
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Streptavidin interfacing as a general strategy to localize fluorescent membrane tension probes in cells†
Antoine
Goujon
,
Karolína
Straková
,
Naomi
Sakai
and
Stefan
Matile
 *
*
School of Chemistry and Biochemistry, National Centre of Competence in Research (NCCR) Chemical Biology, University of Geneva, Geneva, Switzerland. E-mail: stefan.matile@unige.ch; Web: http://www.unige.ch/sciences/chiorg/matile/
First published on 17th October 2018
Abstract
To image the mechanical properties of biological membranes, twisted push–pull mechanophores that respond to membrane tension by planarization in the ground state have been introduced recently. For their application in biological systems, these so-called fluorescent flippers will have to be localized to specific environments of cellular membranes. In this report, we explore streptavidin as a versatile connector between biotinylated flipper probes and biotinylated targets. Fluorescence spectroscopy and microscopy with LUVs and GUVs reveal the specific conditions needed for desthiobiotin-loaded streptavidin to deliver biotinylated flippers selectively to biotinylated membranes. Selectivity for biotinylated plasma membranes is also observed in HeLa cells, confirming the compatibility of this strategy with biological systems. Streptavidin interfacing does not affect the mechanosensitivity of the flipper probes, red shift in the excitation maximum and fluorescence lifetime increase with membrane order and tension, as demonstrated, inter alia, using FLIM.
Introduction
The fluorescence imaging of membrane tension in living cells is one of the more demanding challenges in current biological research that awaits solutions from chemistry.1 The fundamental problem is that forces as such are not directly visualizable, neither in cells nor elsewhere. It is only their consequences that can be imaged. For membrane tension, the consequences are diverse, differ for different membranes, and are often unknown, which is also because reliable fluorescent probes for routine studies have not been available.To image membrane tension in living cells, we have introduced the concept of planarizable push–pull probes,2 also referred to as “fluorescent flippers”.3 The current best mechanophore 1, also called FliptR (fluorescence lipid tension reporter),4 is constructed around two dithienothiophene (DTT)5 “flippers” (Fig. 1).6 They excel with the high surface area needed for high mechanosensitivity and intense monomer fluorescence to keep shining when twisted out of conjugation. This deplanarization is achieved by “chalcogen-bond7 repulsion” between methyls and σ holes next to the twistable bond between the two DTT flippers. The polarization of the twisted mechanophores is achieved by using sulfone acceptors and sulfide donors as bridges in the two DTTs. The former are supported by a cyano acceptor, the latter by an essential thenyl ether, presumably for intramolecular chalcogen bonding. A triazole is used to prevent protonation and the resulting degradation of the thenyl ether.6 The terminal carboxylate is placed to produce amphiphiles that form soluble, non-fluorescent micelles in water.
 | ||
| Fig. 1 (a) Structures of original flipper 1 (with the MEP surface of the planarized conformer; red, electron rich; blue, electron poor), flipper 2 for ganglioside recognition, and flipper 3 introduced in this study; (b) schematic indication of increasing flipper planarization with increasing membrane order, from Ld to So; and (c) the general concept of interfacing with close and remote targets through streptavidin. Relative orientations of ligands are arbitrary. | ||
In apolar solvents, the excitation maximum of flipper 1 is blue shifted. The increasing ground-state planarization of the push–pull probe with increasing order in lipid bilayer membranes shifts the excitation maximum to the red region (Fig. 1b).3 This red shift is accompanied by an increase in fluorescence intensity, i.e., lifetime. These changes in the lifetime are well suited for fluorescence imaging of cells by FLIM (fluorescence lifetime imaging microscopy), a method that is attractive because the readout is independent of probe concentration.4,6 Little change in emission confirms that flippers 1 do not operate in the excited state like most other fluorescent membrane probes,8–14 which function with mechanisms such as solvatochromism,8 TICT (molecular rotors),9 ESIPT,10 PET,11 FRET,12 vibrational unbending,13 and so on. Rather than reporting off-equilibrium on kinetics, that is viscosity, planarizable push–pull probes thus report exclusively on mechanical confinement in space under equilibrium conditions in the ground state.13
Unlike previously proposed optical tension probes,15 FliptR 1 proved compatible with routine imaging of membrane tension in living cells.4 Increasing membrane tension in homogeneous model membranes, applied either by osmotic shock or micropipette aspiration, was found to result in a linearly decreasing fluorescence lifetime. This outcome is consistent with lipid decompression and flipper deplanarization as a response to membrane tension in homogeneous membranes. Increasing membrane tension in phase-separating model membranes as well as cells resulted in a linearly increasing fluorescence lifetime. This is consistent with lipid reorganization, that is the appearance and disappearance of membrane domains, as a dominant response to tension. Tension-induced lipid reorganization has been confirmed to occur in model membranes,4,16 and the FliptR probe has already been used to demonstrate the relevance of tension-induced lipid reorganization for biological function, that is signal transduction.17 Lipid reorganization as a dominant response to membrane tension suggested that other existing membrane probes could, in principle, image membrane tension as well. Considering the many parameters that influence fluorescence response,4 this remains to be confirmed probe by probe, particularly considering that flippers report in the ground state on sterics, whereas other probes report off-equilibrium in the excited state on kinetics, that is viscosity.13
Flipper 1 labels the outer membrane of cells, without strong preferences for different domains. For biological studies, however, it is essential to localize membrane tension probes to specific membrane environments. Preliminary results in this direction have been obtained using a boronic acid containing flipper, 2, which was shown to partition better in ganglioside-enriched lipid domains of mixed-phase vesicles.18 This approach has potential to be extended for selective labeling of cellular organelles, such as mitochondria,19 ER, lysosomes and endosomes,20 through attachment of the well-established targeting units. On the other hand, selective tagging will be necessary to gain higher resolution insight into a particular protein. Compatibility of the Halo tag with molecular rotors has just been demonstrated,21 and SNAP tags22 and native ligand binding23 have been used to label the plasma membrane around surface receptors. In these pioneering studies on targeting, mechanophores were usually expected to report on organelle viscosity, and other probes were used just for labeling; all studied without explicit interest in lipid bilayer membranes, certainly not membrane tension.
In this report, we explore the scope and limitations of streptavidin as a universal connector of tension probes with biotinylated targets (Fig. 1c and 2). Streptavidin–biotin interfacing is one of the best explored methods in biotechnology.24–28 The multivalency of the streptavidin tetramer provides unique versatility; examples extend from the combination of cellular uptake with fluorescent labeling, molecular recognition, self-assembly and catalysis24 to the construction of ordered multicomponent architectures on solid surfaces (Fig. 1c).25 Of particular importance for bioconjugation applications is the AviTag technology, which allows the attachment of biotin ligands at specific positions in proteins of free choice.26 In the following, we introduce biotinylated flipper probe 3 (Fig. 1a) and elaborate on the interfacing with streptavidin 4 to biotinylated lipids in large unilamellar vesicles (LUVs) and giant unilamellar vesicles (GUVs) of different order, and in cells.
