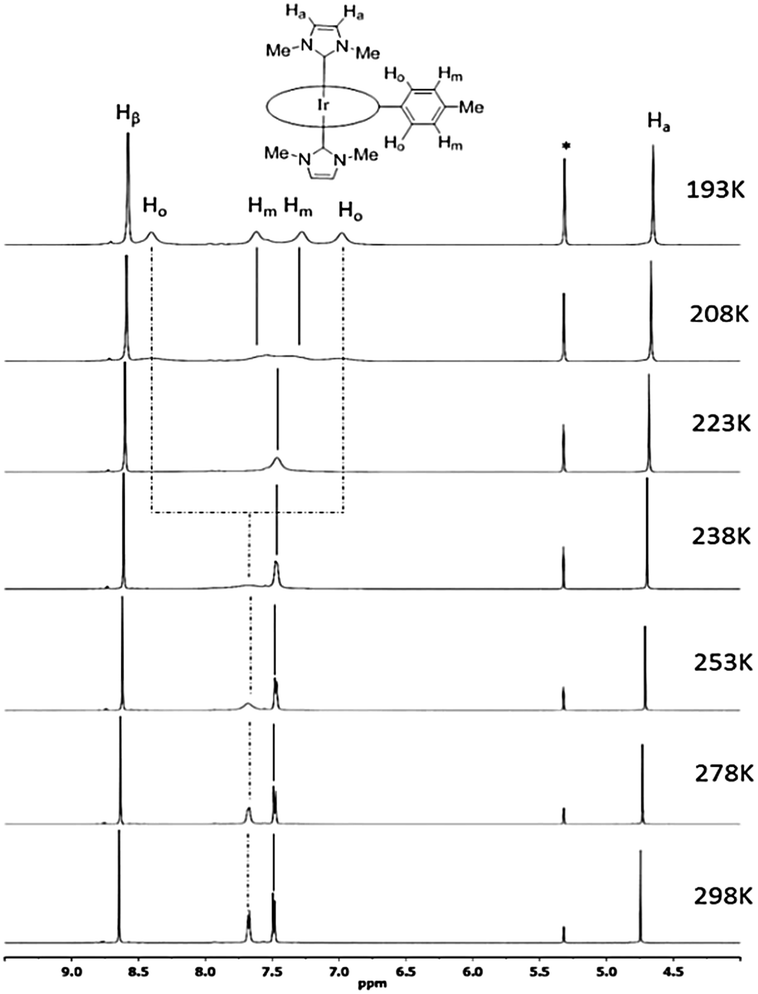
The 1H NMR spectra of all [IrIII(por)(NHC)Cl] and [IrIII(por)(NHC)2]+ complexes show upfield signals of the N-alkyl substituents on the NHC ligands at <0 ppm (TMS reference), revealing the anisotropic effect of the porphyrin ring current upon these alkyl groups. The static crystal structures of 2a, 2d, 2e, and 2f have two-fold symmetry due to the ruffling deformation of the porphyrin ligand. However, all iridium(III) porphyrin complexes in this work display a pseudo-four-fold symmetry in solution; both Hβ (when por = tmp, F20tpp or ttp) and Hmeso (when por = oep) appear to be singlet.
Metalloporphyrins are known to exhibit a variety of conformational dynamics in solution, and the dynamic processes associated with porphyrin ligands include macrocyclic inversion, meso- and β-substituent rotation.21 For the related [RhIII(tpp)(IMe)Cl] and [M(ttp)(IBuMe)Me] (M = RhIII or IrIII) complexes, meso-aryl ring rotation about the Cmeso–CAr bond was suggested to occur with the corresponding ΔGǂROT, which measured 59–63 kJ mol−1.5b,6 In this work, the 1H NMR spectra of 2a from 298 K to 193 K were recorded (Fig. 3) to study its fluxional behavior in solution. At 298 K, the Ho and Hm signals of the meso-tolyl rings appear as two well-resolved doublets, indicating that these nuclei are at their fast exchange limits. As the temperature is lowered, the Ho signal broadens and completely collapses (coalesces) at 223 K. Meanwhile, the Hm signal broadens and coalesces at 208 K. At 193 K, four new sets of signals from Ho and Hm have developed, though they are not well resolved. Notably, the two Ho signals are anomalously split by 1.4 ppm, with one of the Ho signals being farther upfield than the Hm signals. With reference to the related VT 1H NMR study of ruffled NiII–por complexes,9a the observed splitting of the Ho signal at the low-temperature limit reveals a situation in which half of Ho lies above the shielding region of the relatively “frozen” porphyrin ring, which is in a ruffled conformation. The chemical equivalence of Hβ of ttp as well as of Ha and N–Me from two IMe ligands is conserved, as these signals remain singlets throughout the experiment. Considering the symmetric bis-ligation and symmetry of the IMe ligand and the meso-tolyl group, the observed temperature-dependent NMR behavior of 2a is associated with a single dynamic process in which macrocyclic inversion is preferred over axial ligand rotation or meso-tolyl ring rotation.22 From the coalescence temperature and coalescence exchange rate (kexch) of the Ho and Hm signals, the activation barrier of the ring inversion (ΔGǂROT) of 2a is estimated to be 40 ± 1 kJ mol−1. In addition, the reversible binding of NHC ligand(s) to IrIII ion is suggested not to take place at room temperature because the 1H signals from the NHC ligand(s) (i.e., Ha and N–Me) remained unaltered throughout the VT 1H NMR experiment.6 The chemical shifts of Hβ and Hmeso as well as of carbene CNHC are summarized in Table 2. The chemical shift of Hβ or Hmeso is a useful parameter to assess the donor strength of the axial ligands.15c,23 For instance, replacing a π-accepting CO ligand with a σ-donating NHC ligand in [IrIII(tmp)(L)Cl] and [IrIII(F20tpp)(L)Cl] results in an upfield shift of the Hβ signal by 0.30–0.43 ppm. The Hmeso signals for the [IrIII(oep)(L)2]+ complexes differ by ∼0.9 ppm for L = isocyanide ligand (3) and ∼0.8 ppm for L = pyridine ligand (4) versus L = NHC ligand (2c–2e). The Hβ or Hmeso signals for [IrIII(por)(NHC)Cl] and [IrIII(por)(NHC)2]+ complexes bearing axial BIMe ligands are shifted downfield by 0.04–0.08 ppm compared to those of the IMe analogs, showing that the latter is a stronger donor. The effect of the N-alkyl substituent on the NHC ligand was found to be insignificant, as revealed by the Hmeso signals of 2c and 2e.
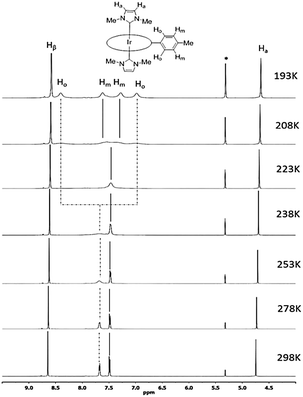 |
| | Fig. 3 Selected region of 1H NMR spectra (500 MHz) of [IrIII(ttp)(IMe)2]+2a in CD2Cl2 from 298 K to 193 K. Asterisks (*) denote residual CHDCl2 signal. | |
Table 2 Chemical shifts of Hβ and Hmeso in CDCl3
| Complex |
|
δHβa (ppm) |
δHmesoa (ppm) |
|
Values reported with reference to δH(Si(CH3)4) = 0 ppm.
Measured in CD2Cl2.
|
| [IrIII(ttp)Cl(CO)] |
|
8.96 |
— |
| [IrIII(tmp)Cl(CO)] |
|
8.66 |
— |
| [IrIII(oep)Cl(CO)] |
|
— |
10.31 |
| [IrIII(F20tpp)Cl(CO)] |
|
9.01 |
— |
| [IrIII(tmp)(IMe)Cl] |
1a
|
8.32 |
— |
| [IrIII(tmp)(BIMe)Cl] |
1b
|
8.36 |
— |
| [IrIII(F20tpp)(IMe)Cl] |
1c
|
8.58 |
— |
| [IrIII(F20tpp)(BIMe)Cl] |
1d
|
8.62 |
— |
| [IrIII(ttp)(IMe)2]+ |
2a
|
8.61 |
— |
| [IrIII(ttp)(BIMe)2]+ |
2b
|
8.69 |
— |
| [IrIII(oep)(IMe)2]+ |
2c
|
— |
9.39 |
| [IrIII(oep)(BIMe)2]+ |
2d
|
— |
9.45 |
| [IrIII(oep)(IiPr)2]+ |
2e
|
— |
9.40 |
| [IrIII(F20tpp)(IMe)2]+b |
2f
|
8.70 |
— |
| [IrIII(oep)(CNPhOMe)2]+ |
3
|
— |
10.31 |
| [IrIII(oep)(py)2]+ |
4
|
— |
10.21 |
Electrochemistry
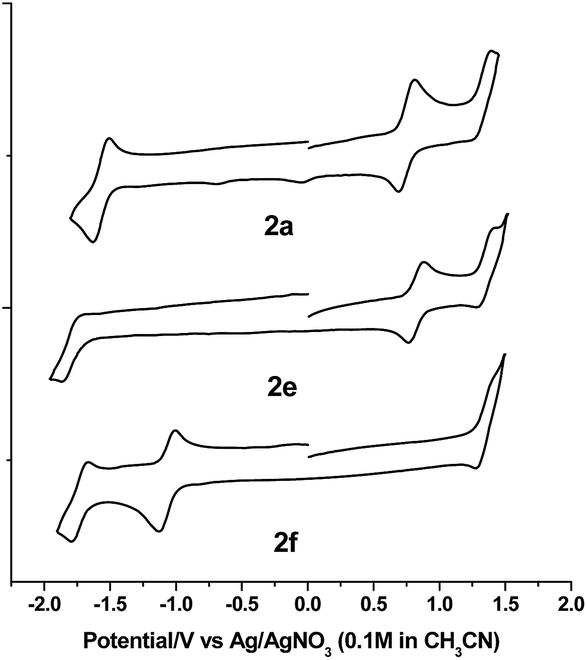
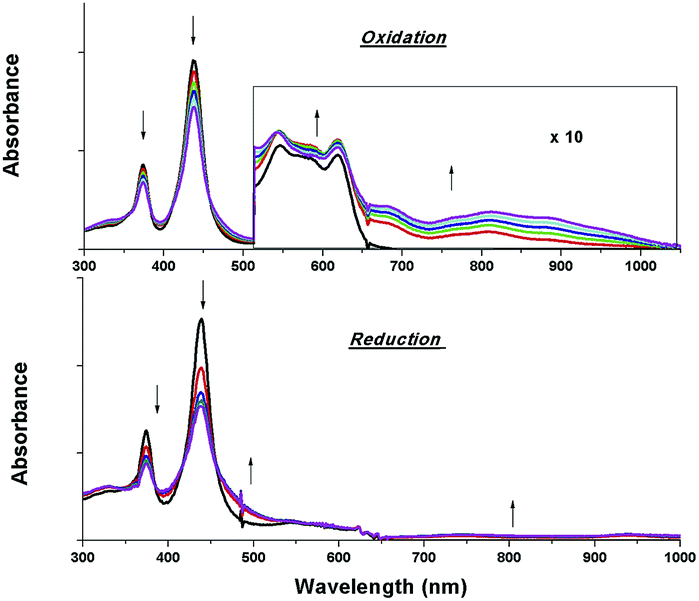
The electrochemical data and cyclic voltammograms of complexes 2a, 2e and 2f are displayed in Table 3 and Fig. 4, respectively. The quasireversible/irreversible reduction waves of these complexes appear at E1/2 = −1.31–2.01 V (vs. Ag/AgNO3). The first reversible oxidation couple occurs at E1/2 = +0.51–+1.12 V, while the second quasireversible/irreversible oxidation takes place at E1/2 = +1.15–+1.45 V. To identify the site of the first oxidation and reduction of 2a, the spectroelectrochemistry was studied. The UV-vis absorption spectra of 2a recorded at various time intervals in the first oxidation and reduction display similar spectral characteristics; the intensity of the Soret bands decrease considerably with the concomitant development of a broad IR-range absorption in the Q bands, ca. 700–1000 nm (Fig. 5). These spectral features are typical of the π-radical cation or anion of the porphyrin ring.24 On the other hand, the Eox − Ered values for the first oxidation and first reduction of the [IrIII(por)(NHC)Cl] and [IrIII(por)(NHC)2]+ complexes fall in the range of 2.33–2.61 V, which are comparable to the conventional HOMO–LUMO gap of metalloporphyrin complexes.25 Thus, both the first oxidation and first reduction are assigned to porphyrin-center processes. The E1/2 values for the IMe complexes (1a, 2a and 2c) are less anodic compared to the corresponding BIMe analogs (1b, 2b and 2d, respectively; Table 5). This difference suggests that IMe is a stronger donor ligand than BIMe, which is similar to the findings from the 1H NMR analysis. Nonplanarity of metalloporphyrins is known to engender cathodic shift of redox potential.9b,d,10a,26 However, the effect of ruffling on redox potential is negligible for 2c–2e because the redox potentials of these complexes are comparable to those of the analogous bis-isocyanide complex (3) and bis-pyridine complex (4), with ΔE being less than 0.08 V.
Table 3 Electrochemical data of 1–4a
| Complex |
|
E
red (V) |
E
ox (V) |
E
ox − Eredc (V) |
|
E
1/2 (V vs. Ag/AgNO3, scan rate = 100 mV s−1) in CH2Cl2 with 0.1 M [nBu4N]PF6 as electrolyte at r.t.; E1/2 (Cp2Fe+/0) = 0.14 V.
E
pa of irreversible wave.
