
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective parallel G-quadruplex recognition by a NIR-to-NIR two-photon squaraine†
Vincenzo
Grande‡
ab,
Chia-An
Shen‡
a,
Marco
Deiana
c,
Marta
Dudek
c,
Joanna
Olesiak-Banska
 c,
Katarzyna
Matczyszyn
*c and
Frank
Würthner
*ab
c,
Katarzyna
Matczyszyn
*c and
Frank
Würthner
*ab
aUniversität Würzburg, Institut für Organische Chemie, Am Hubland, 97074 Würzburg, Germany. E-mail: wuerthner@uni-wuerzburg.de
bCenter for Nanosystems Chemistry & Bavarian Polymer Institute (BPI), Universität Würzburg, Theodor-Boveri-Weg, 97074 Würzburg, Germany
cAdvanced Materials Engineering and Modelling Group, Faculty of Chemistry, Wroclaw University of Science and Technology, Wybrzeze Wyspianskiego 27, 50-370 Wroclaw, Poland. E-mail: katarzyna.matczyszyn@pwr.edu.pl
First published on 14th September 2018
Abstract
Fluorescence imaging probes for specific G-quadruplex (G4) conformations are of considerable interest in biomedical research. Herein, we present the synthesis and the binding properties of a new water-soluble near-infrared (NIR) amphiphilic squaraine dye (CAS-C1) which is capable of selective detection of parallel over non-parallel and non G4 topologies. The striking changes in its linear optical response upon binding to parallel G4s give rise to high fluorescence quantum yields (Φf ≈ 0.7) and one-photon molecular brightness in the far-red-NIR region. The outstanding recognition process of CAS-C1 for parallel G4s via end-stacking provides binding constants in the nanomolar regime (Kb = 107 to 108 M−1) awarding it as one of the most potent parallel G4 binders currently available. Moreover, the CAS-C1-parallel G4 system exhibits large two-photon absorption (TPA) cross-sections and molecular brightness in the second NIR biological transparency window (λ ≈ 1275 nm), making it an ideal candidate for NIR-to-NIR ultrasensitive two-photon procedures.
Introduction
Single-stranded guanine-rich oligonucleotides can fold into noncanonical compact, highly stable four-stranded helices known as G-quadruplexes (from here denoted as G4s).1 Recent studies have demonstrated the occurrence of G4s in human cells and their implication in several key biological events especially those that are disease- and aging-related.2,3 As a consequence, they are recognized as potential drug targets. To date, over 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 G4s have been identified across the human genome,4 especially located in the telomeres at the ends of chromosomes and in the promoter regions of oncogenes. Although indisputable progresses have been made over the past years in the design of G4-binders,5–10 the development of more complex multitasking compounds that combine the optimization of both optical performances (excitation and emission wavelengths > 600 nm and high brightness) and G4 structural selectivity/affinity (G4 over duplex binding and among the G4 topologies, Fig. 1) remains highly challenging.11–13 Indeed, the overwhelming majority of the small optical imaging probes for G4 detection are commonly photoexcited by relatively high-to-moderate energy light (330–600 nm)14,15 in order to generate the emission between the visible and far-red region of the electromagnetic spectrum. Such energies, in particular UV or blue, are in themselves deleterious to cells as they can irreversibly damage DNA and/or induce formation of cytotoxic reactive oxygen species.16 Furthermore, their excitation and emission wavelengths are confined outside the first and second near-infrared (NIR) biological transparency windows (BTWs, NIR-I: 700–950 nm and NIR-II: 1000–1350 nm)17 where absorption and scattering from tissues and oxy/deoxygenated blood is significant, limiting their relevance to 2-D cell cultures and precluding their use in more realistic diagnosis environments, such as in thick tissues or in vivo models. The lack of biocompatible fluorescent organic probes in the BTWs has prevented the use of these highly sensitive spectral ranges for in-depth imaging.17 In addition, the dynamic nature of the G4 structure enable a range of different topologies (e.g. parallel, hybrid and antiparallel) to potentially form under different physiological conditions, making molecular recognition particularly arduous. In our research on G4-probes, we have recently shown that a dicyanovinyl squaraine dye (SQgI, Fig. 1a)18 could behave as G4-specific NIR light-up probe. In the complexed state, SQgl is emissive at 700 nm upon excitation at 661 nm through one-photon absorption (OPA) with an impressive quantum yield of ΦF ≈ 0.6. The probe exhibited good selectivity of G4 over duplex, with the dicyanovinyl squaraine dye capable to impart outstanding selectivity over non-G4 structures and remarkable specificity toward parallel G4 topologies. However, this probe presented inherent optical, practical and affinity limitations including: (i) moderate solubility, (ii) moderate binding constants (Kb values comprised between 105 to 106 M−1)18–20 and (iii) excitation wavelengths outside the BTWs. Thus, we speculated that exploring the chemical space around the squaraine core should enable optimization of both optical and DNA binding performances and we decided to decorate the dicyanovinylene squaraine moiety with bulkier water-soluble brush-type groups.