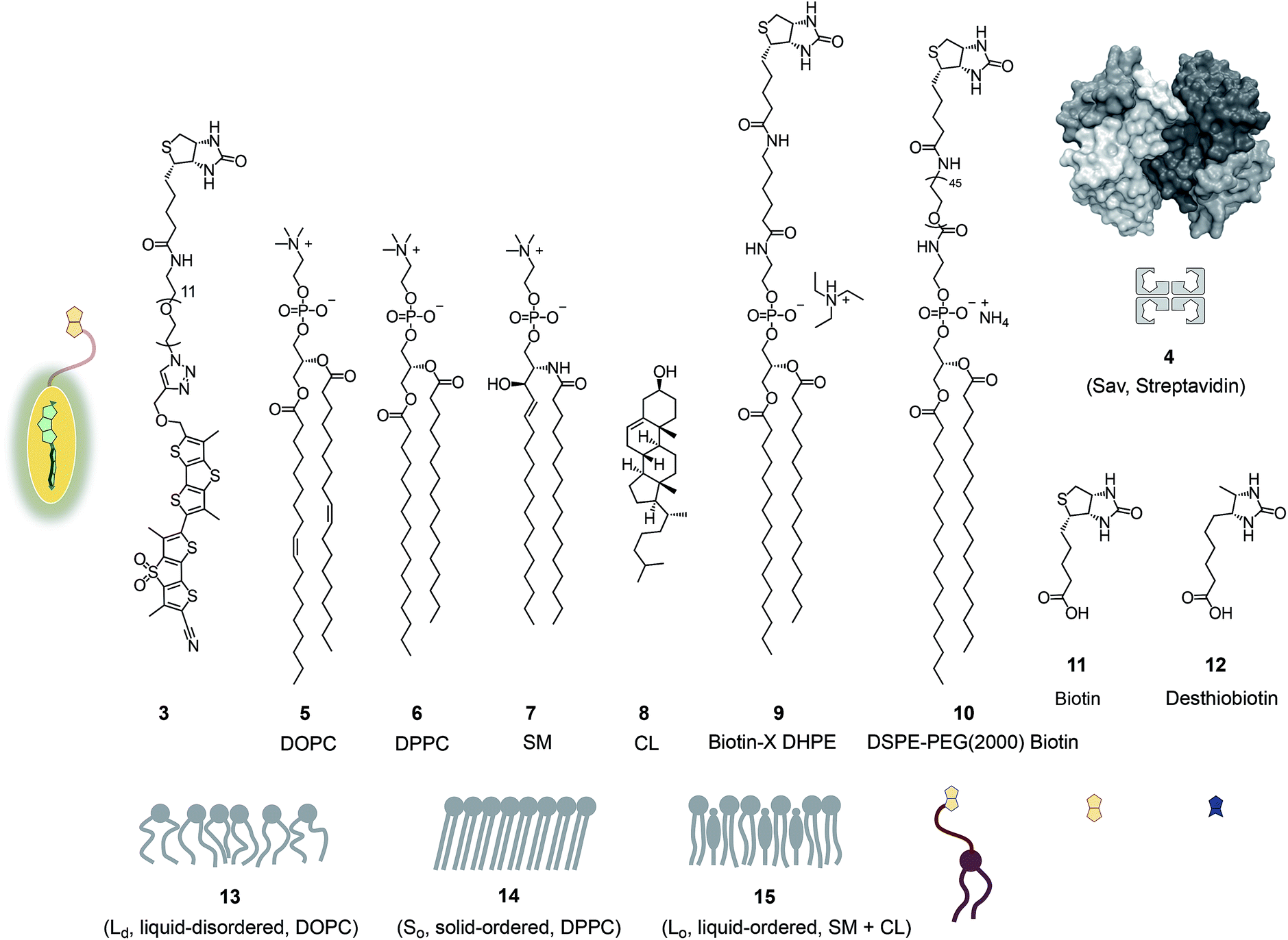 | ||
| Fig. 2 Structures, abbreviations, cartoons and numbering of molecules and molecular systems used in this study. | ||
Results and discussion
Fig. 2 shows a summary of structures, abbreviations, cartoons and numbering of molecules and molecular systems 3–15 used in this study. The syntheses of flipper probes 1 and 2 have been reported.6,18 To prepare biotinylated flipper 3, the CuAAC reaction was performed between the alkyne intermediate and an oligoethyleneglycol containing one azide and one amine terminus, and the resulting product was reacted with the activated NHS ester of biotin 11 (Scheme S1†).Biotinylated flippers
The properties of flipper 3 in lipid bilayer membranes were examined at 25 °C in LUVs of different composition: DOPC 5 (1,2-dioleoyl-sn-glycero-3-phosphocholine) for liquid-disordered (Ld) membranes 13, DPPC 6 (1,2-dipalmitoyl-sn-glycero-3-phosphocholine) for solid-ordered (So) membranes 14, and a mixture of SM 7 (sphingomyelin) and CL 8 (cholesterol) for liquid-ordered (Lo) membranes 15 (Fig. 2). The probe was added to the vesicles (75 μM lipid) in Tris buffer, pH 7.4, 25 °C, to reach final concentrations of 1.0, 0.75, 0.5 and 0.25 μM. When added to So membranes 14, mechanophore 3 gave a broad excitation peak with a maximum at λex = 490 nm (Fig. 3, red, solid). This red shifted λex demonstrated partitioning and planarization of the push–pull probe within the highly ordered membrane 16. The excitation maximum obtained in Lo membranes 17 was nearly the same as in So membrane 16 (Fig. 3, black, dashed). In contrast, the excitation maximum obtained in Ld membranes 18 was clearly blue shifted at λex = 430 nm (Fig. 3, blue, solid). This sensitivity toward membrane order was almost the same as with the original flipper probe 1.6 The increasing planarization of the biotinylated flipper 3 with increasing membrane order fully confirmed its operational mechanosensitivity.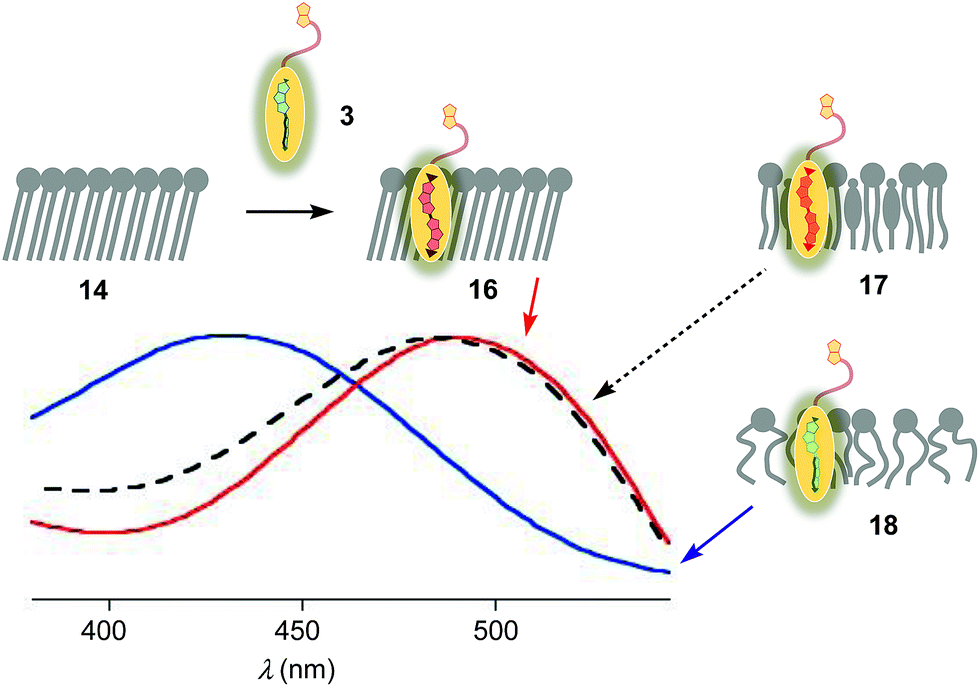 | ||
| Fig. 3 Normalized excitation spectra (λem = 570 nm) of flipper 3 (250 nM) in So (16, red), Lo (17, black) and Ld (18, blue) LUVs at a constant lipid concentration (75 μM) in 10 mM Tris, pH 7.4, 25 °C. | ||
Flipper–streptavidin complex 19 was prepared by adding one equivalent of flipper 3 per wild-type streptavidin tetramer 4 in a buffer at pH 7.4, at room temperature (Fig. 4a). Compared to the very weak fluorescence of the biotinylated flipper 3 in buffer (Fig. 4a, green, dashed), the formation of complex 19 caused an increase in intensity and a red shift of the excitation maximum to λex ∼ 430 nm (Fig. 4a, blue, dotted). The excitation spectra of flipper 3 bound to streptavidin 4 (i.e., 19) and Ld membranes (i.e., 18) were very similar. For the study of membrane-bound flippers, including cellular imaging, this similarity was irrelevant because the 4.8 times higher fluorescence intensity of flippers bound to Ld membranes (i.e., 18) made eventual background contributions from flipper–streptavidin complexes 19 in solution negligible (vide infra). Similar shifts at weaker intensity could suggest that in flipper–streptavidin complexes, the mechanophore interacts weakly with more hydrophobic domains on the protein surface24 to experience similar planarization but more rotational quenching compared to that in Ld membranes. In normalized spectra, eventual binding of 19 to Ld membranes 13 could thus not be detected from shifts of the excitation maxima. For the binding studies in LUVs described in the following, this overlap is irrelevant because the focus is on the more demanding and more informative So DPPC membranes 14. The red shifted λex = 490 nm of planarized mechanophores in these ordered membranes is readily detectable. Results with Lo SM/CL membranes 15 were often very similar to those in So DPPC 14.
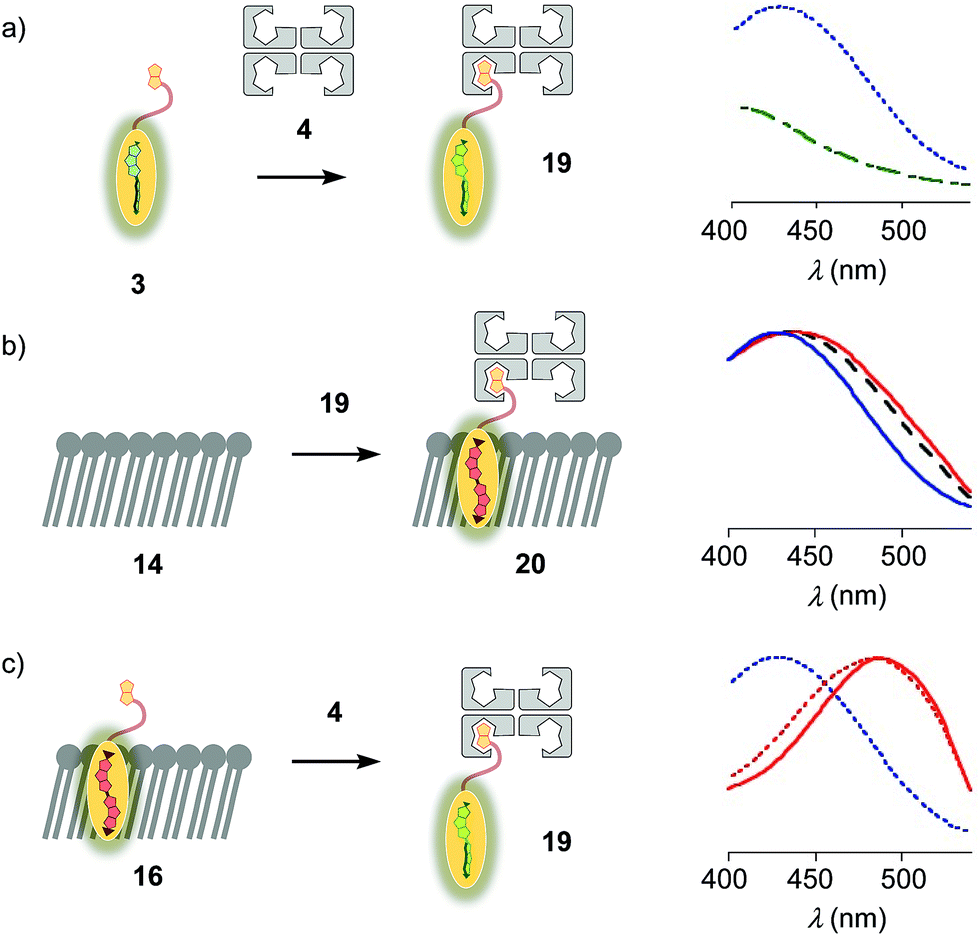 | ||
| Fig. 4 (a) Excitation spectra of flipper 3 before (dashed, green) and after the addition of one equivalent of 4 to yield 19 (dotted, blue). (b) Normalized excitation spectra recorded after the addition of 19 to membranes 13 (DOPC, solid, blue), 14 (DPPC, solid, red) and 15 (SM/CL, dashed, black), showing poor insertion of the probe into the membrane to yield complex 20 or equivalent. (c) Normalized excitation spectra of 16 (solid, red), 19 (dotted, blue) and 16 after addition of 4 (dotted, red), showing poor extraction of the probe from the membrane. | ||
The addition of flipper–streptavidin complex 19 to biotin-free So membranes 14 caused only a small peak broadening toward longer wavelengths in the excitation spectrum (Fig. 4b, red, solid). Spectral deconvolution,18 assuming contributions from membrane bound 20 and the unbound 19 only, suggested that the yield of complex 20 with planarized flippers in So membranes is 16%, while the large majority of flippers bound to streptavidin in complex 19 remain in solution. Similarly poor partitioning could be observed with Lo membranes 15 (13%, Fig. 4b, black, dashed). Reverse addition of streptavidin 4 to flipper–membrane complex 16 did not change much the red shifted excitation maxima of planarized flippers in So membranes (Fig. 4c, red, dashed red). Whereas complexation with streptavidin in 19 thus hindered the insertion of flippers into ordered membranes, streptavidin 4 failed to extract flippers 3 from ordered membranes. These differences did not disappear with time; spectra measured after 15 and 30 min were unchanged. They suggested that the consequences of spatial confinement are important: binding to one partner hinders accessibility to the other. For flipper–streptavidin interactions, this conclusion was consistent with the red shift found for complex 19 compared to unbound flipper 3 (Fig. 4a).