Potential difference between the first oxidation and first reduction.
|
| [IrIII(tmp)(IMe)Cl] |
1a
|
−1.91 |
+0.51, +1.15 |
2.42 |
| [IrIII(tmp)(BIMe)Cl] |
1b
|
−1.87 |
+0.54, +1.19 |
2.41 |
| [IrIII(F20tpp)(BIMe)Cl] |
1d
|
−1.31 |
+1.12, +1.39 |
2.43 |
| [IrIII(ttp)(IMe)2]+ |
2a
|
−1.60 |
+0.73, +1.37b |
2.33 |
| [IrIII(ttp)(BIMe)2]+ |
2b
|
−1.48 |
+0.85,+1.45b |
2.33 |
| [IrIII(oep)(IMe)2]+ |
2c
|
−1.80 |
+0.72, +1.25 |
2.52 |
| [IrIII(oep)(BIMe)2]+ |
2d
|
−1.78 |
+0.79, +1.24b |
2.57 |
| [IrIII(oep)(IiPr)2]+ |
2e
|
−1.82 |
+0.75, +1.29 |
2.57 |
| [IrIII(F20tpp)(IMe)2]+ |
2f
|
−1.77, −1.11 |
+1.31 |
2.42 |
| [Ir(oep)(CNPhOMe)2]+ |
3
|
−1.86 |
+0.72, +1.39b |
2.58 |
| [Ir(oep)(py)2]+ |
4
|
−1.81 |
+0.80, +1.44b |
2.61 |
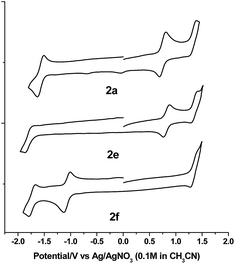 |
| | Fig. 4 Cyclic voltammograms of 2a, 2e and 2f in CH2Cl2 (scan rate = 100 mV s−1). | |
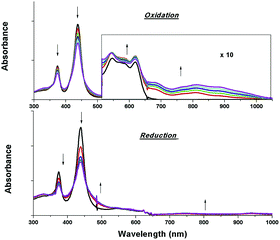 |
| | Fig. 5 UV-vis absorption spectral changes of 2a containing 0.1 M [nBu4N]PF6 during the first oxidation (top) and first reduction (bottom). Asterisk denotes an instrumental artifact. | |
UV-visible absorption spectroscopy
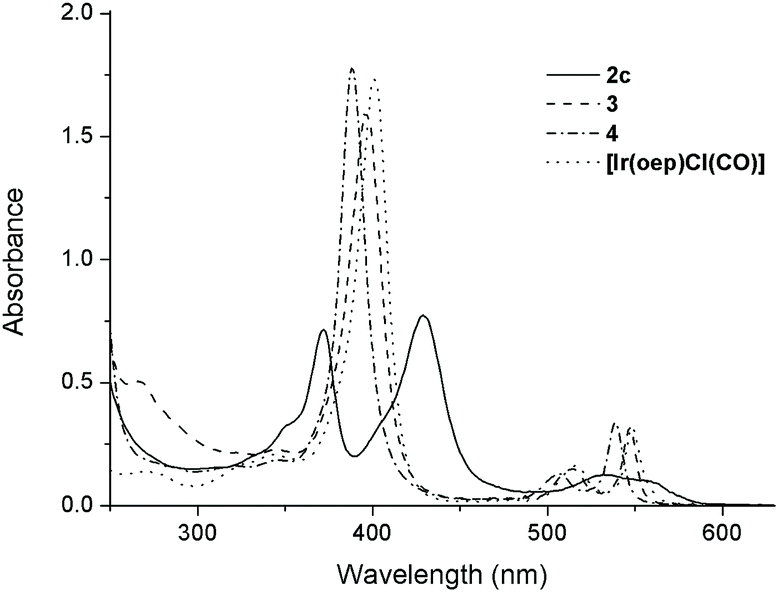
The UV-visible spectral data and photophysical data of 1–4 and their parental [IrIII(por)Cl(CO)] complexes are summarized in Table 4. The UV-vis absorption spectra of complexes 1a–1d, which bear mono-NHC ligands, exhibit a moderately intense absorption band at 367–371 nm in addition to a Soret band at 431 nm (1a and 1b, por = tmp) or 426 nm (1c and 1d, por = F20tpp) with one or two Q band(s) at 530–595 nm (Table 4). All bis(NHC)iridium(III) porphyrin complexes 2 display two intense Soret bands with peaks at 372–375 nm and 428–440 nm, respectively, with λmax of the Q band spanning from 532 to 620 nm (Table 4). The separation of the two Soret bands of 1 and 2 is 54–66 nm. This type of absorption spectrum featuring two Soret bands is classically termed a ‘hyperspectrum’.14,15cFig. 6 compares a representative hyperspectrum from 2c to ‘normal’ spectra from [IrIII(oep)Cl(CO)], 3 and 4. The degree of ‘hyper character’ can be evaluated by Δlogε between the two Soret bands (i.e., the larger Δlogε, the greater the ‘hyper character’ is). Thus, the hyper character of the bis-NHC complexes 2 is generally stronger than that of the mono-NHC complexes 1. Moreover, the hyper character appears to be a function of the porphyrin ligand; Δlogε ranges from 0.36 for 2a and 2b (por = ttp) to 0.11 for 2f (por = F20ttp) and 0–0.06 for 2c–2e (por = oep).
Table 4 UV-visible absorption and emission data of [IrIII(por)Cl(CO)] and 1–4
| Complex |
UV-vis absorption dataa |
Emission dataa |
|
λ
max nm−1 (logε) |
Solution at 298 K |
|
λ
max/nm (τ/μs) |
Φ
em [×10−2] |
|
Measurements were performed in degassed CHCl3.
Complex 1d was unstable for emission measurements.
Broad absorption band spans from ca. 510 to 670 nm.
Measurements were performed in degassed CH2Cl2.
|
| [IrIII(ttp)Cl(CO)] |
|
309 (4.50), 422 (5.58), 533 (4.41), 567 (3.89) |
724 (47.2), 808 (sh) |
0.16 |
| [IrIII(tmp)Cl(CO)] |
|
311 (4.43), 422 (5.58), 533 (4.42), 564 (3.60) |
721 (77.2), 800 (sh) |
0.22 |
| [IrIII(oep)Cl(CO)] |
|
342 (4.44), 403 (5.38), 518 (4.30), 549 (4.62) |
666 (83.3), 722 (sh) |
5.03 |
| [IrIII(F20tpp)Cl(CO)] |
|
329 (4.40), 414 (5.42), 526 (4.45), 557 (3.94) |
676 (101.0), 746 (sh) |
2.92 |
| [IrIII(tmp)(IMe)Cl] |
1a
|
345 (sh) (4.29), 368 (4.56), 431 (5.18), 542 (4.06), 593 (3.40) |
773 (1.8) |
0.05 |
| [IrIII(tmp)(BIMe)Cl] |
1b
|
301 (4.55), 367 (4.61), 431 (5.23), 545 (4.13), 592 (3.92) |
775 (2.3) |
0.06 |
| [IrIII(F20tpp)(IMe)Cl] |
1c
|
346 (sh) (4.44), 371 (4.62), 426 (5.10), 533 (4.12) |
—b |
—b |
| [IrIII(F20tpp)(BIMe)Cl] |
1d
|
296 (4.41), 346 (sh) (4.39), 369 (4.55), 426 (5.10), 535 (4.12) |
729 (1.7), 811 (sh) |
0.29 |
| [IrIII(ttp)(IMe)2]+ |
2a
|
374 (4.76), 438 (5.12), 547 (3.87), 620 (3.82) (br)c |
Nonemissive |
— |
| [IrIII(ttp)(BIMe)2]+ |
2b
|
297 (4.63), 372 (4.63), 437 (4.99), 564 (3.85) (br)d, 611 (3.87) (br)c |
Nonemissive |
— |
| [IrIII(oep)(IMe)2]+ |
2c
|
351 (sh) (4.52), 372 (4.86), 429 (4.89), 532 (4.10), 556 (4.01) |
701 (2.7), 766 (sh) |
0.41 |
| [IrIII(oep)(BIMe)2]+ |
2d
|
297 (4.42), 373, (4.76), 428 (4.82), 533 (4.03), 557 (3.95) |
701 (3.6), 765 (sh) |
0.66 |
| [IrIII(oep)(IiPr)2]+ |
2e
|
353 (sh) (4.49), 375 (4.82), 432 (4.82), 537 (4.09), 560 (4.02) |
701 (1.9), 763 (sh) |
0.03 |
| [IrIII(F20tpp)(IMe)2]+ |
2f
|
355 (sh) (4.63), 374 (4.89), 418 (sh) (4.72), 440 (5.00), 537 (4.13) |
754 (0.5), 829 (sh) |
0.08 |
| [IrIII(oep)(CNPhOMe)2]+ |
3
|
266 (4.70), 344(sh) (4.36), 395 (5.20), 515 (4.17), 547 (4.50) |
663 (124.7), 717(sh) |
6.40 |
| [IrIII(oep)(py)2]+ |
4
|
388 (5.25), 507 (4.11), 539 (4.54) |
651(55.2), 711(sh) |
12.30 |
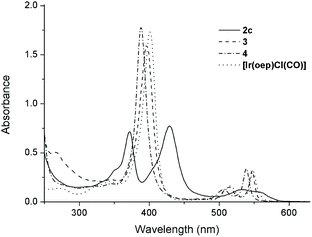 |
| | Fig. 6 UV-vis absorption spectra of 2c, 3, 4 and [Ir(oep)Cl(CO)] in CHCl3 (1 × 10−5 M). | |
In addition to the presence of an extra Soret band, the Soret and Q bands of the NHC complexes are broadened compared to those of their parental [IrIII(por)Cl(CO)]. Moreover, a sizable redshift of the low-energy Soret band is noted. For example, in the oep series, the low-energy Soret band of 2c is redshifted by 26, 34 and 41 nm versus those of [IrIII(oep)Cl(CO)], 3 and 4, respectively. The same trend holds for the cases of 1c, 1d and 2fversus their parental [IrIII(F20tpp)Cl(CO)] complex, where Δλmax (redshift) are 12, 12 and 26 nm, respectively (Fig. S14†).
Complexes 1b, 1d, 2b and 2d, which contain BIMe ligand(s), display an exclusive absorption of moderate intensity (logε = 4.41–4.63) at λmax = 296–301 nm, which can be attributed to π → π* transition of the BIMe ligand. The absorption λmax of the Soret band is relatively insensitive to the NHC ligand (i.e., IMe, BIMe, IPr), and Δλmax is less than 4 nm in all cases for complexes with the same porphyrin scaffold. In addition, solvatochromism was found to be negligible for 2e (Table S2 and Fig. S15†). The variation in λmax of the Soret and Q bands in various solvents is less than 3 nm. These findings indicate predominantly porphyrin-based frontier molecular orbitals (FMOs) for IrIII(por)–NHC complexes despite the nonplanarity of the porphyrin ring.
TD-DFT calculations on split Soret bands
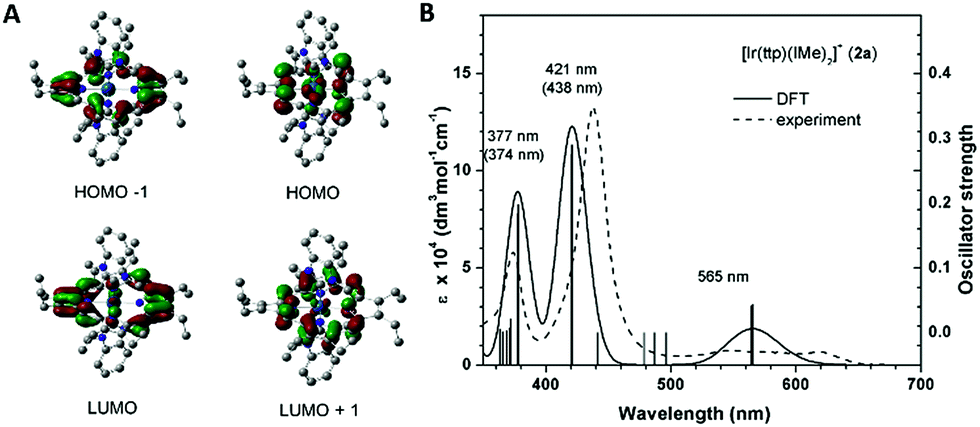
To unveil the nature of the split Soret bands, time-dependent density functional theory (TD-DFT) calculations on the electronic transitions of 2a were performed. The geometry of the ground-state singlet (S0) was optimized, and vertically excited states were obtained via TD-DFT calculations. Notably, the composition and spin density distribution of the FMOs of 2a are in excellent agreement with the classical four-orbital model proposed by Gouterman;14 the contributions from the porphyrin ligand to HOMO−1, HOMO, LUMO and LUMO+1 amount to 99.8%, 95.5%, 97.5% and 97.7%, respectively (Fig. 7A). The simulated UV-vis absorption spectrum of 2a, which shows high resemblance to the one obtained experimentally, is depicted in Fig. 7B. The calculations show that the Q bands at 565 nm originated mainly from HOMO → LUMO (S1, 79%) and HOMO → LUMO+1 (S2, 79%), which can be categorized as IL π → π* transitions of porphyrin. Nevertheless, assigning the nature of the transitions that constitute the low-energy (412 nm, S7/S8) and high-energy Soret bands (377 nm, S9/S10) is not straightforward, as both Soret bands consist of several vertical transitions involving 4 to 6 pairs of molecular orbitals. Accordingly, we examined the natural-transition orbitals (NTOs), which can provide a much more compact description of the excitations than the MOs. As listed in Table S3,† the excitation S7/S8, which relates to the low-energy Soret band, can be described as 68% porphyrin-based π–π* transition and 32% MLCT, while S9/S10, in association with the higher-energy Soret band, are attributed to MLCT transition mixed with intraligand charge transfer (ILCT) character.