000 G4s have been identified across the human genome,4 especially located in the telomeres at the ends of chromosomes and in the promoter regions of oncogenes. Although indisputable progresses have been made over the past years in the design of G4-binders,5–10 the development of more complex multitasking compounds that combine the optimization of both optical performances (excitation and emission wavelengths > 600 nm and high brightness) and G4 structural selectivity/affinity (G4 over duplex binding and among the G4 topologies, Fig. 1) remains highly challenging.11–13 Indeed, the overwhelming majority of the small optical imaging probes for G4 detection are commonly photoexcited by relatively high-to-moderate energy light (330–600 nm)14,15 in order to generate the emission between the visible and far-red region of the electromagnetic spectrum. Such energies, in particular UV or blue, are in themselves deleterious to cells as they can irreversibly damage DNA and/or induce formation of cytotoxic reactive oxygen species.16 Furthermore, their excitation and emission wavelengths are confined outside the first and second near-infrared (NIR) biological transparency windows (BTWs, NIR-I: 700–950 nm and NIR-II: 1000–1350 nm)17 where absorption and scattering from tissues and oxy/deoxygenated blood is significant, limiting their relevance to 2-D cell cultures and precluding their use in more realistic diagnosis environments, such as in thick tissues or in vivo models. The lack of biocompatible fluorescent organic probes in the BTWs has prevented the use of these highly sensitive spectral ranges for in-depth imaging.17 In addition, the dynamic nature of the G4 structure enable a range of different topologies (e.g. parallel, hybrid and antiparallel) to potentially form under different physiological conditions, making molecular recognition particularly arduous. In our research on G4-probes, we have recently shown that a dicyanovinyl squaraine dye (SQgI, Fig. 1a)18 could behave as G4-specific NIR light-up probe. In the complexed state, SQgl is emissive at 700 nm upon excitation at 661 nm through one-photon absorption (OPA) with an impressive quantum yield of ΦF ≈ 0.6. The probe exhibited good selectivity of G4 over duplex, with the dicyanovinyl squaraine dye capable to impart outstanding selectivity over non-G4 structures and remarkable specificity toward parallel G4 topologies. However, this probe presented inherent optical, practical and affinity limitations including: (i) moderate solubility, (ii) moderate binding constants (Kb values comprised between 105 to 106 M−1)18–20 and (iii) excitation wavelengths outside the BTWs. Thus, we speculated that exploring the chemical space around the squaraine core should enable optimization of both optical and DNA binding performances and we decided to decorate the dicyanovinylene squaraine moiety with bulkier water-soluble brush-type groups.
 | ||
| Fig. 1 (a) Synthetic route to CAS-C1. (b) Schematic illustration of the multiphotonic selective detection mechanism: the squaraine shows amphiphilic assembly in water into micelles. The formation of a stable complex with parallel G-quadruplex (left) is able to displace the squaraine from the micellar aggregate and trigger the fluorescent emission in the NIR-I biological window upon both one-photon excitation in the NIR-I and two-photon excitation in the NIR-II. CAS-C1 bestows high selectivity of interactions over non-quadruplex and non-parallel G4 structures (right). | ||
Herein, we present the first example of a water-soluble amphiphilic squaraine dye (hereafter CAS-C1, Fig. 1a) exhibiting G4-selective fluorescence light-up upon photoexcitation both within the NIR-I (λmax ≈ 720 nm and ΦF ≈ 0.7) through OPA, and within the NIR-II (two-photon absorption cross-section, σ2, of ≈ 300 GM and two-photon molecular brightness, σ2 × ΦF, of ≈ 200 GM at 1275 nm) through TPA. Moreover, the unprecedented nonlinear optical properties of CAS-C1 are combined with the outstanding selectively for parallel over non-parallel G4 architectures and duplex morphologies of any length and composition awarding it as the best two-photon parallel G4-specific probe currently available.