Biotinylated lipids
The schematic structures of all streptavidin complexes including 19 show the molar ratios of the components used. In reality, the multivalency of streptavidin complicates the situation.26–28 Without cooperativity effects, the complex stoichiometries reflecting the substrate ratio should dominate clearly.27 Conflicting reports suggest that cooperativity depends on the nature of the biotin ligand,27 and that, if desired, stoichiometries and structures of the complex can be controlled with mutants.26,28 It is generally accepted that the second biotin binds preferentially at the distant (≈3.5 nm) trans binding site rather than at the nearby (≈2.0 nm) cis site. Binding of the third and the fourth biotin ligands, inevitably at the cis positions of the first two ligands, could suffer from steric or charge repulsion. Anchoring of streptavidin on the bilayer surface requires divalent binding in the cis or trans orientation, and the remaining free sites can interact with more ligands.25 The availability of free binding sites could eventually be of interest to efficiently interface flipper probes with membrane proteins, either through biotinylated ligands of these receptors or strategically bioengineered biotin tags.26The biotinylated lipid 9 has been used previously to, for example, immobilize liposomes on streptavidin-coated surfaces, probe phosphoinositide–protein interactions, or assemble liposomes (Fig. 2).29 The addition of flipper–streptavidin–lipid complex 21 with, on average, one flipper 3 and three lipids 9 to So membranes 14 afforded complex 22 with planarized mechanophores in only 30%, according to spectral deconvolution (Fig. 5a, red, solid). Partitioning of complex 21 into Lo membranes 15 was even poorer (17%, Fig. 5a, black, dashed).
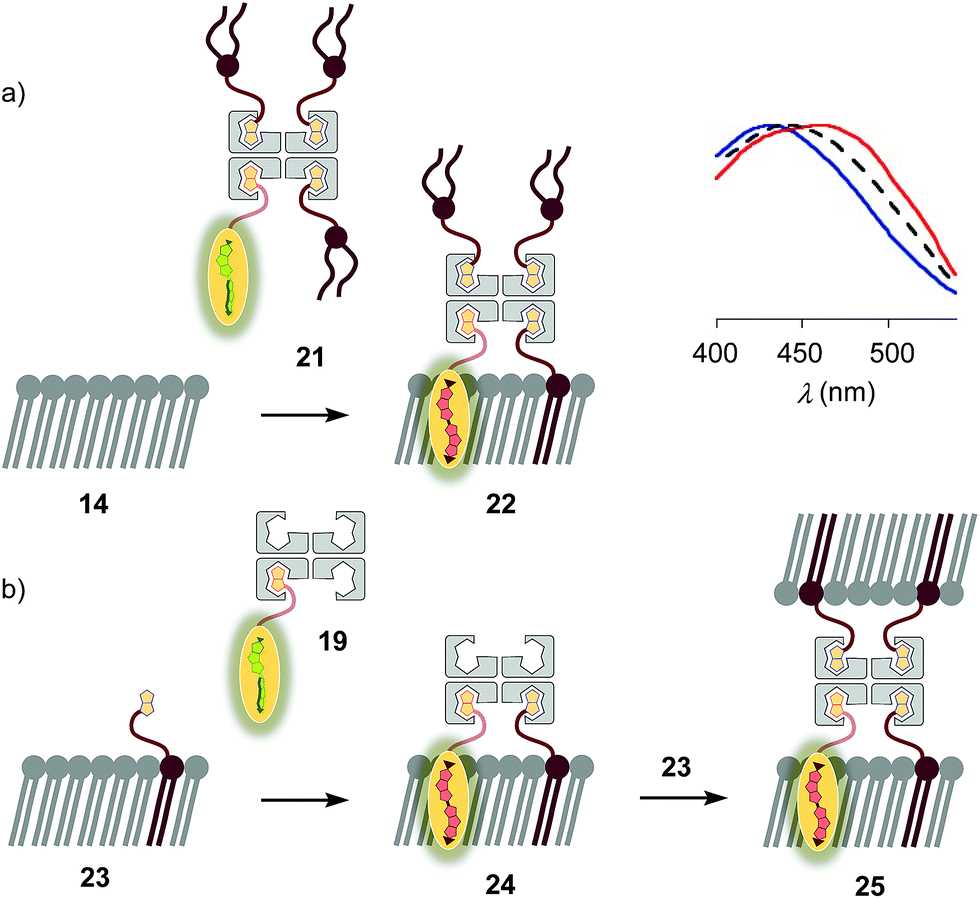 | ||
| Fig. 5 (a) Normalized excitation spectra recorded after the addition of complex 21 to membranes 13 (DOPC, blue), 15 (SM/CL, black) and 14 (DPPC, red; according to spectral deconvolution, formation of complex 22 occurred in 30%). (b) Fast vesicle precipitation resulted after the addition of complex 19 to biotinylated membranes 23. | ||
The complementary addition of flipper–streptavidin complex 19 to biotinylated So DPPC membranes 23 caused intense precipitation (5 mol% 9, Fig. 5b). Dominant precipitation from complex 24 was consistent with the crosslinking of vesicles through streptavidin binding to biotins in different membranes (Fig. 1c) to form complex 25.29c
To inhibit the formation of insoluble aggregates 25, the free binding sites in flipper–streptavidin complex 19 were “protected” with desthiobiotin 12 (Fig. 2 and 6). Desthiobiotin 12 has a high affinity for the binding pocket of streptavidin 4, but lower than that of biotin 11 itself (KD (11) = 40 fM, KD (12) = 500 fM).30 Upon addition of flipper–streptavidin–desthiobiotin complex 26 to biotinylated So membranes 23, precipitation was not observed, even after fifteen minutes. The red shifted excitation maximum was consistent with flipper planarization in So membranes, that is the successful formation of the desired interfaced architectures 27 (Fig. 6, red, solid). Similar flipper planarization was observed for the corresponding complex 28 in SM/CL Lo vesicles (Fig. 6, black, dashed), whereas the spectral signature of the interfaced complex 29 was consistent with that of the twisted flippers in Ld vesicles (Fig. 6, blue, solid).