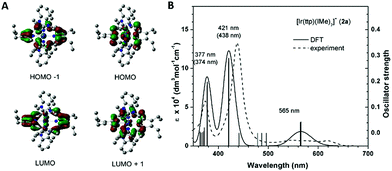 |
| | Fig. 7 (A) Frontier molecular orbitals (HOMO−1, HOMO, LUMO and LUMO+1), (B) simulated (solid line) and experimental (dashed line) UV-vis absorption spectra of 2a (number in parenthesis is the experimental abs λmax). | |
Resonance Raman spectroscopy
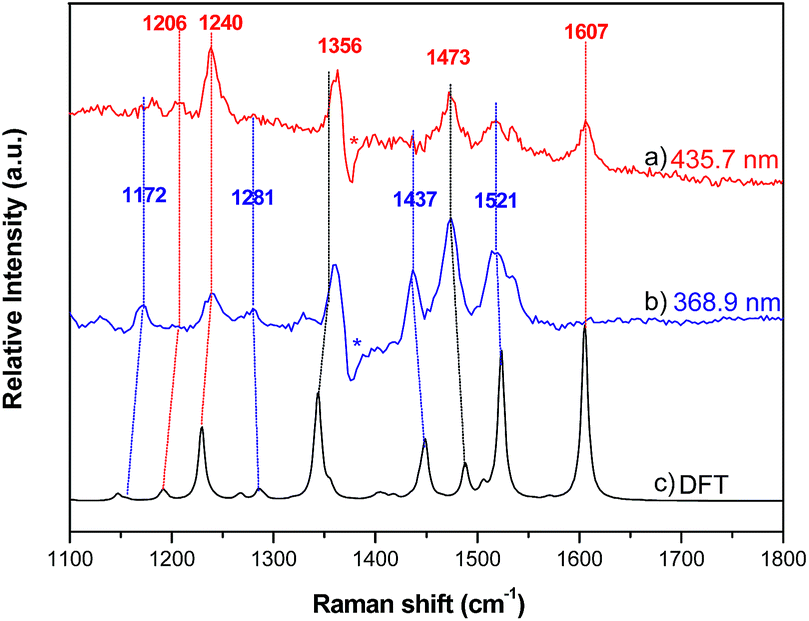
Resonance Raman (RR) spectroscopy is an effective method to probe the nature of an electronic transition because the vibrations associated with the resonant chromophore can be selectively enhanced.27 To help verify the parentage of the split Soret bands, the RR spectra of 2a were recorded using excitation at 368.9 and 435.7 nm in MeCN. The former excitation wavelength is within the higher-energy Soret band, while the latter excitation wavelength is covered by the lower-energy Soret band. The experimental RR spectra and simulated ground-state Raman spectrum of 2a from 1100–1800 cm−1 are shown in Fig. 8, and the assignment of vibrational bands is listed in Table S4.† The calculated Raman frequencies match well with the experimental ones. The RR spectra exhibit different intensity enhancements in response to excitation at 435.7 and 368.9 nm. The band at 1607 cm−1, which results from C–C bond stretching within the tolyl rings and rocking of their hydrogen atoms without any contribution from the pyrrole rings, appears only under excitation at 435.7 nm. The other two enhancements at 1240 and 1206 cm−1 also result largely from the vibrational modes of the tolyl rings. The resonance in the excited state associated with the low-energy Soret band (435.7 nm) enhances the vibrational modes associated with tolyl rings more than those associated with pyrrole rings. In the case of excitation at 368.9 nm, four vibrational bands are relatively enhanced, namely, those at 1172, 1281, 1437 and 1521 cm−1. Vibrational mode analysis shows that these four Raman bands all contain major components of the vibrational modes within the pyrrole rings. For example, the Raman band at 1437 cm−1 is mainly from symmetric stretching of the Cα–Cmeso–Cα bonds and bending deformation of pyrrole rings, including rocking of the hydrogen atoms within the pyrrole rings. Thus, the resonance in the excited state associated with the higher-energy Soret band enhances the vibrational modes in pyrrole rings more than those in tolyl rings. The Raman bands that appear at 1356 and 1473 cm−1 under both excitation wavelengths (435.7 nm and 368.9 nm) are attributed to vibrational modes from both tolyl rings and pyrrole rings.
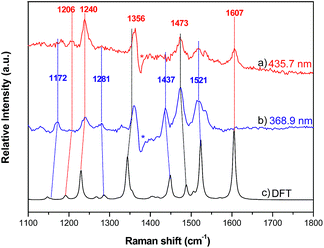 |
| | Fig. 8 Resonance Raman spectra of 2a with excitation wavelengths of (a) 435.7 nm and (b) 368.9 nm; (c) Raman spectrum of 2a calculated by DFT. | |
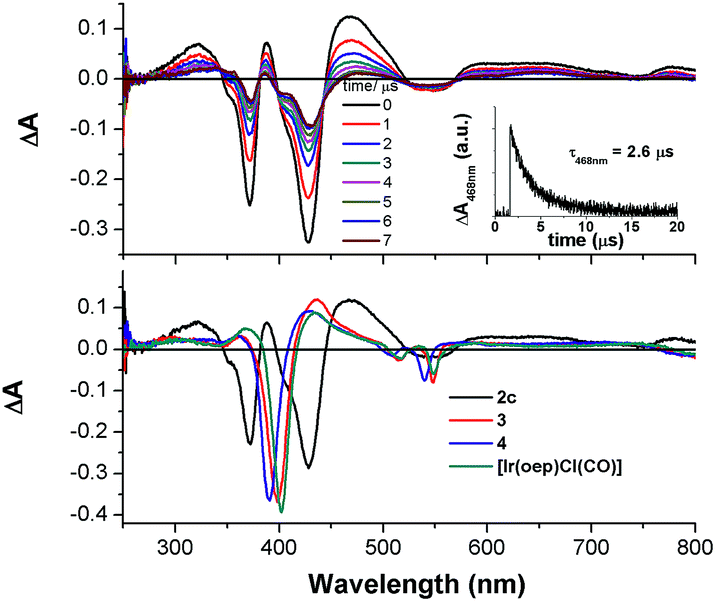
Nanosecond time-resolved absorption spectroscopy
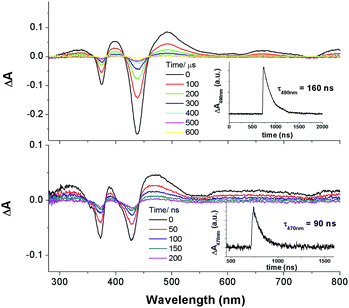
The nanosecond time-resolved absorption difference (ns-TA) spectrum of 2c, as a representative of bis-NHC complexes, is examined, and the following spectral features are noted (Fig. 9, top): (1) there are two intense positive absorption peaks at 388 and 468 nm, respectively, which are redshifted from the ground-state double Soret bands; (2) a broad, moderately intense positive absorption lies from ca. 550 nm to 800 nm. Indeed, despite a marked redshift of the excited-state Soret band by over 30 nm and a shorter-lived triplet state decay (τ468nm = 2.6 μs), the ns-TA spectrum of 2c resembles the spectral profile of its counterparts 3, 4, and [IrIII(oep)Cl(CO)] (Fig. 9, bottom).
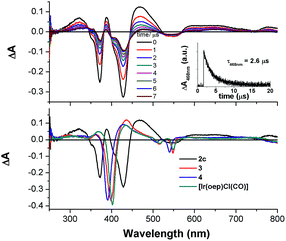 |
| | Fig. 9 (Top) The ns-TA spectra of 2c evolved from 0 to 7 μs; inset: Decay kinetics of absorption difference at 468 nm. (Bottom) Overlaid ns-TA spectra of 2c, 3, 4, and [Ir(oep)Cl(CO)] recorded at 0 μs. | |
Emission spectroscopy
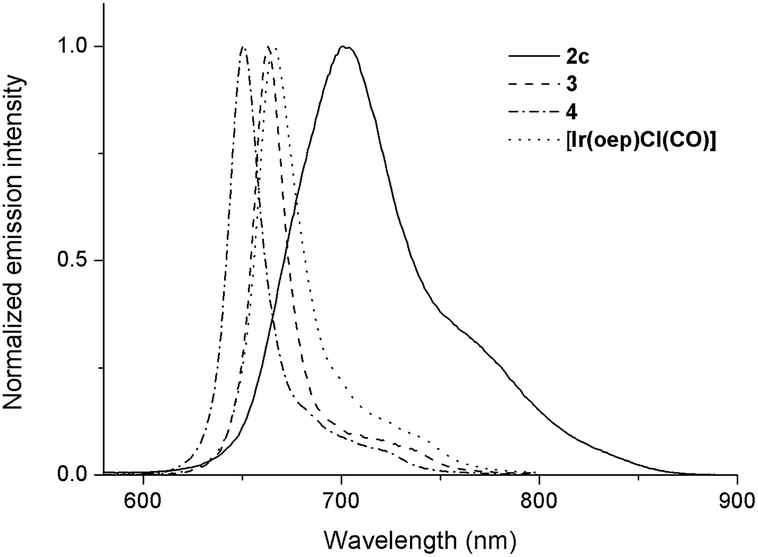
Upon photoexcitation, complexes 1 and 2 (except 2a and 2b) produce NIR emission with λmax spanning from 701 to 775 nm in degassed CHCl3 solution (degassed CH2Cl2 for 2f) at room temperature. The emission is assigned to the 3(π, π*) state of porphyrin; the emission spectra generally feature a broad though indistinct vibronic shoulder, akin to that of typical phosphorescent metalloporphyrins.16,28 Several characteristics can be noted for the emissions of complexes 1 and 2. (1) Their emission bands are much broader and are redshifted with respect to those of their counterparts. For instance, the Full-Width-at-Half-Maximum (FWHM) for 2c is two to three times greater than those of [IrIII(oep)Cl(CO)], 3 and 4 (i.e., 66 nm vs. 26, 19 and 20 nm, respectively). In addition, the emission λmax of 2c is 701 nm, which is redshifted by more than 35 nm compared to those of its counterparts (Fig. 10). (2) The solution emission quantum yields and lifetimes at room temperature are substantially lower and shorter, respectively. For example, the Φem values of 2c–2e supported by an oep ligand are only 0.03–0.66%, and τ ranges from 1.9 to 3.6 μs. This result is in stark contrast to the observations of [IrIII(oep)Cl(CO)] and of the bis-ligand counterparts 3 and 4, all of which show long-lived (τ = 55.2–124.7 μs) and strongly emissive 3(π, π*) states of porphyrin (Φem = 5.03–12.30%). Likewise, these observations hold for complexes supported by F20tpp ligands 1d and 2fversus their parent (Φem = 0.08–0.29% vs. 2.92%; τ = 0.5–1.7 vs. 101.0 μs). (3) Modification of NHC ligand exerts a negligible effect on both the emission profile and emission λmax, as demonstrated for 2c–2e (Fig. S16†). (4) Increasing the number of NHC ligands, as shown in the case of [IrIII(F20tpp)Cl(CO)], 1d and 2f, results in a progressive redshift in the emission λmax from 676 to 754 nm (Fig. S17†). (5) Varying the porphyrin ligand causes the emission λmax to range from 701 to 775 nm, and λmax was found to increase in the order oep < F20tpp < ttp ≈ tmp. Similar findings have previously been reported for PdII- and PtII-porphyrin complexes.28
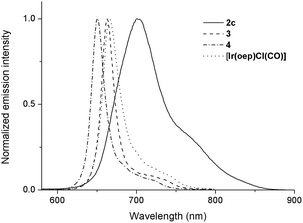 |
| | Fig. 10 Normalized emission spectra of 2c, 3, 4 and [IrIII(oep)Cl(CO)] in degassed CHCl3. | |
Photochemistry
Recently, a panel of IrIII–porphyrin16d and IrIII–corrole29 complexes were found to be efficient singlet oxygen photosensitizers (Table 5).
Table 5 The quantum yield of singlet oxygen production (Φso) by 2a, 2c and reported IrIII–porphyrin and corrole complexes
| Complex |
Φ
so
|
|
Measured in CHCl3 with reference to the 1O2 emission intensity of H2tpp.
Measured in CH2Cl2 with reference to the photooxidation of 1,3-diphenylisobenzofuran by methylene blue.