Results and discussion
Design and synthesis
With the aim to increase the binding selectivity and the solubility of ligand SQgl, we decorated the dicyanovinylene squaraine core scaffold with brush substituents carrying six triethylene glycol chains (synthesis and characterization in ESI). The massive presence of oligoethylene glycol chains is not only beneficial for the solubility of the ligand, but it is also known to strengthen the affinity of the ligand toward the parallel G4s upon noncovalent interactions within the grooves. This approach is reported in the literature and has been exploited in the design of selective G4-ligands.21–23 The molecular design of dicyanovinyl squaraine revealed a suitable choice in terms of affinity and selectivity with G4s.18 Additionally, the donor–acceptor–donor (D–A–D) nature of the chromophores24 appeared very promising for multi-photon absorption processes at long-infrared wavelengths. The synthesis of the newly designed squaraine according to the route shown in Fig. 1a starts from the reported brush precursor 1 that was brominated to give the corresponding bromide 2, which underwent a nucleophilic substitution with methyl-benzothiazole to give 3. Following the reported synthetic procedure for dicyanovinyl squaraines,24CAS-C1 was obtained after condensation of the squaric ester 4 with two equivalents of methyl-benzothiazolium 3. Squaraine CAS-C1 revealed fairly good solubility in various organic solvents. The absorption and emission spectra in these solvents revealed a moderate negative solvatochromism (Fig. S2†) and can be assigned to monomeric species (Fig. S1†).Aggregation in water
In aqueous solution, CAS-C1 absorption spectra reveal a different band shape and a strongly blue-shifted absorption maximum at 632 nm with an apparent extinction coefficient![[small epsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c6.gif) max = 6.2 × 104 M−1 (Fig. 2a, red line) which are first evidences for an H-type exciton coupling between aggregated dyes.25
max = 6.2 × 104 M−1 (Fig. 2a, red line) which are first evidences for an H-type exciton coupling between aggregated dyes.25
 | ||
| Fig. 2 (a) UV/Vis absorption spectra of an aqueous CAS-C1 solution (c = 3.0 × 10−6 M, 25 °C, red line) and upon addition of increasing amounts of SDS. The inset shows the incorporation of the squaraine dye in the SDS micelle upon addition of the surfactant with a critical concentration of 2.1 × 10−3 M. (b) AFM height images on mica after spin coating the CAS-C1 aqueous solution (3.0 × 10−6 M). Z scale is 1.2 nm. (c) Cross-section from yellow dashed lines 1–1′. | ||
Atomic force microscopy (AFM) revealed the presence of small particles with an irregular diameter of 10–30 nm and a height of 0.6–3.0 nm (Fig. 2b) which suggests the formation of micellar structures. The blue-shifted absorption band indicates a co-facial stacking of CAS-C1 molecules, most likely in an antiparallel fashion in order to cancel the dyes' large dipole moment.26 Such arrangement explains well the strong H-type excitonic coupling and the quenched fluorescence emission. Due to the massive presence of ethylene glycol chains, CAS-C1 is particularly suited to form such micellar structure where the hydrophilic parts are predominantly directed to the outside (Fig. 1b).
Upon heating to 95 °C, CAS-C1 micellar aggregates become disfavored for entropic reasons leading to a new band at 676 nm with isosbestic point at 653 nm. These changes are reversible and are ascribed to the formation of the monomeric species in water, with the typical fluorescence emission peaked at 699 nm (Fig. S3A†). Global fitting of the temperature-dependent measurement in the range 550–750 nm is in good agreement with an isodesmic aggregation mechanism (Fig. S3B†). Despite the fact that the pure monomeric state is not fully reached in water, the extrapolation of the fitting at 676 nm predicts a reasonable value for monomeric CAS-C1 of max = 9.0 × 104 M−1 cm−1. In accordance with the disaggregation model, a fluorescence emission in water lights-up at 699 nm (Fig. S3A†, dotted line). In contrast, the emission profile of CAS-C1 in its aggregated form is fully quenched due to nonradiative relaxation pathways in its H-aggregate state. In order to investigate the photophysical properties of the excited states, time-correlated single photon counting (TCSPC) measurements of CAS-C1 in water at 80 °C were performed. The time-dependent decay fitted better with the biexponential function (eqn S2, ESI†) suggesting the coexistence of two different species: one with longer lifetime (τ1 = 4.76 ns), consistently ascribed to the excited state of the free monomer in solution; the other one with shorter lifetime (τ2 = 1.35 ns), ascribed to the species aggregated in the micellar structure (Fig. S4†). The dramatic decrease of the lifetime and emission intensity is, therefore, in accordance with the formation of additional radiationless pathways in the aggregated form.