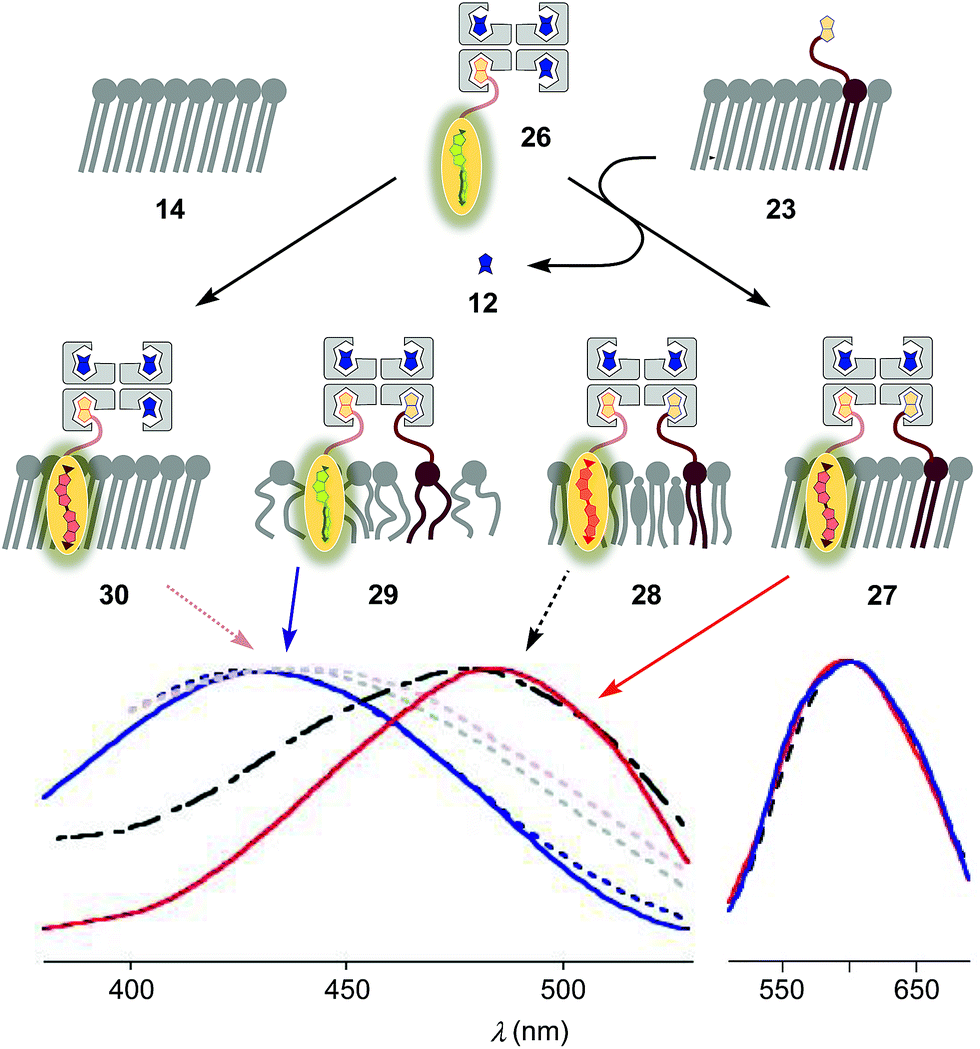 | ||
| Fig. 6 Normalized excitation (left) and emission spectra (right) recorded after the addition of complex 26 (250 nM) to Ld, Lo and So (23) membranes with 5 mol% biotinylated lipid 9 to afford 29 (blue, solid), 28 (black, dashed) and 27 (red, solid), respectively. Control experiments show that addition of 26 to non-biotinylated Ld (blue, dotted), Lo (grey, dotted) and So (14, red, dotted) membranes results in poor binding (e.g., 30). 75 μM lipid, 10 mM Tris, pH 7.4, 25 °C. | ||
Control experiments with non-biotinylated So membranes 14 gave insignificant red shifts upon addition of flipper–streptavidin–desthiobiotin complexes 26, confirming that the formation of complex 30 is negligible (Fig. 6, red, dotted). Control experiments with non-biotinylated Lo and Ld membranes gave similarly poor partitioning (Fig. 6, grey and blue, dotted). These consistent trends confirmed that desthiobiotins in complex 26 are efficiently displaced by the biotinylated lipids in membranes 23 and equivalent, leading to the insertion of the mechanosensitive probes into the membrane and formation of the correctly interfaced architectures 27–29.
The emission maxima of the interfaced flipper complexes 27–29 were almost identical (Fig. 6). This was as with non-interfaced flippers3,6 and confirmed that mechanosensitivity originates from planarization in the ground state in response to sterics.
At a constant biotin level in Lo membranes, the dependence of fluorescence intensity on flipper concentration was roughly linear up to at least 800 nM (Fig. 7a). Similar observations were made for Ld membranes (Fig. S1a†), while progressive saturation was monitored at higher concentrations in So membranes (Fig. S2a†). With ≈2 μM biotin available on the LUV surfaces, these results were consistent with the need for less than two equivalents of biotinylated lipids to anchor the streptavidin complex. The absence of evident saturation at submicromolar concentrations further supported that the interfaced flipper complexes 27 and equivalent operate as monomers and do not aggregate under these conditions.
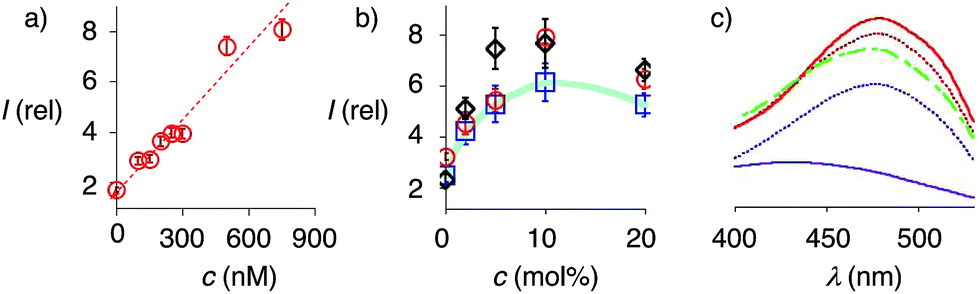 | ||
| Fig. 7 (a) Dependence of fluorescence intensity on the concentration of complex 26 after addition to Lo SM/CL LUVs containing 5 mol% 9. (b) Relative fluorescence intensity of complex 26 (250 nM) after addition to DOPC (blue squares), DPPC (red circles) and SM/CL (black diamonds) LUVs containing 0 to 20 mol% 9. (c) Excitation spectra of complex 26 recorded after addition to SM/CL LUVs with 0 (purple, solid), 2 (purple, dotted), 5 (red, dotted), 10 (red, solid) and 20 mol% 9 (green, dashed). Shown are the mean values ± standard errors from three independent experiments. | ||
At constant flipper concentration, the dependence of the fluorescence intensity, i.e., the formation of the interfaced complexes 27–29, on the concentration of biotinylated lipids 9 was bell-shaped for all membranes tested, with a maximum around 10 mol% (Fig. 7b, c, S1b and S2b†). Several explanations for this saturation and ultimately decrease at higher mole fractions were conceivable. The decreasing intensity coincided with peak broadening at a shorter wavelength, indicating hindered flipper partitioning and/or planarization due to the disturbed organization of these over-biotinylated membranes (Fig. 7c, green).
Substitution of desthiobiotin 12 in complex 26 by biotin 11 in flipper–streptavidin–biotin complex 31 hindered efficient flipper interfacing (Fig. 8a). The addition of complex 31 to biotinylated So membranes 23 gave a broad excitation peak with a maximum at 480 nm but a shoulder extending to 430 nm that is characteristic of incomplete flipper planarization. Spectra measured after 15 min were unchanged. Spectral deconvolution suggested that only 30% of complex 32 was formed with vesicles that were biotinylated with 5 mol% of lipid 9. This incomplete formation of complex 32 supported that the displacement of biotin or the biotinylated flipper is slow, and rapid desthiobiotin–biotin exchange is essential for correct and efficient interfacing.
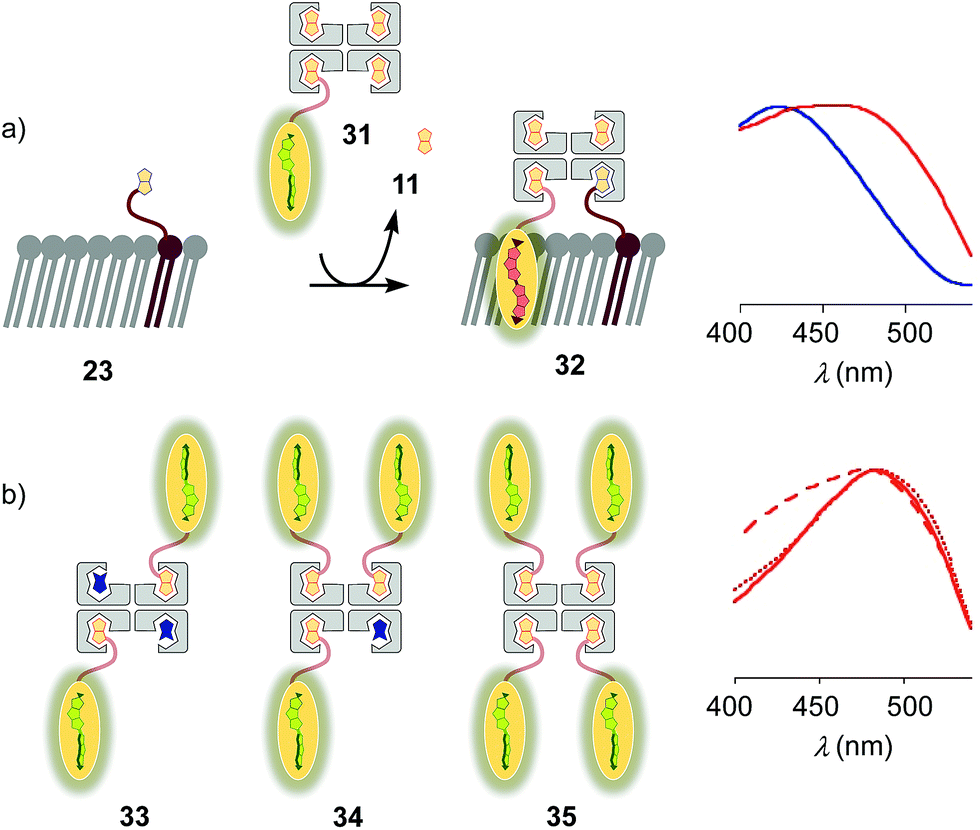 | ||
| Fig. 8 (a) Normalized excitation spectra of complex 31 after addition to DOPC (blue) and DPPC (red) LUVs prepared with 5 mol% 9. (b) Normalized excitation spectra of complex 26 (solid), 33 (dashed) and 34 (dotted) recorded after addition to 23 (i.e., DPPC LUVs prepared with 5 mol% 9). Recorded in LUVs at constant lipid concentration in 10 mM Tris, pH 7.4, 25 °C, with a concentration of 1 μM of probe 3. | ||
With increasing flipper content from 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 complex 33 to 3:1 complex 34 and 4:0 complex 35, the spectral signature of the target complex 27 did not improve with regard to flipper planarization, i.e., red shift (Fig. 8b). Spectral deconvolution revealed 77% insertion for 33 and 93% for 34 compared to 26. Red shift recovery from 33 to 34 was likely due to the displacement of a flipper 3 upon binding with 9, which then directly partitions into the membrane and increases the proportion of the planarized probe in the spectra with contributions from non-interfaced flippers 16 (Fig. 3). Further increasing flipper content in the pure 4:0 flipper–streptavidin complex 35 caused intense and instantaneous precipitation, possibly due to the partitioning of cis and trans flippers into different vesicles (as outlined for lipids in 25, Fig. 5).