|
|
2a
|
0.64a |
|
2c
|
0.88a |
| [IrIII(corrole)(pyridine)2] |
0.09–0.15a29 |
| [IrIII(ttp)(aza-BODIPY)] |
0.79–0.85b16d |
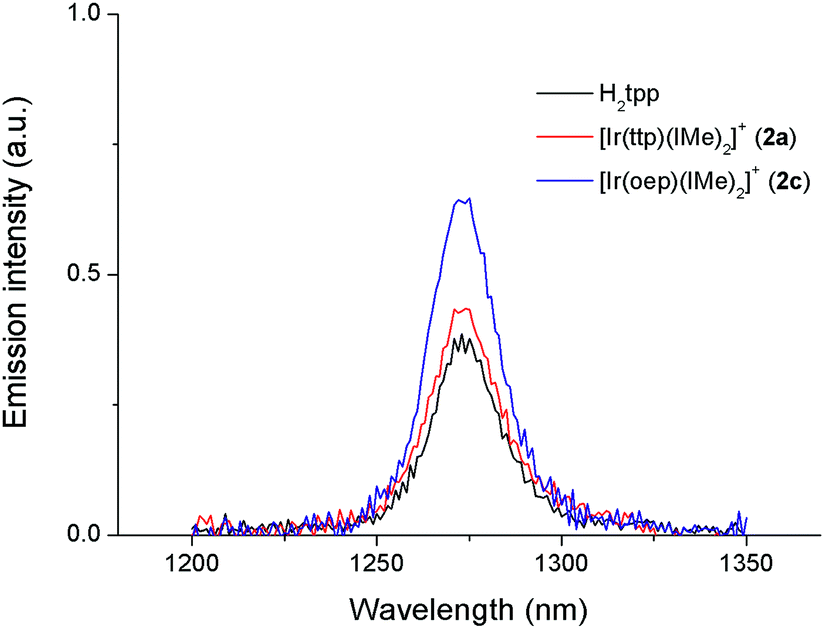
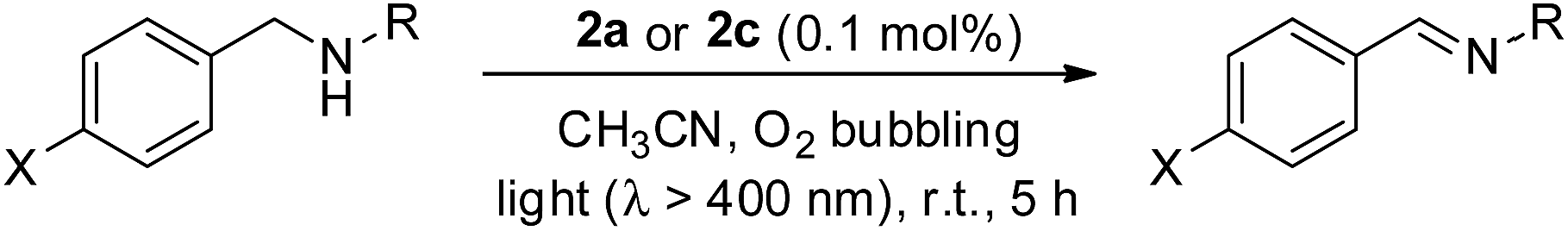
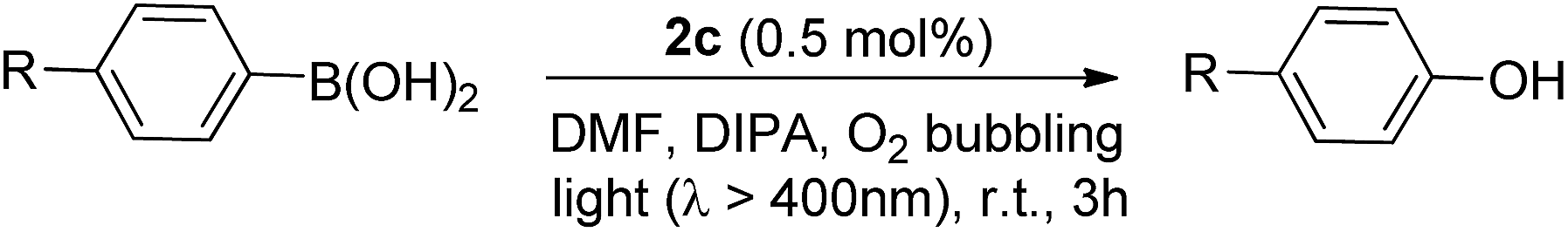
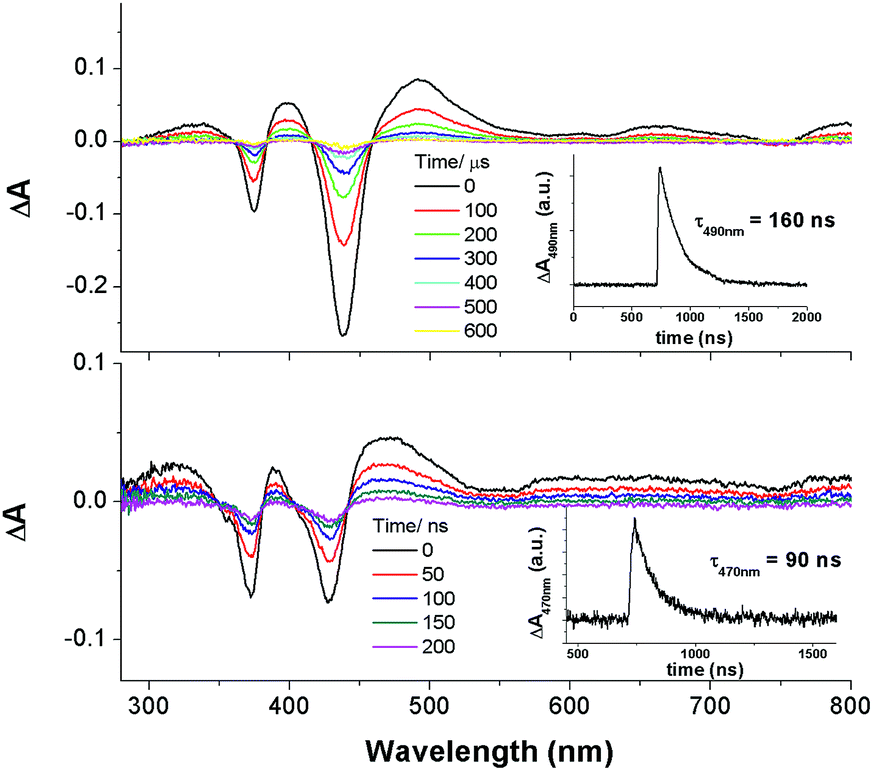
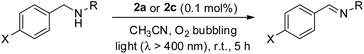
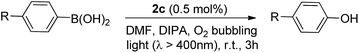
In particular, the quantum yield of singlet oxygen production (Φso) by BODIPY conjugates of IrIII-ttp upon excitation at 690 nm is up to 0.85. In this work, [IrIII(por)(NHC)2]+ complexes were also found to be highly efficient photosensitizers for singlet oxygen production. With reference to the 1O2 emission intensity (λmax = 1270 nm) of H2tpp (Φso = 0.55 in CHCl3),30 the Φso values of oxygen-saturated CHCl3 solutions of 2a and 2c were found to be 0.64 and 0.88, respectively, upon excitation at their Soret bands (Fig. 11). As described in the previous section, upon bis-ligation of the NHC ligand to iridium porphyrin, complexes 2 show redshifted and broadened Soret and Q bands, resulting in broad coverage of the visible light spectral region. This spectral feature and the high singlet oxygen quantum yield enable 2 to act as a photosensitizer for catalytic aerobic oxidation reactions and prompted us to examine the aerobic oxidation of secondary amines and arylboronic acids. When a solution of dibenzylamine treated with 0.1 mol% of 2a was bubbled with O2 under light irradiation (λ > 400 nm) at room temperature for 5 h, the corresponding imine was produced in 92% yield with complete substrate conversion (entry 1, Table 6). Changing the photosensitizer to 2c led to a higher product yield of 99% (entry 2, Table 6). The reaction also worked for substrates with halogen/methyl substituted benzyl groups and tert-butyl groups, furnishing the respective imine products in 96–99% yields (entry 3–5, Table 6). In another reaction, irradiating (λ > 400 nm) solutions of para-substituted arylboronic acids in the presence of diisopropylamine and 0.5 mol% of 2c for 3 h at room temperature gave rise to the corresponding aryl alcohols in yields of 82 to 99% (Table 7). The control experiments indicated the necessity of 2a or 2c for both reactions (entry 6, Table 6; entry 6, Table 7). The mechanisms of the photochemical reactions were investigated by experiments on the quenching of 2a or 2c. The presence of a large excess (1000 equiv.) of amine (i.e., dibenzylamine and diisopropylamine) or arylboronicacid (i.e., 4-chlorophenylboronic acid) did not have significant effect on the lifetimes of the photogenerated 2a* and 2c* under degassed conditions (Fig. S19†). Nevertheless, the lifetimes of 2a* and 2c* were considerably reduced to 0.16 μs and 0.09 μs in oxygen-saturated solution, respectively (Fig. 12). On the basis of these findings, both photoinduced aerobic amine oxidation and arylboronic acid oxidation reactions begin with the oxygen quenching of photogenerated 2a*/2c* by energy transfer to give 1O2, which subsequently oxidizes the secondary amines to imines or the arylboronic acids to aryl alcohols.
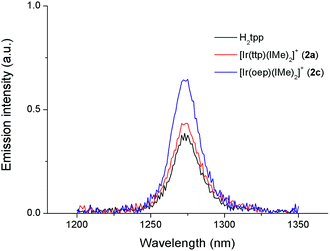 |
| | Fig. 11 Singlet oxygen emission spectra of H2tpp, 2a and 2c in O2-saturated CHCl3. | |
Table 6 Light-induced aerobic oxidation of secondary amine to imine, catalyzed by 2a or 2ca
|
|
| Entry |
PS |
X/R |
Convn.b (%) |
Yieldc (%) |
|
Reaction conditions: dibenzylamine (0.1 mmol) and catalyst (0.1 μmol) in MeCN (1.5 mL), O2 bubbling, xenon lamp (>400 nm).
Determined by 1H NMR analysis of the crude reaction mixture.
Determined by 1H NMR analysis using 1,1-diphenylethylene as an internal standard.
n.d. = not determined.
|
| 1 |
2a
|
H/Bn |
100 |
92% |
| 2 |
2c
|
H/Bn |
100 |
99% |
| 3 |
2c
|
Cl/tBu |
100 |
99% |
| 4 |
2c
|
Br/tBu |
100 |
99% |
| 5 |
2c
|
Me/tBu |
100 |
96% |
| 6 |
— |
H/Bn |
<5% |
n.d.d |
Table 7 Light-induced aerobic oxidation of arylboronic acid catalyzed by 2ca
|
|
| Entry |
R |
Convn. (%)c |
Yield (%)d |
|
Reaction conditions: arylboronic acid (0.1 mmol), diisopropylamine (0.4 mmol) and 2c (0.5 μmol) in DMF (1.5 mL), O2 bubbling, xenon lamp (>400 nm).
No 2c added.
Determined by 1H NMR analysis of the crude reaction mixture.
Determined by 1H NMR analysis using 1,1-diphenylethylene as an internal standard.
n.d. = not determined.
|
| 1 |
Cl |
100 |
92% |
| 2 |
Br |
100 |
98% |
| 3 |
CHO |
100 |
98% |
| 4 |
Ph |
100 |
82% |
| 5 |
CN |
100 |
99% |
| 6b |
Br |
<5% |
n.d.e |
 |
| | Fig. 12 Nanosecond time-resolved absorption difference spectra of 2a in O2-saturated MeCN (top) and 2c in O2-saturated DMF (bottom). Inset: decay kinetic of absorption difference at 490 nm (top) and 470 nm (bottom). | |
Anticancer properties
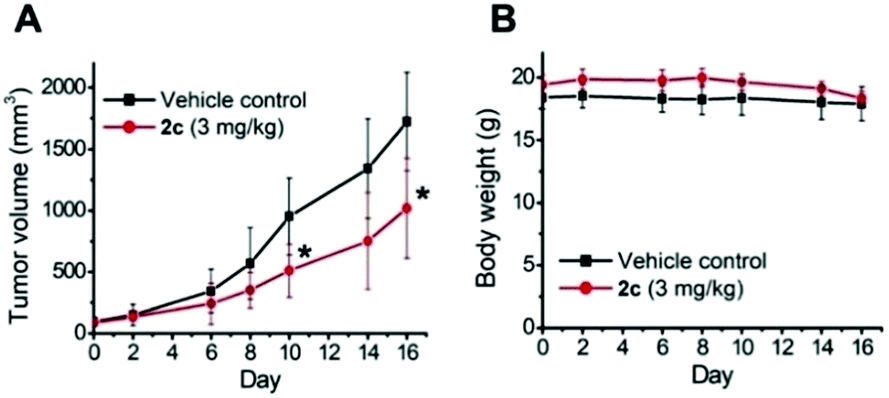
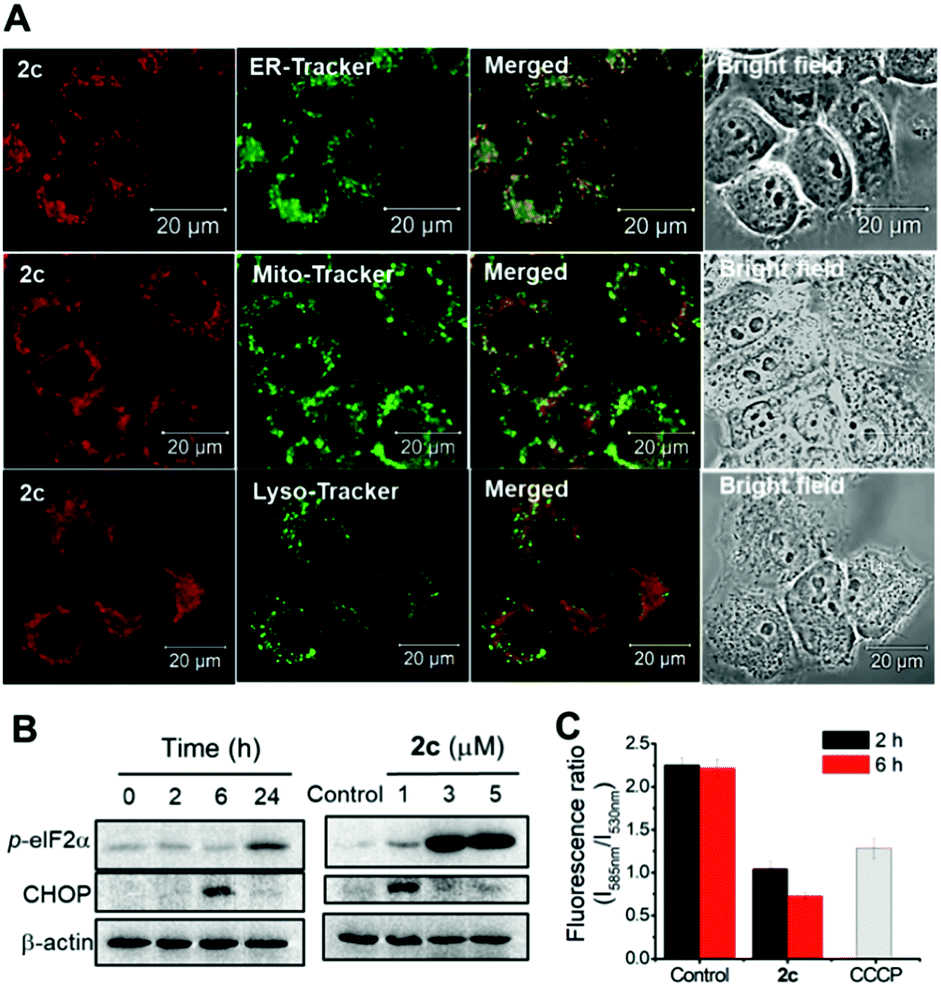
We have previously reported various cationic metalloporphyrins, particularly gold(III) porphyrins, and metal–NHC complexes that exhibit promising anticancer and antitumor activities.31 In this work, iridium(III) porphyrin complexes bearing NHC ligand(s) were found to be stable against ligand exchange with DMSO or glutathione up to an 24 h incubation, as determined by UPLC-QTOF mass spectrometry (Fig. S21 and S22†). DMSO-substituted forms (ca. 8%) were found for 3 (Fig. S21f†). All of the cationic iridium(III) complexes exhibit potent cytotoxicity towards a panel of human cancer cell lines with submicromolar IC50 values, while 3 shows generally reduced cytotoxicity (Table 8). Among the bis-NHC complexes, the higher cellular uptake of 2c (with octaethylporphyrin) than that of 2a (with meso-tetratolylporphyrin) and 2f (with meso-tetrakis(pentafluorophenyl)porphyrin) results in higher cytotoxicity towards different cancer cell lines. In addition, the charge-neutral [IrIII(oep)Cl(CO)], which showed the lowest cellular uptake and lipophilicity, is relatively noncytotoxic, with an IC50 value >50 μM to all the cancer cell lines. These findings highlight that the cationic character and porphyrin scaffold of iridium(III) bis-NHC complexes are crucial for facilitating efficient accumulation in cells for anticancer activities. On the basis of the potent in vitro cytotoxicity of 2c, its in vivo antitumor properties were examined. Nude mice bearing NCI-H460 human non-small cell lung cancer xenografts were administered 2c (3 mg kg−1) via intravenous injection thrice weekly. The tumor size was reduced by 41% after a 16 day treatment without apparent toxicity, including body weight loss and death (Fig. 13). In view of the advantageous photophysical properties (including high emission quantum yield and long-lived electronic excited state) of 2c among the bis-NHC iridium(III) complexes, cellular imaging of 2c was performed to examine its subcellular localization in NCI-H460 cells. As shown in Fig. 14A, the resulting images clearly revealed that the red-emitting 2c mainly colocalized with the green-emitting stain of the endoplasmic reticulum (ER-Tracker) with a high Pearson's correlation coefficient (R) of 0.903. In contrast, a relatively poor overlap was observed between the fluorescence images of the complex and Mito-Tracker green or Lyso-Tracker green, with an R-value of 0.548 and 0.210. These findings indicated that 2c accumulated mainly in the ER and only somewhat in the mitochondria or lysosome in NCI-H460 cells. We therefore examined if 2c induced ER stress to the NCI-H460 cells.32 By the western blot analysis, the ER stress-associated proteins including CHOP and phosphorylated eIF2α were found to be dose- and time-dependently upregulated upon treatment with 2c compared to the DMSO vehicle (Fig. 14B), confirming the induction of ER stress. The impact of 2c on mitochondria was also examined by monitoring the changes in mitochondrial membrane potential (MMP, ΔΨm). With the use of a MMP-dependent ratiometric fluorescence probe (JC-1), time-dependent decrease in MMP of NCI-H460 cells was shown upon the treatment of 2c, as indicated by the progressive change in fluorescence from red to green compared with DMSO vehicle (Fig. 14C).