:2 complex 33 to 3:1 complex 34 and 4:0 complex 35, the spectral signature of the target complex 27 did not improve with regard to flipper planarization, i.e., red shift (Fig. 8b). Spectral deconvolution revealed 77% insertion for 33 and 93% for 34 compared to 26. Red shift recovery from 33 to 34 was likely due to the displacement of a flipper 3 upon binding with 9, which then directly partitions into the membrane and increases the proportion of the planarized probe in the spectra with contributions from non-interfaced flippers 16 (Fig. 3). Further increasing flipper content in the pure 4:0 flipper–streptavidin complex 35 caused intense and instantaneous precipitation, possibly due to the partitioning of cis and trans flippers into different vesicles (as outlined for lipids in 25, Fig. 5).
The addition of biotinylated insulin 36 to the operational, correctly interfaced target complex 27 caused a gradual broadening of the excitation maxima of the planarized flippers toward the blue region (Fig. 9a). This result was consistent with the formation of first complex 37 with four different ligands bound to the tetravalent streptavidin, followed by flipper extraction from the membrane with complex 38 or similar, with two insulins and maybe also lipid 9 displaced by another insulin 36. Spectral deconvolution gave 41% flipper removal in the presence of ten equivalents of insulin 36 (Fig. 9b). This reluctant flipper removal from So membranes implied the formation of non-interfaced flipper 16via displacement of biotinylated flipper 3 in complex 37 by biotinylated insulin 36.
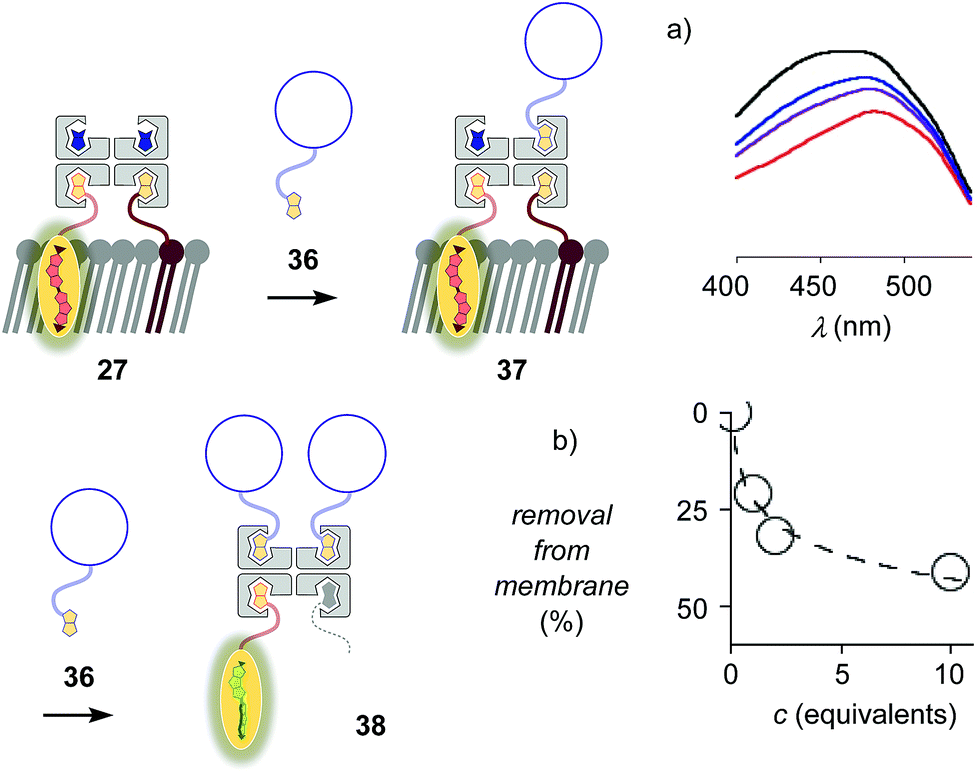 | ||
| Fig. 9 (a) Excitation spectra of complex 27 after the addition of 0 (red), 1 (purple), 2 (blue) and 10 (black) equivalents of biotinylated insulin 36. (b) Flipper removal from the membrane with increasing concentration of insulin, obtained from deconvolution of spectra in (a). | ||
Fluorescence imaging in GUVs
The lessons learned in LUVs were applied to imaging flipper interfacing in GUVs. For convenience only, the studies were carried out mostly in Lo SM/CL membranes 15 (Fig. 2). Confocal laser scanning microscopy (CLSM) images of biotinylated Lo membranes 39 (5 mol% 9) after addition of flipper–streptavidin complex 26 with exchangeable desthiobiotin 12 in the extra binding sites showed cleanly labeled GUVs without any precipitation i.e. the desired target complex 28 (Fig. 10a). The same flipper–streptavidin–desthiobiotin complex 26 failed to label Lo SM/CL membranes 15 without biotin on their surface (Fig. 10b). Moreover, the flipper–streptavidin complex 31 with poorly exchangeable biotin 11 rather than the readily substituted desthiobiotin 12 failed to label biotinylated Lo SM/CL membranes 39 (Fig. 10c). Finally, the addition of flipper–streptavidin complex 19 with neither desthiobiotin 12 nor biotin 11 in the extra binding sites produced labeled GUVs together with small and also very large precipitates, i.e., architectures 40 and 41 (Fig. 10d).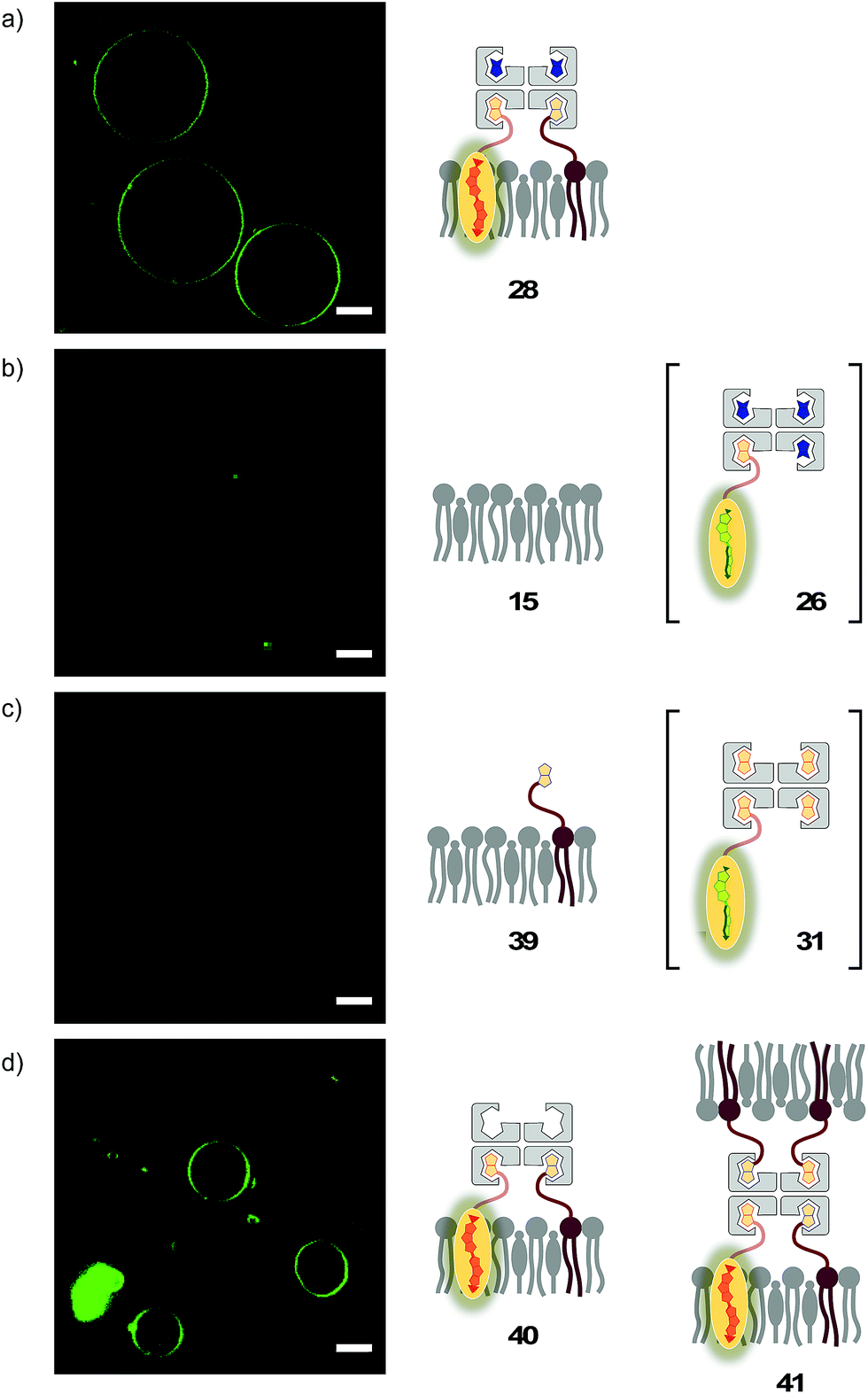 | ||
| Fig. 10 CLSM images of complex 26 added to (a) biotinylated (yielding 28) and (b) non-biotinylated Lo SM/CL GUVs, and (c) complex 31 and (d) complex 19 added to biotinylated Lo SM/CL GUVs. Pictures were taken 2 min after the addition of complexes, 500 nM 3, λex = 488 nm, λem = 600 ± 50 nm, 30% laser power, scale bar 10 μm. | ||
Biotin-free Lo SM/CL GUVs 15 loaded with sulforhodamine 101 (SR101) were imaged together with biotinylated Lo SM/CL GUVs 39 after the addition of the flipper–streptavidin–desthiobiotin complex 26. Consistent with the formation of the target complex 28, the membrane of biotinylated GUVs 39 could be clearly observed (in green), whereas the non-biotinylated but SR101-loaded GUVs 15 (in red) did not show fluorescently labeled membranes (Fig. 11a). This series of CLSM images confirmed that the correctly interfaced complex 28 is accessible exclusively by the addition of the flipper–streptavidin–desthiobiotin complex 26 to biotinylated membranes 39 (Fig. 10a and 11a) because the presence of desthiobiotin is essential (Fig. 10c and d) and, most importantly, non-biotinylated membranes are not labeled (Fig. 10b and 11a). The results with GUVs were in full agreement with the spectroscopic analysis in LUVs (Fig. 4b, 5b, 6, and 8a). GUV imaging thus validated the addition of the flipper–streptavidin–desthiobiotin complex 26 to biotinylated targets as the winning strategy for operational interfacing.