Table 8
In vitro cytotoxicity of the selected iridium(III) porphyrin complexes against a panel of human cancer cell linesabc
|
|
IC50 (μM)b |
IC50 (μM) |
PIc |
Uptaked |
Log Pe |
| HeLa |
HepG2 |
MCF-7 |
HCT116 |
HCC827 |
NCI-H460 (dark) |
NCI-H460 (light) |
|
HeLa, cervical epithelial carcinoma; HepG2, hepatocellular carcinoma; MCF-7, breast carcinoma; HCT-116, colorectal carcinoma; HCC827, non-small cell lung carcinoma; NCI-H460, non-small cell lung carcinoma.
IC50 values were examined by MTT assay after incubation of drugs for 72 h.
PI = IC50(dark)/IC50(light).
Cellular uptake was determined by the iridium content (μg) in the cellular proteins (g) after treatment of the NCI-H460 cells with each complex (1 μM) for 2 h.
Lipophilicity was determined by measuring the ratio of the amount of iridium (μg) in each complex partitioned in n-octanol and saline solution (0.9%, w/v).
|
|
1d′
|
0.37 ± 0.10 |
1.60 ± 0.21 |
0.71 ± 0.04 |
0.42 ± 0.06 |
0.55 ± 0.11 |
0.46 ± 0.10 |
0.04 ± 0.003 |
11.5 |
235.9 ± 9.81 |
2.67 |
|
2a
|
0.17 ± 0.1 |
2.1 ± 0.3 |
0.65 ± 0.2 |
0.14 ± 0.04 |
2.29 ± 0.50 |
1.22 ± 0.03 |
0.11 ± 0.08 |
11.1 |
147.6 ± 6.4 |
3.03 |
|
2c
|
0.03 ± 0.01 |
0.93 ± 0.1 |
0.16 ± 0.1 |
0.14 ± 0.1 |
1.1 ± 0.3 |
0.15 ± 0.05 |
0.009 ± 0.004 |
16.7 |
309.7 ± 46.7 |
2.96 |
|
2e
|
0.10 ± 0.1 |
2.4 ± 0.2 |
0.73 ± 0.5 |
0.11 ± 0.03. |
0.69 ± 0.01. |
0.16 ± 0.04 |
0.006 ± 0.002 |
26.7 |
158.7 ± 29.5 |
3.23 |
|
2f
|
0.10 ± 0.1 |
0.94 ± 0.1 |
0.26 ± 0.1 |
0.4 ± 0.2 |
1.23 ± 0.10 |
0.31 ± 0.04 |
0.03 ± 0.002 |
10.3 |
131.3 ± 10.2 |
2.76 |
|
3
|
7.9 ± 0.5 |
>100 |
>100 |
23 ± 3.3 |
50 ± 3.6 |
2.9 ± 0.02 |
0.12 ± 0.02 |
24.2 |
116.2 ± 20.6 |
3.11 |
|
4
|
0.10 ± 0.01 |
1.32 ± 0.10 |
0.12 ± 0.02 |
0.25 ± 0.08 |
1.27 ± 0.32 |
0.11 ± 0.005 |
0.005 ± 0.001 |
23.1 |
305.1 ± 57.2 |
2.83 |
| [IrIII(oep)Cl(CO)] |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
19.70 ± 1.0 |
>2.5 |
34.1 ± 2.8 |
2.38 |
| Cisplatin |
3.80 ± 0.51 |
6.18 ± 0.82 |
13.20 ± 1.03 |
6.94 ± 0.54 |
9.61 ± 0.81 |
3.96 ± 0.48 |
4.61 ± 0.31 |
0.86 |
— |
— |
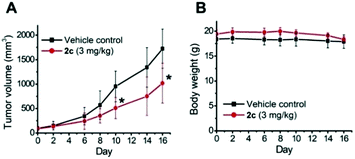 |
| | Fig. 13
In vivo antitumor activity of complex 2c in nude mouse model bearing NCI-H460 xenograft. (A) Tumor volume and (B) body weight of mice. Data are expressed as the mean ± standard error; *p < 0.05. | |
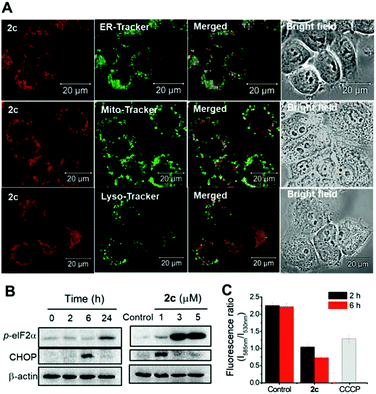 |
| | Fig. 14 (A) Confocal microscopy imaging of NCI-H460 cells treated with complex 2c (1 μM; λex = 555 nm, λem = 650–750 nm) for 2 h and organelle trackers, including ER-Tracker green, Mito-Tracker green and Lyso-Tracker green (50 nM, λex = 488 nm, λem = 500–530 nm) for 15 min. The merged images of red and green channels, and the bright field were also shown. Scale bar: 20 μm. (B) Western blot analysis of ER stress protein markers in NCI-H460 cells after treatment with 2c in time- and dose-dependent manner. (C) Effect of 2c (1 μM) on mitochondrial membrane potential (MMP) analyzed by the fluorescence ratio of NCI-H460 cells stained with JC-1 (λex = 488 nm, λem = 530 nm (green) and 585 nm (red)). Treatments with carbonylcyanide m-chlorophenylhydrazone (CCCP, 50 μM, 10 min) and DMSO were served as positive and vehicle controls, respectively. | |
Photocytotoxicity
Prompted by the finding that the iridium(III) porphyrin complexes are excellent singlet oxygen photosensitizers, we examined their photocytotoxicity. NCI-H460 lung cancer cells incubated with the complexes were exposed to a low dose of visible light irradiation (2.8 mW cm−2) for 1 h. The cytotoxicity of the iridium(III) porphyrin complexes increased markedly by 10- to 27-fold upon irradiation, and [IrIII(oep)(IiPr)2]+ (2e) showed the largest enhancement among the complexes examined (Table 8 and Fig. S23†). [IrIII(oep)(IMe)2]+ (2c), [IrIII(oep)(IiPr)2]+ (2e) and [IrIII(oep)(py)2]+ (4) exhibited very potent cytotoxicity with nanomolar IC50 values. For the mono-NHC complex [IrIII(F20tpp)(BIMe)(NH3)]+ (1d′), significant potentiation of the cytotoxicity was also found under the same conditions. The cytotoxicity of [IrIII(oep)Cl(CO)] was also evaluated for comparison. This complex is relatively noncytotoxic in the dark with an IC50 value >50 μM and increased to 19.7 ± 1.0 μM upon visible light irradiation. For cisplatin, the difference in the phototoxicity index (PI) was <1, indicating an absence of photoinduced cytotoxicity under our experimental conditions. Thus, the cytotoxicity of the iridium(III) porphyrin complexes with the incorporation of axial bis-carbene ligands is significantly enhanced under light irradiation.
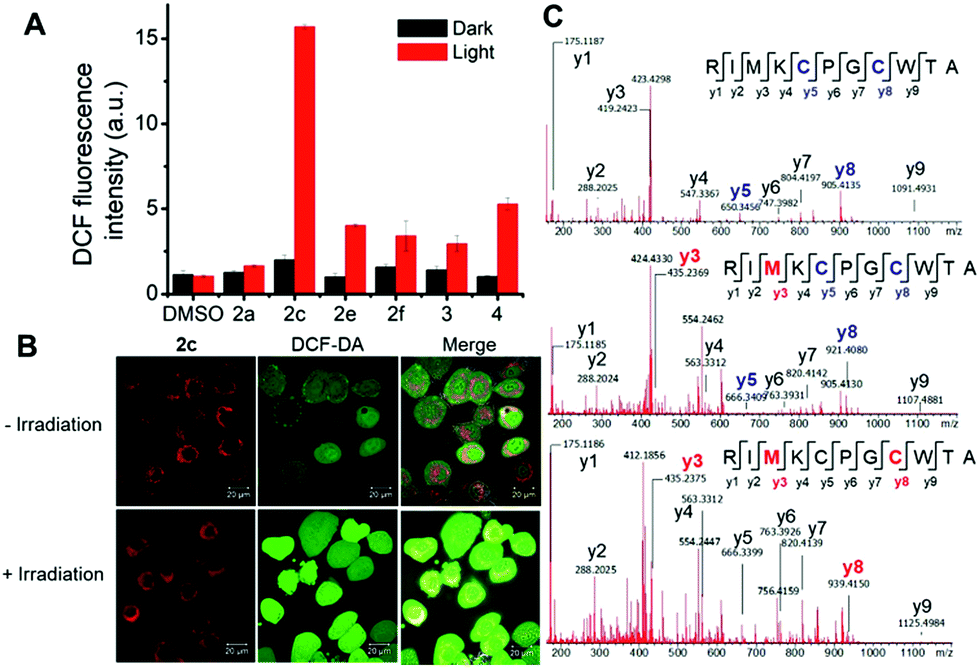
To examine the relationship between photocytotoxicity and the photosensitizing properties of the complexes, we measured the cellular reactive oxygen species (ROS) generation using the ROS probe H2DCF-DA after treatment of cancer cells with the iridium(III) porphyrin complexes. As shown in Fig. 15A, no significant change in DCF fluorescence was observed in NCI-H460 lung cancer cells incubated with the complexes in the dark. Exposure to visible light irradiation resulted in increased DCF fluorescence intensity, revealing elevated cellular ROS levels. In particular, a 10-fold elevation in ROS levels in cells treated with 2c was observed. The result was further corroborated by the strong green fluorescence observed from DCF when cells were treated with 2c followed by light irradiation (Fig. 15B). These results confirmed the generation of ROS, possibly 1O2, in NCI-H460 cells treated with Ir(III) complexes upon visible light irradiation. We further examined the possibility of protein oxidation as a consequence of photoinduced oxidative stress caused by 2c. A peptide (RIMKCPGCWTA) from thioredoxin (Trx) was employed as a model to investigate the possible oxidative modification sites by electrospray ionization tandem mass spectrometry (ESI-MS/MS). The peptide was found to undergo oxidative modifications in the presence of 2c upon irradiation. Three different types of oxidized products, including the formation of a disulfide bridge and the oxidation of the sulfur atoms of cysteine and methionine, were characterized (Fig. 15C, S25 and Tables S6–S8†). The peptide alone as a control was shown to remain unchanged when incubated in the dark or upon light irradiation (Fig. S24 and Table S5†). As shown in Fig. 15C and S25,† three triplycharged species were detected with mass differences of −0.671 (top), +4.660 (middle) and +10.662 (bottom) Da, respectively, from the unmodified peptide. Further MS/MS analysis revealed that the most intense peak (m/z 421.8638; Fig. 15C (top), Table S6†) corresponds to the formation of an intramolecular disulfide bond, in which the resulting fragment y5 and y8 ions bearing free cysteine and cysteine thioaldehyde residues, respectively, were observed when comparing with the peptide alone (Fig. S26†).
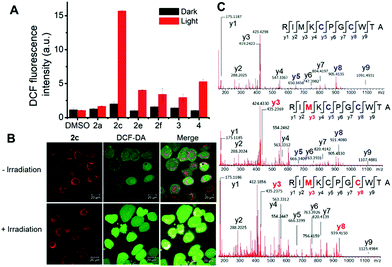 |
| | Fig. 15 (A) Generation of ROS examined by DCF fluorescence measurement in NCI-H460 cells treated with or without iridium(III) porphyrin complexes (1 μM) for 2 h with or without irradiation by visible light. (B) Confocal microscopy imaging of ROS generation in cells incubated with complex 2c with or without irradiation by visible light. Scale bar: 20 μm. (C) Photooxidation of peptide from thioredoxin by 2c. ESI-MS/MS spectra of the triply charged disulfide-bridged peptide (m/z 421.8638; top) and oxidized peptides (m/z 427.1954; middle, m/z 433.1978; bottom). Fragments with blue labels indicate sites of disulfide bond formation, and red labels represent oxidative modification sites. | |
Moreover, additional oxidative modification of the methionine residue to give sulfoxide (y3) with a shift of +16 Da was also found on the disulfide-bridged peptide (m/z 427.1954; Fig. 15c (middle), Table S7†). In addition, another oxidation product (m/z 433.1978) with a mass increment of +10.662 in a triply charged state is ascribed to the addition of two oxygen atoms (+32 Da) in the singly charged species. MS/MS sequence analysis displayed the identified y-ions, which are attributed to the oxidized methionine (y3; O-Met, Met +16 Da) and cysteine (y8; O-Cys, Cys +16 Da) of the peptide (Fig. 15c (bottom) and Table S8†). These results show that treatment with 2c combined with light activation promoted the oxidation of cysteine and methionine residues through a singlet oxygen-mediated mechanism.
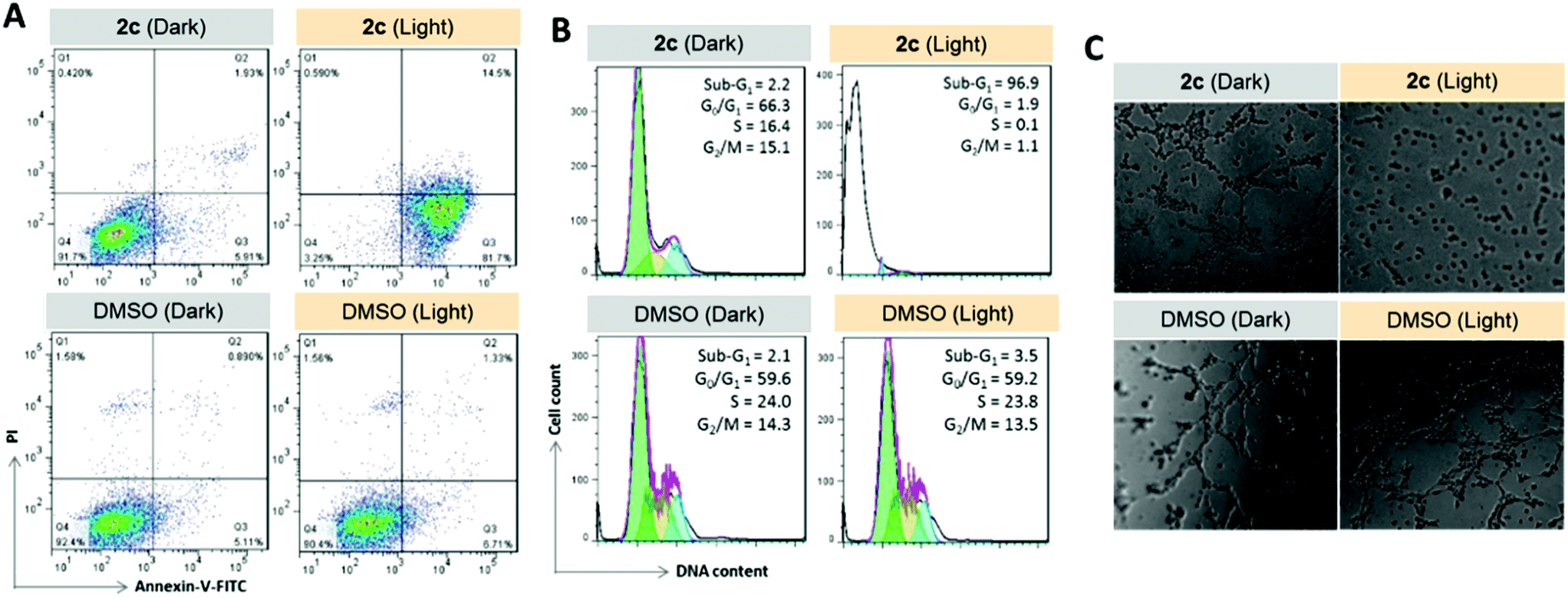
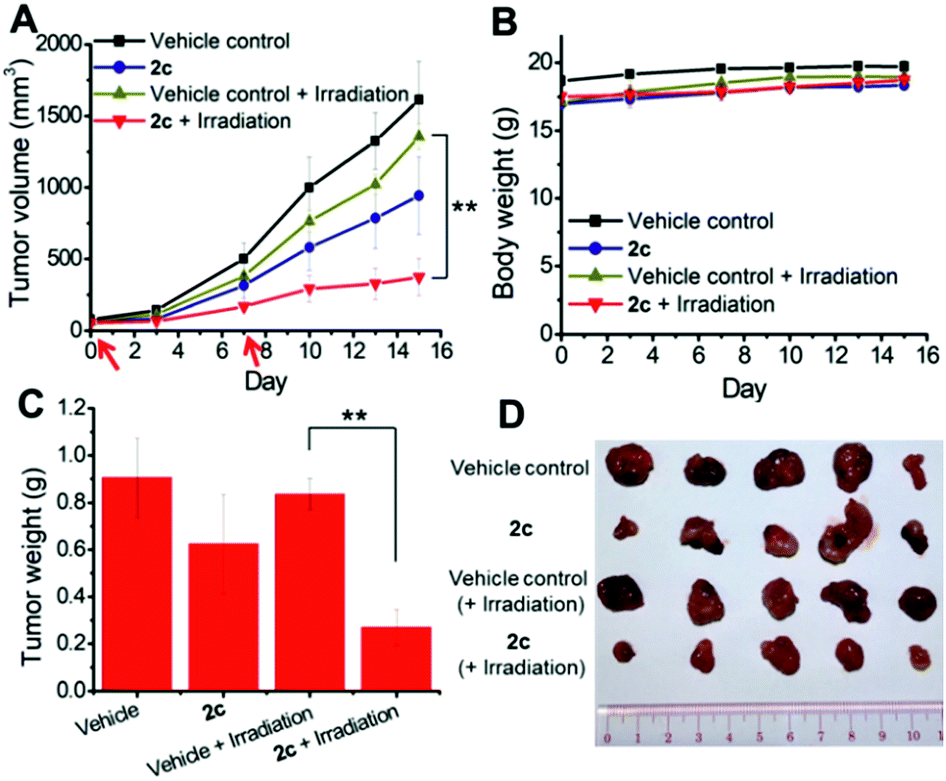
To examine the photoinduced cytotoxicity, the apoptotic cell death and cell cycle progression of the cancer cells treated with 2c and visible light irradiation were analyzed by flow cytometry. NCI-H460 cells were incubated with 2c at 0.1 μM, a concentration that could not lead to strong antiproliferative effects in the dark. Upon exposure to light irradiation, the proportion of cells treated with 2c undergoing apoptotic cell death increased from 5.9% to 81.7%, as shown by the annexin-V-FITC/propidium iodide assay (Fig. 16A). On the other hand, complex 2c (0.1 μM) did not exhibit a marked effect on the progression of the cell cycle in the dark, showing only a mild increase in the G0/G1-population from 59.6% to 66.3%. In agreement with the annexin-V/propidium iodide flow cytometry results above, visible light irradiation led to a significant increase in the sub-G1 population (from 2.2% to 96.9%) as a result of extensive DNA fragmentation due to cell death (Fig. 16B). We also examined the antiangiogenic property of 2c in the inhibition of endothelial cell tube formation. As shown in Fig. 16C, moderate inhibition of MS-1 cell tube formation was observed after treatment of the complex in the dark, while tube formation was completely abrogated upon light irradiation. The above results showed that a low dose of 2c causes very low-level cellular damage in the dark but could induce pronounced apoptosis and inhibition of angiogenesis upon visible light irradiation. Noticeably, other iridium porphyrin complexes were also observed to exert similar effects (Fig. S27–S29†). The in vivo photoinduced therapeutic efficacy of 2c was also examined. NCI-H460 tumor-bearing mice were divided into four groups, vehicle control, 2c (3.0 mg kg−1), vehicle control with irradiation, and 2c (3.0 mg kg−1) with irradiation, and subjected to two intratumoral injections of the compounds in a period of 15 days. Mice in the irradiation groups were injected with 2c or solvent vehicle followed by exposure of the tumor site to white light (400–800 nm) at a power density of 110 mW cm−2 for 30 min on the first and seventh days. The other two groups of mice received the same treatment without irradiation as a control. As shown in Fig. 17A, the tumor growth of mice treated with 2c and irradiation was markedly decreased by 72% in comparison to that of the vehicle group with light irradiation after only two injections. In contrast, treatment with 2c without irradiation was able to inhibit tumor growth by only 41%. All of the mice displayed negligible changes in body weight throughout the treatment period (Fig. 17B). Moreover, the tumor weight of 2c-treated mice in the photoirradiation group was found to be much lower than that in the dark control group (Fig. 17C and D), demonstrating the markedly enhanced antitumor efficacy of the combination of dark and photoinduced tumor inhibitory activities.
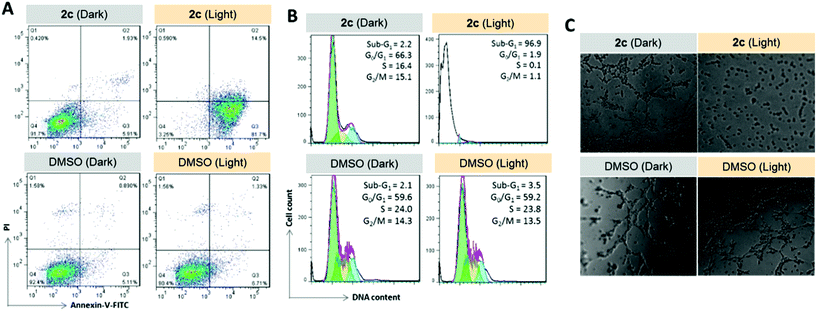 |
| | Fig. 16 Anticancer properties of 2c (0.1 μM) upon visible light irradiation. (A) Apoptosis/necrosis in NCI-H460 cells as examined by flow cytometry of annexin-V-FITC/propidium iodide-stained cells. (B) Cell cycle progression in NCI-H460 cells as examined by flow cytometry. (C) Inhibition of angiogenesis of MS-1 endothelial cells. | |
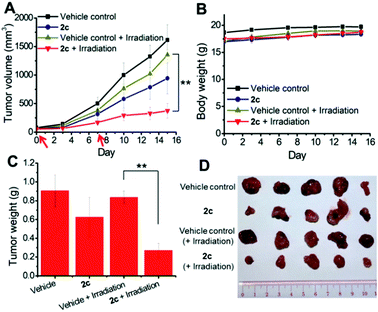 |
| | Fig. 17
In vivo antitumor effects of complex 2c in nude mouse model bearing NCI-H460 lung cancer xenografts. (A) Tumor volume. (B) Body weight of mice. (C) Tumor weight after the experiment. Red arrows indicate the injection of the mice with 2c together with irradiation on day 0 and day 7. (D) Photo of tumors of each group after treatment for 15 days. Data are expressed as the mean ± standard error; **p < 0.01. | |
Discussion
The coordination of the NHC ligand to IrIII–porphyrins causes several major alterations to the spectral and photophysical parameters compared to those of the [IrIII(por)Cl(CO)] and [Ir(por)L2]+ (L = isocyanide, pyridine or imidazole) complexes.16a
Split Soret band
All NHC complexes in this work feature two distinct Soret bands with comparable intensity. Metalloporphyrins showing split Soret bands are classically termed hyperporphyrins.14,15c The valence of the metal ions and the nature of the axial ligand(s) are known to dictate this spectral attribute. Hyperporphyrins can be divided into p-type and d-type. In d-type hyperporphyrins, which are relevant to our case, the metal ions are generally trivalent or at a higher oxidation state with low metal reduction potential and partially filled dπ orbitals. As a result, the extra Soret band is attributed to the ring-to-metal LMCT transition of a1u(π),a2u(π) → eg(dπ) character. Metalloporphyrins of FeIII, MnIII and CrIII ions are canonical examples of this type.14 However, this scenario is not applicable to 1 and 2, which contain a d6 IrIII ion, because all nominal dπ (dxz, dyz) orbitals are fully occupied, and no such LMCT transition can take place in the UV-visible spectral region. Notably, the double Soret band found in d6 metalloporphyrin is not unique to our Ir–NHC system; a few IrIII- and RhIII-porphyrin complexes bearing phenyl(alkoxycarbonyl)carbene or NHC with a trans methyl ligand, recently described by Woo and co-workers, also exhibit this spectral attribute.6,33 Woo and co-workers suggested that orbital mixing with relatively low-energy doubly excited states, [eg(dπ)]3[a1u(π),a2u(π)]3[eg(π*)]2, may be responsible for the extra Soret band. Our TD-DFT calculations on 2a indicated that the high-energy (extra) Soret band mainly comes from electronic transitions from the hybrid of the iridium dxy orbital and the π orbitals of the porphyrin's peripheral tolyl groups to the core π* porphyrin orbitals (pyrrole rings + mesocarbons), which is best described as an MLCT transition with mixed ILCT character, while the low-energy (conventional) Soret band mainly comes from π → π* IL transitions within the porphyrin ligand. Based on RR experiments, the independent excitation of 2a at the extra Soret band (435.7 nm) and at the conventional Soret band (368.9 nm) results in the selective enhancement of different vibrational modes, corroborating that the excited states associated with the two bands have different characteristics. In particular, the former (MLCT) excitation primarily enhances vibrational modes involving the core skeleton to which the Ir(III) ion is directly linked, while the latter (π–π*) excitation enhances mainly the vibrational modes within the tolyl rings to which the porphyrin core is connected. Overall, the experimental findings from the RR study of 2a concur with the conclusion from DFT calculations that the extra Soret band is associated with an Ir-to-porphyrin MLCT excitation.
Interestingly, the intensity of the MLCT band is found to vary with the number of NHC ligands, as exemplified in the series [IrIII(F20tpp)Cl(CO)] (logεMLCT = 0), [IrIII(F20tpp)(IMe)Cl] (1c) (logεMLCT = 4.62) and [IrIII(F20tpp)(IMe)2]+ (2f) (logεMLCT = 4.89). Thus, NHC ligands play an indispensable role in enhancing the allowedness of the MLCT transition. Notably, the extra Soret band (LMCT band) of high-spin [MnIII(tpp)]+ complexes, typical d-type hyperporphyrins, also displays comparable intensity to their ‘conventional’ π–π* Soret band.14,34 This observation was explained by the fact that the high-lying dπ(dxz, dyz) orbitals of the MnIII ion can undergo substantial interaction with the porphyrin eg(π*) orbitals due to their energetic proximity, which leads to strong mixing of the π(por) → dπ(MnIII) LMCT and Soret π–π* states.14,33 Thus, we propose that in our case, the strongly electron-donating NHC ligand(s) can boost the Ir dπ orbitals to a higher level at which they can mix efficiently with the porphyrin π* orbitals. This phenomenon in turn enables borrowing of the oscillator strength from the Soret π–π* transition to enhance the allowedness of the MLCT transition. In fact, the intensity of the MLCT Soret band increases at the expense of the π–π* Soret band from [IrIII(F20tpp)Cl(CO)], 1c to 2f with logεMLCT/logεSoret values of 0/5.42, 4.62/5.10 and 4.89/5.00, respectively. Incidentally, the possibility that ruffling deformation also contributes to enhancing the MLCT band intensity cannot be ruled out, as all reported IrIII- and RhIII-porphyrin complexes with such an extra Soret band were found or calculated to have a ruffled porphyrin ring.5b,6,33
Spectral redshift, reduced emission quantum yield and lifetime
The ruffled porphyrin ring, as revealed by X-ray crystallographic analysis, is a key structural feature of [MIII(por)-NHC] (M = Ir and Rh) complexes.5b,6 Here, the extent of ruffling increases with the number of NHC ligands owing to the enhanced steric congestion. In the literature, out-of-plane ring deformation in the form of either ruffling or saddling is well documented to engender a spectral redshift in both free base porphyrins and metalloporphyrins; the extent of redshift is usually proportional to the magnitude of distortion, albeit in a nonlinear fashion.8 In addition, metalloporphyrins with deformed porphyrin rings are known to undergo rapid macrocyclic inversion in both the ground state and the excited state. This inversion generally leads to diminished luminescence and emission lifetime compared to those of planar metalloporphyrins as a result of the surging nonradiative decay activities in the form of low-energy out-of-plane vibrations in the excited state.8a,e,9c Indeed, the mono- and bis-NHC complexes of Ir(III)-porphyrin in this work exhibit the aforementioned photophysical attributes of nonplanar metalloporphyrins; their Soret and Q bands and their porphyrin-based 3(π, π*) emissions are markedly redshifted by up to 80 nm with respect to the parental [IrIII(por)Cl(CO)] complexes and bis-ligand analogs (Table 4; Fig. 6 and 10). Furthermore, the emission quantum yield and lifetime drop to less than 0.7% and 3.6 μs respectively, with the bis-NHC analogs suffering more severely. As revealed by the excited-state photophysical parameters in Table 9, the increase in the nonradiative decay constant of the emissive excited state of up to 200 times underlies the decrease in emission quantum yield and lifetime, although the radiative decay rate constants increase slightly by 3–14-fold.