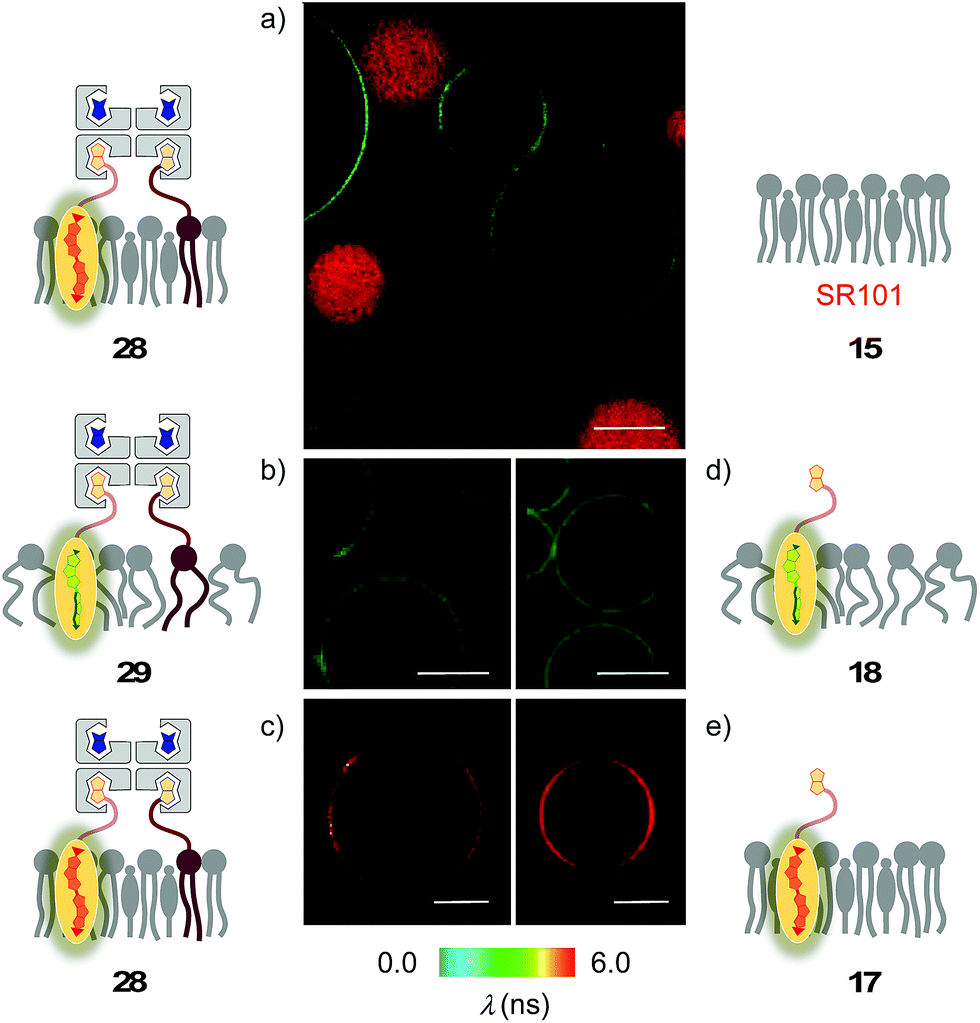 | ||
| Fig. 11 (a) CLSM images of complex 26 added to a mixture of SM/CL/9 (5%) GUVs (yielding 28, green) and SR101-loaded SM/CL GUVs (red). (b–e) FLIM images of complex 26 added to (b) DOPC/9 (5%) GUVs (yielding 29) and (c) SM/CL/9 (5%) GUVs (yielding 28), and of flipper 3 added to (d) DOPC GUVs (yielding 18) and (e) SM/CL GUVs (yielding 17). Pictures were taken after 2 min, 500 nM 3, λex = 488 nm, λem = 600 ± 50 nm, 30% laser power, scale bar 10 μm. | ||
FLIM was performed after the addition of the flipper–streptavidin complex 26 to biotinylated Lo SM/CL GUVs 39 (Fig. 11c). The fluorescence lifetime τLo = 5.7 ns obtained for the interfaced Lo complex 28 was in the range between τLo = 6.1 ns measured after the addition of flipper 3 to Lo SM/CL GUVs, i.e., complex 17 (Fig. 11e), and τLo = 5.8 ns reported6 for the original flipper 1 in Lo membranes. Consistent with the lifetime of the original flipper 1 in Ld membranes, the fluorescence lifetimes obtained with flipper 3 in Ld DOPC GUVs, i.e., complex 18, were much shorter (τLd = 3.8 ns, Fig. 11d). FLIM images of flipper–streptavidin–desthiobiotin complex 26 added to biotinylated Ld DOPC GUVs, i.e., the interfaced Ld complex 29, gave the same lifetime (τLd = 3.8 ns, Fig. 11b). These trends confirmed that the fluorescence property of planarized flippers in adequately interfaced Lo architectures 28 and deplanarized flippers in Ld architectures 29 is similar to that of flippers in the absence of streptavidin. In other words, streptavidin interfacing did not disturb the operation of flipper probes and is thus compatible with FLIM imaging of rationally localized membrane tension in cells.
Fluorescence imaging in cells
Cell surface biotinylation was achieved by growing HeLa Kyoto cells for three days in the presence of DSPE-PEG(2000) biotin 10 (Fig. 2). This lipid has been used routinely to, for example, immobilize GUVs on the surface, quantify the uptake of viruses in cells, or study the partitioning of synthetic lipids in mixed phase GUVs.4,31 The biotinylated lipid 9 used in vesicles could not be used in cells because it was not soluble enough in solvents miscible with water (methanol, DMSO, etc.) to be added to the cellular growth medium.The addition of the operational flipper–streptavidin–desthiobiotin complex 26 to the biotinylated HeLa cells selectively stained the plasma membrane with little background signal (Fig. 12a). In contrast, the addition of complex 26 to HeLa cells without biotinylation did not result in any significant fluorescence (Fig. 12b). FLIM experiments with interfaced flipper 3 in the biotinylated plasma membrane of HeLa cells gave a fluorescence lifetime of τ = 5.5 ns (Fig. 12c). This lifetime was similar to the one measured for the original flipper 1 in the plasma membrane of HeLa cells.4 Under hyperosmotic conditions, the lifetime of the interfaced flipper 3 in the plasma membrane of HeLa cells decreased significantly to τ = 4.95 ns (Fig. 12d). This response to the reduction of membrane tension was as with the original flipper 1.4 The decrease of flipper lifetime with tension has been proposed to originate from tension-induced lipid reorganization dominated by the disappearance of highly ordered domains with long-lived, strongly emitting planarized flipper probes.