Table 9
k
r and knr values of IrIII(por) complexes in CHCl3 solutions at 298 K
| Complex |
|
Φ
em [×10−2] |
τ
em/μs |
k
r
[×10]/s |
k
nr
[×103]/s |
|
Radiative decay rate constant determined by kr = Φem/τ.
Nonradiative decay rate constant determined by knr = (1 − Φem)/τ.
Data in CH2Cl2.
|
| [IrIII(tmp)Cl(CO)] |
|
0.22 |
77.2 |
2 |
13 |
| [IrIII(oep)Cl(CO)] |
|
5.03 |
83.3 |
60 |
11 |
| [IrIII(F20tpp)Cl(CO)] |
|
2.92 |
101.0 |
29 |
10 |
| [IrIII(tmp)(IMe)Cl] |
1a
|
0.05 |
1.8 |
28 |
555 |
| [IrIII(tmp)(BIMe)Cl] |
1b
|
0.06 |
2.3 |
26 |
435 |
| [IrIII(F20tpp)(BIMe)Cl] |
1d
|
0.29 |
1.7 |
171 |
587 |
| [IrIII(oep)(IMe)2]+ |
2c
|
0.41 |
2.7 |
152 |
368 |
| [IrIII(oep)(BIMe)2]+ |
2d
|
0.66 |
3.6 |
183 |
276 |
| [IrIII(oep)(IiPr)2]+ |
2e
|
0.03 |
1.9 |
16 |
526 |
| [IrIII(F20tpp)(IMe)2]+c |
2f
|
0.08 |
0.5 |
160 |
1998 |
| [IrIII(oep)(CNPhOMe)2]+ |
3
|
6.40 |
124.7 |
52 |
8 |
| [IrIII(oep)(py)2]+ |
4
|
12.30 |
55.3 |
222 |
16 |
Photosensitized singlet oxygen generation
Metalloporphyrins and their derivatives, such as metallocorroles, are important classes of photochemical singlet oxygen sensitizers that exhibit good biocompatibility, strong visible light absorption, high triplet-state quantum yields and long triplet-state lifetimes.29,35 They have been extensively studied as sensitizers for photodynamic therapy and photodriven aerobic oxidation reactions.28c,36,37 Several iridium(III)-porphyrin and corrole complexes have recently been found to exhibit such favorable inherent properties and are capable of generating singlet oxygen photochemically.16d,29 In particular, two σ-bonded aza-BODIPY complexes of IrIII(ttp) were reported to have Φso values of 0.79 and 0.85, which are more than 100 times greater than those of the respective free aza-BODIPY ligands, revealing the effectiveness of the iridium(III) porphyrin moiety in promoting singlet oxygen sensitization.
Two cationic bis-NHC complexes examined in this work, 2a and 2c, were found to be efficient photosensitizers for the photoinduced aerobic oxidation of secondary amines and arylboronic acids. Quenching experiments suggested that singlet oxygen generated by quenching of the excited state of 2a or 2c is presumably the active oxidant for these photooxidation reactions. This finding is in good agreement with their high Φso values of 0.64 and 0.88, respectively. The singlet oxygen quantum yield of a photosensitizer is determined by the triplet-excited-state quantum yield and the efficiency of energy transfer from the triplet excited state to molecular oxygen.35a Considering that the heavy iridium ion with its large spin orbit coupling constant (ξIr = 3909 cm−1) often confers an ultrafast intersystem crossing rate and triplet-excited-state formation yield close to unity,38 the higher singlet oxygen formation quantum yield of 2c than of 2a is attributed to the former having a longer triplet-state lifetime (τ = 2.3 vs. 0.4 μs) (Fig. S19†).
Stability, cytotoxicity and photochemical activities
Our previous works showed that porphyrin ligand can stabilize gold(III) ion against demetallation under physiological conditions rendering gold(III) porphyrins to display effective anticancer activity against multiple cancer cell types in vitro and in vivo.31a–d We have also found that as a result of strong σ-donating property of NHC ligand, pincer gold(III) and platinum(II) complexes containing NHC auxiliary ligand are stable against ligand exchange reaction under physiological conditions and these complexes display potent antitumor properties.31e,39 By using both porphyrin and NHC ligands altogether, we prepared a panel of stable octahedral iridium(III) porphyrin complexes containing NHC ligand that exhibit potent in vitro anticancer activities and show significant inhibition of tumor growth in vivo upon photo-irradiation.
The Ir(III) porphyrin complexes bearing bis-NHC or mono-NHC axial ligand(s) display a good stability against ligand exchange with DMSO solvent or glutathione (Fig. S21 and S22†). Complex 3 bearing bis-isocyanide ligands is more substitution labile (Fig. S21f†). Most of the [IrIII(por)(NHC)2]+ complexes exhibit cytotoxicity to different cancer cell lines with IC50 values down to submicromolar concentrations (Table 8). Incorporation of NHC ligand(s) into iridium porphyrin increases lipophilicity of the complexes hence their cellular uptake and accumulation in cancer cells. Complex 2f with meso-tetrakis(pentafluorophenyl)porphyrin ligand displays the lowest cellular uptake and lipophilicity among the bis-NHC complexes. Cationic iridium porphyrin complexes show more efficient cellular internalization and hence increase in anticancer activity as indicated by lower IC50 values compared to those of the neutral complex [IrIII(oep)Cl(CO)] and the isoelectronic [RuII(oep)(CO)] reported by Bogoeva and co-workers.40 Complex 2c accumulates preferentially in cellular ER and somewhat in mitochondria as shown by the confocal laser microscopy images in the co-staining experiments, which could account for the subsequent induction of ER stress and mitochondrial membrane depolarization by this complex.
Incorporation of bis-NHC ligands to iridium(III) porphyrin also endows the complexes to display remarkable singlet oxygen production capability. Complex 2c (with octaethylporphyrin) has the highest Φso compared to 2a (with meso-tetratolylporphyrin) and other iridium(III) complexes ([IrIII(corrole)(pyridine)2] and [IrIII(ttp)(aza-BODIPY)]) without bis-NHC ligands; it exhibits potent cytotoxicity with an IC50 value in the nano-molar range and triggers the generation of ROS upon visible light irradiation. The photo-induced production of ROS (e.g., 1O2) was found to lead to oxidation of sulfur-containing residues of peptides, which is presumably a cytotoxic, protein-damaging mechanism. The photo-cytotoxic effects led to induction of apoptotic cell death and inhibition of angiogenesis. The in vivo photo-induced antitumor effect of 2c was demonstrated in a mouse xenograft model of human lung cancer. Overall, given the synergetic anticancer properties of the prototype bis-NHC iridium(III) porphyrin complexes in the dark and upon light activation as well as the flexibility in derivatization of NHC ligand, cationic [IrIII(por)(NHC)n]+ (H2por = porphyrin; n = 1 or 2) complexes have great potential for development as a new class of photo-activated anticancer agents.
Conclusions
We synthesized a panel of luminescent ruffled iridium(III) porphyrin complexes bearing mono- and bis-NHC ligands, four of which were structurally characterized by X-ray crystallography. The unique photophysical properties resulting from the coordination of NHC ligands, including split Soret bands, broadened and redshifted absorption and emission spectra and substantially reduced emission quantum yield and lifetime, were investigated by spectroscopic and theoretical means and discussed. In spite of the low triplet-state energy, which limits their use as a photoredox mediator, the bis-NHC complexes are versatile photochemical singlet oxygen generators that can efficiently catalyze the photoinduced aerobic oxidation of secondary amines and arylboronic acids. In addition, the bis-NHC complexes are stable in physiological conditions and exhibit potent cytotoxicity and photocytotoxicity, presumably via the generation of ROS, against a panel of cancer cell lines, and one was demonstrated to have significant inhibitory effects against tumor growth in a nude mouse model.
Author contributions
Chi-Ming Che designed and initiated this research project. Chun-Nam Lok designed the biological experiments. Tsz-Lung Lam synthesized and characterized all complexes in this work, performed the spectroscopic and electrochemical measurements and carried out the photocatalytic reactions. Tsz-Lung Lam, Chun-Nam Lok, and Chi-Ming Che wrote the manuscript. Ka-Chung Tong and Wai-Lun Kwong carried out the biological experiments. Chen Yang was responsible for X-ray crystal structure determinations and assisted the time-resolved spectroscopic measurements. Ming-De Li and David Lee Phillips carried out the resonance Raman spectroscopic measurements. Xianguo Guan, Vanessa Kar-Yan Lo, and Sharon Lai-Fung Chan were responsible for the DFT/TDDFT calculations on UV-vis absorption spectra as well as resonance Raman spectra and the associated vibrational mode assignments. All authors reviewed the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Faisal Mehmood and Pui-Yan Lee for the help on biological studies. This work is financially supported by Hong Kong Research Grants Council, General Research Fund (17303815), Innovation and Technology Fund (ITS/130/14FP) and Basic Research Program-Shenzhen Fund (JCYJ20160229123546997 and JCYJ20170412140257516).
Notes and references
- Selected reviews:
(a)
N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH, New York, 2006 Search PubMed;
(b)
N-Heterocyclic Carbenes in Transition Metal Catalysis, ed. F. Glorius, Springer-Verlag, Berlin, Heidelberg, 2007, vol. 21 Search PubMed;
(c) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef;
(d) L.-A. Schaper, S. J. Hock, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2013, 52, 270–289 CrossRef CAS PubMed;
(e) A. Nasr, A. Winkler and M. Tamm, Coord. Chem. Rev., 2016, 316, 68–124 CrossRef CAS;
(f) D. Janssen-Müller, C. Schlepphorst and F. Glorius, Chem. Soc. Rev., 2017, 46, 4845–4854 RSC.
- Selected reviews:
(a) L. Meres and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903–1912 RSC;
(b) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC;
(c) M. Elie, J.-L. Renaud and S. Gaillard, Polyhedron, 2018, 140, 158–168 CrossRef CAS.
- Selected reviews:
(a) K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859–3884 CrossRef CAS PubMed;
(b) M.-L. Teyssot, A.-S. Jarrousse, M. Manin, A. Chevry, S. Roche, F. Norre, C. Beaudoin, L. Morel, D. Boyer, R. Mahiou and A. Gautier, Dalton Trans., 2009, 6894–6902 RSC;
(c) L. Oehninger, R. Rubbiani and I. Ott, Dalton Trans., 2013, 42, 3269–3284 RSC;
(d) W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC;
(e) F. Cisnetti and A. Gautier, Angew. Chem., Int. Ed., 2013, 52, 11976–11978 CrossRef CAS;
(f) C. Hu, X. Li, W. Wang, R. Zhang and L. Deng, Curr. Med. Chem., 2014, 21, 1220–1230 CrossRef CAS;
(g) W. Liu and R. Gust, Coord. Chem. Rev., 2016, 329, 191–213 CrossRef CAS.
- Selected reviews:
(a) P. L. Arnold and I. J. Casely, Chem. Rev., 2009, 109, 3599–3611 CrossRef CAS;
(b) H. Jacobsen, A. Correa, A. Poater, C. Costabile and L. Cavallo, Coord. Chem. Rev., 2009, 253, 687–703 CrossRef CAS;
(c) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS PubMed;
(d) P. G. Edwards and F. E. Hahn, Dalton Trans., 2011, 40, 10278–10288 RSC;
(e) S. J. Hock, L.-A. Schaper, W. A. Herrmann and F. E. Kühn, Chem. Soc. Rev., 2013, 42, 5073–5089 RSC;
(f) C. Fliedel, G. Schnee, T. Avilés and S. Dagorne, Coord. Chem. Rev., 2014, 275, 63–86 CrossRef CAS;
(g) S. Bellemin-Laponnaz and S. Dagorne, Chem. Rev., 2014, 114, 8747–8774 CrossRef CAS;
(h) V. Charra, P. de Friémont and P. Braunstein, Coord. Chem. Rev., 2017, 341, 53–176 CrossRef CAS.
-
(a) M. Albrecht, P. Maji, C. Häusl, A. Monney and H. Müller-Bunz, Inorg. Chim. Acta, 2012, 380, 90–95 CrossRef CAS;
(b) J. Olguin, H. Müllen-Bunz and M. Albrecht, Chem. Commun., 2014, 50, 3488–3490 RSC.
- B. J. Anding, A. Ellern and L. K. Woo, Organometallics, 2014, 33, 2219–2229 CrossRef CAS.
- K.-H. Chan, X. Guan, V. K.-Y. Lo and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 2982–2987 CrossRef CAS.
-
(a) C. M. Drain, C. Kirmaier, C. J. Medforth, D. J. Nurco, K. M. Smith and D. Holten, J. Phys. Chem., 1996, 100, 11984–11993 CrossRef CAS;
(b) J. A. Shelnutt, X.-Z. Song, J.-G. Ma, S.-L. Jia, W. Jentzen and C. J. Medforth, Chem. Soc. Rev., 1998, 27, 31–42 RSC;
(c) A. B. Parusel, T. Wondimagegn and A. Ghosh, J. Am. Chem. Soc., 2000, 122, 6371–6374 CrossRef CAS;
(d) R. E. Haddad, S. Gazeau, J. Pecaut, J.-C. Marchon, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2003, 125, 1253–1268 CrossRef CAS;
(e) A. Y. Lebedev, M. A. Filatov, A. V. Cheprakov and S. A. Vinogradov, J. Phys. Chem. A, 2008, 112, 7723–7733 CrossRef CAS;
(f) F. Nifiatis, W. Su, J. E. Haley, J. E. Slagle and T. M. Copper, J. Phys. Chem. A, 2011, 115, 13764–13772 CrossRef CAS;
(g) M. O. Senge, S. A. MacGowan and J. M. O'Brien, Chem. Commun., 2015, 51, 17031–17063 RSC.
-
(a) C. J. Medforth, M. O. Senge, K. M. Smith, L. D. Sparks and J. A. Shelnutt, J. Am. Chem. Soc., 1992, 114, 9859–9869 CrossRef CAS;
(b) K. M. Kadish, M. Lin, E. Van Caemelbecke, G. De Stefano, C. J. Medforth, D. J. Nurco, N. Y. Nelson, B. Krattinger, C. M. Muzzi, L. Jaquinod, Y. Xu, D. C. Shyr, K. M. Smith and J. A. Shelnutt, Inorg. Chem., 2002, 41, 6673–6687 CrossRef CAS;
(c) A. Rosa, G. Ricciardi, E. J. Baerends, M. Zimin, M. A. J. Rodgers, S. Matsumoto and N. Ono, Inorg. Chem., 2005, 44, 6609–6622 CrossRef CAS;
(d) N. Grover, M. Sankar, Y. Song and K. M. Kadish, Inorg. Chem., 2016, 55, 584–597 CrossRef CAS;
(e) J. Schindler, S. Kupfer, A. A. Ryan, K. J. Flanagan, M. O. Senge and B. Dietzek, Coord. Chem. Rev., 2018, 360, 1–16 CrossRef CAS.
-
(a) F. D'Souza, M. E. Zandler, P. Tagliatesta, Z. Ou, J. Shao, E. V. Caemelbeck and K. M. Kadish, Inorg. Chem., 1998, 37, 4567–4572 CrossRef;
(b) B. Röder, M. Büchner, I. Rückmann and M. O. Senge, Photochem. Photobiol. Sci., 2010, 9, 1152–1158 RSC.
-
(a) R. Guilard, K. Perié, J.-M. Barbe, D. J. Nurco, K. M. Smith, E. Van Caemelbecke and K. M. Kadish, Inorg. Chem., 1998, 37, 973–981 CrossRef CAS;
(b) I. Akira and N. Mikio, Chem. Lett., 2005, 34, 1046–1047l CrossRef;
(c) A. Ikezaki and M. Nakamura, J. Porphyrins Phthalocyanines, 2016, 20, 1–13 CrossRef.
-
(a) T. Sakai, Y. Ohgo, A. Hoshino, T. Ikeue, T. Saitoh, M. Takahashi and M. Nakamura, Inorg. Chem., 2004, 43, 5034–5043 CrossRef CAS;
(b) M. Nakamura, Coord. Chem. Rev., 2006, 250, 2271–2294 CrossRef CAS;
(c) A. Tozuka, A. Ikezaki, M. Taniguchi and M. Nakamura, Inorg. Chem., 2010, 49, 10400–10408 CrossRef CAS;
(d) M. Li, A. G. Oliver, T. J. Neal, C. E. Schulz and W. R. Scheidt, J. Porphyrins Phthalocyanines, 2013, 17, 118–124 CrossRef CAS;
(e) D. Sahoo and S. P. Rath, Chem. Commun., 2015, 51, 16790–16793 RSC.
-
(a) R. Salzmann, C. J. Ziegler, N. Godbout, M. T. McMahon, K. S. Suslick and E. Oldfield, J. Am. Chem. Soc., 1998, 120, 11323–11334 CrossRef CAS;
(b) Z. Yao, C. E. Schulz, N. Zhan and J. Li, Inorg. Chem., 2017, 56, 12615–12624 CrossRef CAS.
-
M. Gouterman, in The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1978, vol. 3, pp. 1–165 Search PubMed.
-
(a) L. K. Hanson, M. Gouterman and J. C. Hanson, J. Am. Chem. Soc., 1973, 95, 4822–4829 CrossRef CAS;
(b) A. Antipas, J. W. Buchler, M. Gouterman and P. D. Smith, J. Am. Chem. Soc., 1978, 100, 3015–3024 CrossRef CAS;
(c) A. Antipas, J. W. Buchler, M. Gouterman and P. D. Smith, J. Am. Chem. Soc., 1980, 102, 198–207 CrossRef CAS;
(d) L. M. A. Levine and D. Holten, J. Phys. Chem., 1998, 92, 714–720 CrossRef.
-
(a) K. Koren, S. M. Borisov, R. Saf and I. Klimant, Eur. J. Inorg. Chem., 2011, 1531–1534 CrossRef CAS;
(b) K. Koren, R. I. Dmitriev, S. M. Borisov, D. B. Papkovsky and I. Klimant, ChemBioChem, 2012, 13, 1184–1190 CrossRef CAS;
(c) B.-B. Wang, H. Zho, J. Mack, P. Majumdar, T. Nyokong, K. S. Chan and Z. Shen, J. Porphyrins Phthalocyanines, 2015, 19, 973–982 CrossRef CAS;
(d) J. Zhou, L. Gai, Z. Zhou, J. Mack, K. Xu, J. Zhao, H. Qiu, K. S. Chan and Z. Shen, RSC Adv., 2016, 6, 72115–72120 RSC.
- Selected examples of coordinating NHC ligand to iridium metal center via transmetallation of [Ag(NHC)2]+ complexes:
(a) S. Burling, M. F. Mahon, S. P. Reade and M. K. Whittlesey, Organometallics, 2006, 25, 3761–3767 CrossRef CAS;
(b) L. N. Appelhans, C. D. Incarvito and R. H. Crabtree, J. Organomet. Chem., 2008, 693, 2761–2766 CrossRef CAS;
(c) U. Hintermair, U. Englert and W. Leitner, Organometallics, 2011, 30, 3726–3731 CrossRef CAS.
- C.-M. Che and W.-Y. Yu, Pure Appl. Chem., 1999, 71, 281–288 CAS . Oxidation state marker band of [IrIII(tmp)Cl(CO)]: ν = 1022 cm−1; [IrIII(F20tpp)Cl(CO)]: ν = 1026 cm−1; [IrIII(ttp)Cl(CO)]: ν = 1017 cm−1; [IrIII(oep)Cl(CO)]: ν = 1022 cm−1.
-
(a) X. Song and K. S. Chan, Organometallics, 2007, 26, 965–970 CrossRef CAS;
(b) C. W. Cheung and K. S. Chan, Organometallics, 2008, 27, 3043–3055 CrossRef CAS;
(c) C. W. Cheung and K. S. Chan, Organometallics, 2011, 30, 4269–4283 CrossRef CAS.
- Selected examples:
(a) J. M. Meredith, R. Robinson Jr, K. I. Goldberg, W. Kaminsky and D. M. Heinekeym, Organometallics, 2012, 31, 1879–1887 CrossRef CAS;
(b) M. C. Lehman, P. D. Boyle, R. D. Sommer and E. A. Ison, Organometallics, 2014, 33, 5081–5084 CrossRef CAS;
(c) S. Sabater, M. Baya and J. A. Mata, Organometallics, 2014, 33, 6830–6839 CrossRef CAS;
(d) L. Rubio-Pérez, M. Iglesias, J. Munárriz, V. Polo, V. Passarelli, J. J. Pérez-Torrente and L. A. Oro, Chem. Sci., 2017, 8, 4811–4822 RSC.
-
C. J. Medforth, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, Boston, 2000, vol. 5, pp. 1–80 Search PubMed.
-
(a) J. R. Polam, T. K. Shokhireva, K. Raffii, U. Simonis and F. A. Walker, Inorg. Chim. Acta, 1997, 263, 109–117 CrossRef CAS;
(b) C. J. Medforth, C. M. Muzzi, K. M. Smith, R. J. Abraham, J. D. Hobbs and J. A. Shelnutt, J. Chem. Soc., Chem. Commun., 1994, 1843–1844 RSC;
(c) C. J. Medforth, R. E. Haddad, C. M. Muzzi, N. R. Dooley, L. Jaquinod, D. C. Shyr, D. J. Nurco, M. M. Olmstead, K. M. Smith, J.-G. Ma and J. A. Shelnutt, Inorg. Chem., 2003, 42, 2227–2241 CrossRef CAS.
-
(a)
H. Scheer and J. J. Katz, in Porphyrins and Metalloporphyrins, ed. K. M. Smith, Elsevier, Amsterdam, 1975, pp. 399–524 Search PubMed;
(b) Y. Li, J.-S. Huang, G.-B. Xu, N. Zhu, Z.-Y. Zhou, C.-M. Che and K.-Y. Wong, Chem.–Eur. J., 2004, 10, 3486–3502 CrossRef CAS;
(c) C.-M. Che, C.-M. Ho and J.-S. Huang, Coord. Chem. Rev., 2007, 251, 2145–2166 CrossRef CAS.
- C. Swistak, J.-L. Cornillon, J. E. Anderson and K. M. Kadish, Organometallics, 1987, 6, 2146–2150 CrossRef CAS.
-
(a)
R. H. Felton, in The Porphyrins, ed. D. Dolphin, Academic Press, San Diego, 1978, vol. 5, pp. 53–126 Search PubMed;
(b) P. Chen, O. S. Finikova, Z. Ou, S. A. Vinogradow and K. M. Kadish, Inorg. Chem., 2012, 51, 6200–6210 CrossRef CAS.
- C.-J. Liu, W.-Y. Yu, S.-M. Peng, T. C. W. Mak and C.-M. Che, J. Chem. Soc., Dalton Trans., 1998, 1805–1812 RSC.
- R. Horvath and K. C. Gordon, Coord. Chem. Rev., 2010, 254, 2505–2518 CrossRef CAS.
-
(a) S.-W. Lai, Y. J. Hou, C.-M. Che, H.-L. Pang, K.-Y. Wong, C. K. Chang and N. Zhu, Inorg. Chem., 2004, 43, 3724–3732 CrossRef CAS PubMed;
(b) D. B. Papkovsky and T. C. O'Riordan, J. Fluoresc., 2005, 15, 569–584 CrossRef CAS;
(c) W.-P. To, Y. Liu, T.-C. Lau and C.-M. Che, Chem.–Eur. J., 2013, 19, 5654–5664 CrossRef CAS.
- W. Sinha, L. Ravotto, P. Ceroni and S. Kar, Dalton Trans., 2015, 44, 17767–17773 RSC.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1993, 22, 113–262 CrossRef CAS.
-
(a) C.-M. Che, R. W.-Y. Sun, W.-Y. Yu, C.-B. Ko, N.-Y. Zhu and H. Z. Sun, Chem. Commun., 2003, 1718–1719 RSC;
(b) C.-T. Lum, Z. F. Yang, H.-Y. Li, R. W.-Y. Sun, S.-T. Fan, R. T.-P. Poon, M. C.-M. Lin, C.-M. Che and H.-F. Kung, Int. J. Cancer, 2006, 118, 1527–1538 CrossRef CAS PubMed;
(c) Y. Wang, Q. Y. He, R. W.-Y. Sun, C.-M. Che and J.-F. Chiu, Cancer Res., 2005, 65, 11553–11564 CrossRef CAS;
(d) R. W.-Y. Sun, C. K.-L. Li, D.-L. Ma, J. J. Yan, C.-N. Lok, C.-H. Leung, N. Zhu and C.-M. Che, Chem.–Eur. J., 2010, 16, 3097–3113 CrossRef CAS;
(e) R. W.-Y. Sun, A. L.-F. Chow, X.-H. Li, J. J. Yan, S. S.-Y. Chui and C.-M. Che, Chem. Sci., 2011, 2, 728–736 RSC;
(f) C.-T. Lum, R. W.-Y. Sun, T. T. Zou and C.-M. Che, Chem. Sci., 2014, 5, 1579–1584 RSC;
(g) C. Y. Chung, S. K. Fung, K.-C. Tong, P.-K. Wan, C.-N. Lok, Y. Huang, T. Chen and C.-M. Che, Chem. Sci., 2017, 8, 1942–1953 RSC;
(h) S. K. Fung, T. Zou, B. Cao, P.-Y. Lee, E. Y. M. Fung, D. Hu, C.-N. Lok and C.-M. Che, Angew. Chem., Int. Ed., 2017, 56, 3892–3896 CrossRef CAS.
- T. T. Zou, C.-N. Lok, Y.-M. Fung and C.-M. Che, Chem. Commun., 2013, 49, 5423–5425 RSC.
- B. J. Anding, J. Brgoch, G. J. Miller and L. K. Woo, Organometallics, 2012, 31, 5586–5590 CrossRef CAS.
- L. J. Boucher, Coord. Chem. Rev., 1972, 7, 289–329 CrossRef CAS.
-
(a) M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS;
(b) H.-G. Jeong and M.-S. Choi, Isr. J. Chem., 2016, 56, 110–118 CrossRef CAS;
(c) M. Soll, K. Sudhakar, N. Fridman, A. Müller, B. Röder and Z. Gross, Org. Lett., 2016, 18, 5840–5843 CrossRef CAS.
-
(a)
G. S. D. Tyler, Y.-Y. Huang and H. R. Michael, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, London, 2014, vol. 27, pp. 255–301 Search PubMed;
(b) S. Sunaina, A. Aggarwal, N. V. D. K. Bhupathiraju, G. Arianna, K. Tiwari and C. M. Drain, Chem. Rev., 2015, 115, 10261–10306 CrossRef.
- Y.-Z. Chen, Z. U. Wang, H. Wang, J. Lu, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2017, 139, 2035–2044 CrossRef CAS PubMed.
-
(a) L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef CAS;
(b) Y. You and W. Nam, Chem. Soc. Rev., 2012, 41, 7061–7084 RSC.
- J. J. Yan, A. L.-F. Chow, C.-H. Leung, R. W.-Y. Sun, D.-L. Ma and C.-M. Che, Chem. Commun., 2010, 46, 3893–3895 RSC.
- V. Bogoeva, M. Siksjø, K. G. Sæterbø, T. B. Melø, A. Bjørkøy, M. Lindgren and O. A. Gederaas, Photodiagn. Photodyn. Ther., 2016, 14, 9–17 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, Tables S1–S8, Fig S1–S27. CCDC 1846458–1846461. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02920b |
| ‡ Deceased 16 July 2017. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Ka-Chung
Tong
a,
Chen
Yang
a,
Ka-Chung
Tong
a,
Chen
Yang