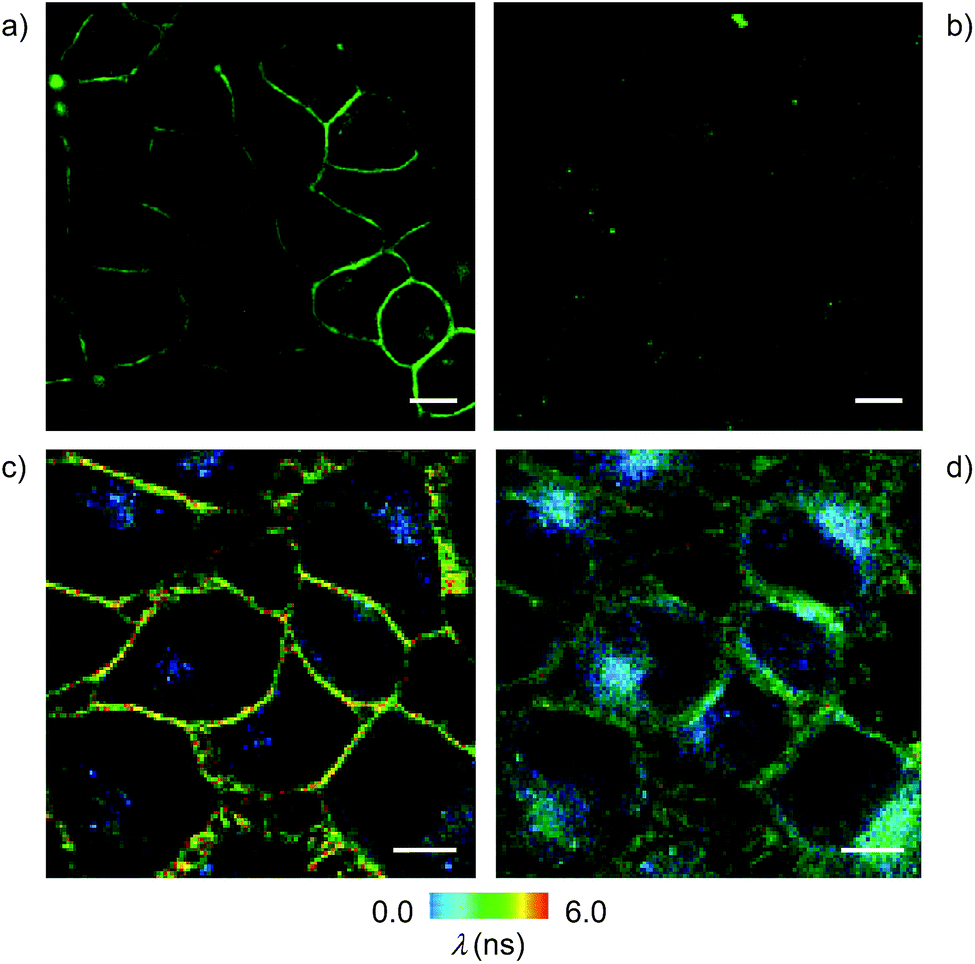 | ||
| Fig. 12 CLSM images of HeLa Kyoto cells with (a) and without (b) pre-incubation with biotinylated lipid 10 (25 μg mL−1) for 3 days, followed by washing and incubation with 26 (2 μM, 5 min; scale bar: 10 μm). FLIM images of HeLa cells incubated with 10 (25 μg mL−1) for 3 days followed by washing and incubation with 26 (2 μM, 5 min) under (c) isoosmotic and (d) hyperosmotic conditions. | ||
In sharp contrast to the original flipper probe 1, hyperosmotic shock not only reduced the lifetime of interfaced flippers in the plasma membrane but also caused rapid partial internalization and the appearance of punctate spots with a very short lifetime: τ = 2.8 ns. Although explanations on their origin remain to be found, the different responses to membrane tension observed with the original flipper 1 and interfaced flipper 3 beautifully forecast the specific information that will become available with the introduction of interfacing strategies to rationally localize flipper mechanophores within cells.
Conclusions
With fluorescent probes for the routine imaging of membrane tension in cells in hand,4 the next milestone will be the development of a universal interfacing strategy to measure membrane tension at any place in any living cell. This report suggests that streptavidin interfacing can meet this important challenge. The key to success was to protect extra binding sites in the tetrameric interface with exchangeable desthiobiotin (Fig. 6). Streptavidin complexes with a biotinylated flipper and desthiobiotin exchangers are shown to specifically label membranes that contain biotinylated lipids. As shown by fluorescence spectroscopy and FLIM, the probe retains its mechanosensitive properties even if it is part of such large supramolecular architectures, and can reveal unique characteristics of the target. Preliminary results for staining biotinylated plasma membranes are promising (Fig. 12). Compatibility with further interfacing to membrane proteins, either through biotinylated ligands or engineered AviTags,26 is demonstrated.The potential identified in this paper will have to be validated in studies on real biological problems and compared to other approaches such as Halo tags,21 SNAP tags22 or IEDDA ligation with artificial amino acids in engineered proteins.32 The unique versatility of a connector with four similar but non-identical binding sites is advantageous and disadvantageous at the same time. One disadvantage is the presence of mixtures of complexes with different stoichiometries. However, our findings suggest that this unsatisfactory heterogeneity has surprisingly little relevance when it comes to the specific labeling of biotinylated membranes in practice (Fig. 11 and 12). A general advantage of tetravalent interfacing is access to multiple functionalities. For example, membrane interfacing can be coupled with protein interfacing (Fig. 9), or with cellular delivery vehicles.24
Experimental section
See ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank J. Mareda (U Geneva) for computational modeling, A. Roux and A. Colom for assistance with cellular imaging, the NMR, MS and bioimaging platforms for services, and the University of Geneva, the Swiss National Centre of Competence in Research (NCCR) Molecular Systems Engineering, the NCCR Chemical Biology and the Swiss NSF for financial support.Notes and references
- (a) A. Diz-Muñoz, O. D. Weiner and D. A. Fletcher, Nat. Phys., 2018, 14, 648–652 Search PubMed; (b) B. Pontes, P. Monzo and N. C. Gauthier, Semin. Cell Dev. Biol., 2017, 71, 30–41 CrossRef CAS; (c) P. Sens and J. Plastino, J. Phys.: Condens. Matter, 2015, 27, 273103 CrossRef; (d) A. Anishkin, S. H. Loukin, J. Teng and C. C. Kung, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7898–7905 CrossRef CAS PubMed.
- A. Fin, A. Vargas Jentzsch, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2012, 51, 12736–12739 CrossRef CAS.
- (a) M. Dal Molin, Q. Verolet, A. Colom, R. Letrun, E. Derivery, M. Gonzalez-Gaitan, E. Vauthey, A. Roux, N. Sakai and S. Matile, J. Am. Chem. Soc., 2015, 137, 568–571 CrossRef CAS; (b) M. Macchione, M. Tsemperouli, A. Goujon, A. R. Mallia, N. Sakai, K. Sugihara and S. Matile, Helv. Chim. Acta, 2018, 101, e1800014 CrossRef.
- A. Colom, E. Derivery, S. Soleimanpour, C. Tomba, M. Dal Molin, N. Sakai, M. Gonzalez-Gaitan, S. Matile and A. Roux, Nat. Chem. DOI:10.1038/s41557-018-0127-3 , in press.
- (a) M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036–3140 CrossRef CAS; (b) G. Barbarella and F. Di Maria, Acc. Chem. Res., 2015, 48, 2230–2241 CrossRef CAS.
- S. Soleimanpour, A. Colom, E. Derivery, M. Gonzalez-Gaitan, A. Roux, N. Sakai and S. Matile, Chem. Commun., 2016, 52, 14450–14453 RSC.
- (a) B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS; (b) N. Biot and D. Bonifazi, Chem.–Eur. J., 2018, 24, 5439–5443 CrossRef CAS PubMed; (c) S. Benz, C. Besnard and S. Matile, Helv. Chim. Acta, 2018, 101, e1800075 CrossRef.
- (a) A. S. Klymchenko and R. Kreder, Chem. Biol., 2014, 21, 97–113 CrossRef CAS; (b) T. Baumgart, G. Hunt, E. R. Farkas, W. W. Webb and G. W. Feigenson, Biochim. Biophys. Acta, 2007, 1768, 2182–2194 CrossRef CAS; (c) I. A. Karpenko, M. Collot, L. Richert, C. Valencia, P. Villa, Y. Mély, M. Hibert, D. Bonnet and A. S. Klymchenko, J. Am. Chem. Soc., 2015, 137, 405–412 CrossRef CAS.
- (a) M. A. Haidekker and E. A. Theodorakis, J. Mater. Chem. C, 2016, 4, 2707–2718 RSC; (b) Z. Yang, Y. He, J.-H. Lee, N. Park, M. Suh, W.-S. Chae, J. Cao, X. Peng, H. Jung, C. Kang and J. S. Kim, J. Am. Chem. Soc., 2013, 135, 9181–9185 CrossRef CAS; (c) F. Liu, T. Wu, J. Cao, S. Cui, Z. Yang, X. Qiang, S. Sun, F. Song, J. Fan, J. Wang and X. Peng, Chem.–Eur. J., 2013, 19, 1548–1553 CrossRef CAS; (d) P. Sherin, I. López-Duarte, M. R. Dent, M. Kubánková, A. Vyšniauskas, J. A. Bull, E. S. Reshetnikova, A. S. Klymchenko, Y. P. Tsentalovich and M. K. Kuimova, Chem. Sci., 2017, 8, 3523–3528 RSC; (e) R. Guo, J. Yin, Y. Ma, Q. Wang and W. J. Lin, J. Mater. Chem. B, 2018, 6, 2894–2900 RSC; (f) L.-L. Li, K. Li, M.-Y. Li, L. Shi, Y.-H. Liu, H. Zhang, S.-L. Pan, N. Wang, Q. Zhou and X.-Q. Yu, Anal. Chem., 2018, 90, 5873–5878 CrossRef CAS; (g) A. Vysniauskas, M. Balaz, H. L. Anderson and M. K. Kuimova, Phys. Chem. Chem. Phys., 2015, 17, 7548–7554 RSC; (h) A. Vysniauskas, D. Ding, M. Qurashi, I. Boczarow, M. Balaz, H. L. Anderson and M. K. Kuimova, Chem.–Eur. J., 2017, 23, 11001–11010 CrossRef CAS.
- (a) A. Klymchenko, S. Oncul, P. Didier, E. Schaub, L. Bagatolli, G. Duportail and Y. Mély, Biochim. Biophys. Acta, 2009, 1788, 495–499 CrossRef CAS; (b) J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC.
- R. U. Kulkarni and E. W. Miller, Biochemistry, 2017, 56, 5171–5177 CrossRef CAS.
- (a) Y. Xiong, A. Vargas Jentzsch, J. Osterrieth, E. Sezgin, I. V. Sazanovich, K. Reglinski, S. Galiani, A. W. Parker, C. Eggeling and H. L. Anderson, Chem. Sci., 2018, 9, 3029–3040 RSC; (b) Y. Wang, Y. Liu, H. A. DeBerg, T. Nomura, M. T. Hoffman, P. R. Rohde, K. Schulten, B. Martinac and P. R. Selvin, eLife, 2014, 3, e01834 CrossRef.
- H. V. Humeniuk, A. Rosspeintner, G. Licari, V. Kilin, L. Bonacina, E. Vauthey, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2018, 57, 10559–10563 CrossRef CAS.
- N. Sakai and S. Matile, J. Am. Chem. Soc., 2018, 140, 11438–11443 CrossRef CAS.
- (a) Y. Liu, K. Galior, V. P.-Y. Ma and K. Salaita, Acc. Chem. Res., 2017, 50, 2915–2924 CrossRef CAS; (b) Y.-L. Zhang, J. A. Frangos and M. Chachisvilis, Biochem. Biophys. Res. Commun., 2006, 347, 838–841 CrossRef CAS; (c) D. T. Warshaviak, M. J. Muellner and M. Chachisvilis, Biochim. Biophys. Acta, 2011, 1808, 2608–2617 CrossRef CAS; (d) N. P. Kamat, Z. Liao, L. E. Moses, J. Rawson, M. Therien, I. J. Dmochowski and D. A. Hammer, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13984–13989 CrossRef CAS; (e) T. Muraoka, K. Umetsu, K. V. Tabata, T. Hamada, H. Noji, T. Yamashita and K. Kinbara, J. Am. Chem. Soc., 2017, 139, 18016–18023 CrossRef CAS; (f) R. H. Templer, S. J. Castle, A. R. Curran, G. Rumbles and D. R. Klug, Faraday Discuss., 1999, 111, 41–53 RSC.
- (a) J. C. S. Ho, P. Rangamani, B. Liedberg and A. N. Parikh, Langmuir, 2016, 32, 2151–2163 CrossRef CAS; (b) D. Chen and M. M. Santore, Biochim. Biophys. Acta, 2014, 1838, 2788–2797 CrossRef CAS PubMed; (c) T. Hamada, Y. Kishimoto, T. Nagasaki and M. Takagi, Soft Matter, 2011, 7, 9061–9068 RSC.
- M. Riggi, K. Niewola-Staszkowska, N. Chiaruttini, A. Colom, B. Kusmider, S. Soleimanpour, M. Stahl, S. Matile, A. Roux and R. Loewith, Nat. Cell Biol., 2018, 20, 1043–1051 CrossRef CAS.
- K. Strakova, S. Soleimanpour, M. Diez-Castellnou, N. Sakai and S. Matile, Helv. Chim. Acta, 2018, 101, e1800019 CrossRef.
- A. Jiménez-Sánchez, E. K. Lei and S. O. Kelley, Angew. Chem., Int. Ed., 2018, 57, 8891–8895 CrossRef.
- (a) H. Zhu, J. Fan, J. Du and X. Peng, Acc. Chem. Res., 2016, 49, 2115–2126 CrossRef CAS; (b) F. Hu and B. Liu, Org. Biomol. Chem., 2016, 14, 9931–9944 RSC; (c) N. Wagner, M. Stephan, D. Höglinger and A. Nadler, Angew. Chem., Int. Ed., 2018, 57, 13339–13343 CrossRef CAS.
- J. E. Chambers, M. Kubánková, R. G. Huber, I. López-Duarte, E. Avezov, P. J. Bond, S. J. Marciniak and M. K. Kuimova, ACS Nano, 2018, 12, 4398–4407 CrossRef CAS.
- E. Prifti, L. Reymond, M. Umebayashi, R. Hovius, H. Riezman and K. Johnsson, ACS Chem. Biol., 2014, 9, 606–612 CrossRef CAS.
- I. A. Karpenko, M. Collot, L. Richert, C. Valenca, P. Villa, Y. Mély, M. Hibert, D. Bonnet and A. S. Klymchenko, J. Am. Chem. Soc., 2015, 137, 405–412 CrossRef CAS.
- (a) F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142–231 CrossRef CAS; (b) T. R. Ward, Acc. Chem. Res., 2011, 44, 47–57 CrossRef CAS; (c) Y. Okamoto, R. Kojima, F. Schwizer, E. Bartolami, T. Heinisch, S. Matile, M. Fussenegger and T. R. Ward, Nat. Commun., 2018, 9, 1943 CrossRef; (d) Y. Cotelle, V. Lebrun, N. Sakai, T. R. Ward and S. Matile, ACS Cent. Sci., 2016, 2, 388–393 CrossRef CAS; (e) P. Morelli, E. Bartolami, N. Sakai and S. Matile, Helv. Chim. Acta, 2018, 101, e1700266 CrossRef; (f) C. M. Dundas, D. Demonte and S. Park, Appl. Microbiol. Biotechnol., 2013, 97, 9343–9353 CrossRef CAS.
- (a) S. A. Darst, M. Ahlers, P. H. Meller, E. W. Kubalek, R. Blankenburg, H. O. Ribi, H. Ringsdorf and R. D. Kornberg, Biophys. J., 1991, 59, 387–396 CrossRef CAS; (b) L. Välimaa, K. Pettersson, M. Vehniäinen, M. Karp and T. Lövgren, Bioconjugate Chem., 2003, 14, 103–111 CrossRef; (c) G. V. Dubacheva, C. Araya-Callis, A. Geert Volbeda, M. Fairhead, J. Codée, M. Howarth and R. P. Richter, J. Am. Chem. Soc., 2017, 139, 4157–4167 CrossRef CAS PubMed.
- (a) M. Howarth and A. Y. Ting, Nat. Protoc., 2008, 3, 534–545 CrossRef CAS PubMed; (b) Y. Li and R. Sousa, Protein Expression Purif., 2012, 82, 162–167 CrossRef CAS.
- (a) A. Loosli, U. E. Rusbandi, J. Gradinaru, K. Bernauer, C. W. Schlaepfer, M. Meyer, S. Mazurek, M. Novic and T. R. Ward, Inorg. Chem., 2006, 45, 660–668 CrossRef CAS; (b) H. J. Gruber, M. Marek, H. Schindler and K. Kaiser, Bioconjugate Chem., 1997, 8, 552–559 CrossRef CAS.
- (a) M. Howarth, D. J.-F. Chinnapen, K. Gerrow, P. C. Dorrestein, M. R. Grandy, N. L. Kelleher, A. El-Husseini and A. Y. Ting, Nat. Methods, 2006, 3, 267–273 CrossRef CAS; (b) X. Sun, D. Montiel, H. Li and H. Yang, Bioconjugate Chem., 2014, 25, 1375–1380 CrossRef CAS; (c) M. Fairhead, D. Krndija, E. D. Lowe and M. Howarth, J. Mol. Biol., 2014, 426, 199–214 CrossRef CAS.
- (a) B. Pignataro, C. Steinem, H.-J. Galla, H. Fuchs and A. Janshoff, Biophys. J., 2000, 78, 487–498 CrossRef CAS; (b) A. Knödler and P. Mayinger, BioTechniques, 2005, 38, 858–862 CrossRef; (c) S. Chiruvolu, S. Walker, J. Israelachvili, F.-J. Schmitt, D. Leckband and J. A. Zasadzinski, Science, 1994, 264, 1753–1756 CrossRef CAS; (d) E. T. Kisak, M. T. Kennedy, D. Trommeshauser and J. A. Zasadzinski, Langmuir, 2000, 16, 2825–2831 CrossRef CAS; (e) P. Vermette, S. Taylor, D. Dunstan and L. Meagher, Langmuir, 2002, 18, 505–511 CrossRef CAS.
- (a) J. D. Hirsch, L. Eslamizar, B. J. Filanoski, N. Malekzadeh, R. P. Haugland, J. M. Beechem and R. P. Haugland, Anal. Biochem., 2002, 308, 343–357 CrossRef CAS; (b) Z. Parandoosh, S. K. Knowles, X. Y. Xiao, C. Zhao, G. S. David and M. P. Nova, Comb. Chem. High Throughput Screening, 1998, 1, 135–142 CAS.
- (a) C. Lv, Y. Lin, A.-A. Liu, Z.-Y. Hong, L. Wen, Z. Zhang, Z.-L. Zhang, H. Wang and D.-W. Pang, Biomaterials, 2016, 106, 69–77 CrossRef CAS; (b) N. Momin, S. Lee, A. K. Gadok, D. J. Busch, G. D. Bachand, C. C. Hayden, J. C. Stachowiak and D. Y. Sasaki, Soft Matter, 2015, 11, 3241–3250 RSC.
- B. Oller-Salvia, G. Kym and J. W. Chin, Angew. Chem., Int. Ed., 2018, 57, 2831–2834 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures and results for all reported experiments. See DOI: 10.1039/c8sc03620a |
| This journal is © The Royal Society of Chemistry 2019 |