A more efficient approach to disassemble CAS-C1 into monomeric dyes is provided by the addition of surfactant molecules. Thus, we performed titration with sodium dodecyl sulfate (SDS) (Fig. 2a). The surfactant affects moderately the formation of the micelles at lower SDS concentration. Above the critical concentration of 2.1 × 10−3 M, SDS induces a marked breakup of CAS-C1 micelles by dissolving the CAS-C1 monomer within the SDS micelles (Fig. 2a).
G4 binding
In a similar fashion to the disaggregation induced by SDS, molecular recognition of CAS-C1 to suitable π-surfaces should also cause the disassembly of the micelle. This idea prompted us to study the recognition process of the squaraine toward a large panel of biologically relevant G4 topologies such as those found in the human telomeres (Tel-22) and minisatellite (Ceb25), in the proto-oncogene involved in the tumorigenesis of urinary bladder cancer (hRAS1), in the promoter region of myc, kit, KRAS, bcl2, and vav genes (c-myc, c-kit2, c-kit87up, KRAS 32, KRAS 22, bcl2 VAV1), in the vascular endothelial growth factor (VEGF) gene, in the telomeric transcripts (TERRA) and the thrombin-binding DNA aptamer (TBA) (see ESI† for details concerning the G4 topologies and their folding process). Indeed, upon addition of parallel G4s (e.g. VAV-1, c-myc and VEGF) the disappearance of the absorption band of CAS-C1 aggregate along with the formation of a new bathochromically shifted band centered at ≈ 700 nm indicate the formation of G4 (parallel):CAS-C1 complexes (Fig. 3, S6 and S7†). Concomitant with absorption changes, the steady-state emission of the squaraine fluorophore upon excitation at λexc = 660 nm, almost fully quenched in its aggregated unbound state (λem = 698 nm), lights-up in the NIR-I at λem ≈ 720 nm (Fig. 3a, S8 and S9†).Notably, both the absorption and the emission bands are ≈ 20 nm redshifted compared to the spectrum of the monomeric squaraine in water (Fig. S3†). This is a clear indication of a direct interaction of the cyanine π-surface of CAS-C1 with the guanines of the parallel quadruplexes. The excitation spectra and the lifetime decays recorded for CAS-C1 in the presence of G4s indicate that the strong photoluminescence observed arises mainly from the CAS-C1:G4 complex through geometrical and structural restrictions which inhibit the nonradiative relaxation pathways, hence the increased lifetime and ΦF (Fig. S10–S12†). TCSPC decay trace of the CAS-C1:G4 excited states (Fig. S1 and S12†) showed the presence of only one species, ascribed to the complexed squaraine, in accordance with the monoexponential function (eqn S1, ESI†). The singlet state lifetimes obtained by fitting of time-resolved fluorescence decays of the CAS-C1:G4 complexes are generally longer than the ones in organic solvents and comprised in the range of 5–6 ns (Fig. S2B†). This relatively long lifetimes with outstanding ΦF in the 700–800 nm range makes CAS-C1 particularly intriguing candidate as a fluorescence lifetime-based probe for G4 live-cell visualization by fluorescence lifetime imaging microscopy (FLIM).
Comparison of absorption and fluorescence responses between parallel and antiparallel G4s as well as single- and double-stranded DNA of any length and composition indicates a clear-cut binding preference of CAS-C1 for G4 parallel topologies (Fig. 3b). This was further confirmed by duplex DNA competition titration assays (Fig. S19 and S20†) which revealed very modest interference of interaction with c-myc and VAV-1, even in the presence of 50 eq. of the ds-DNA.
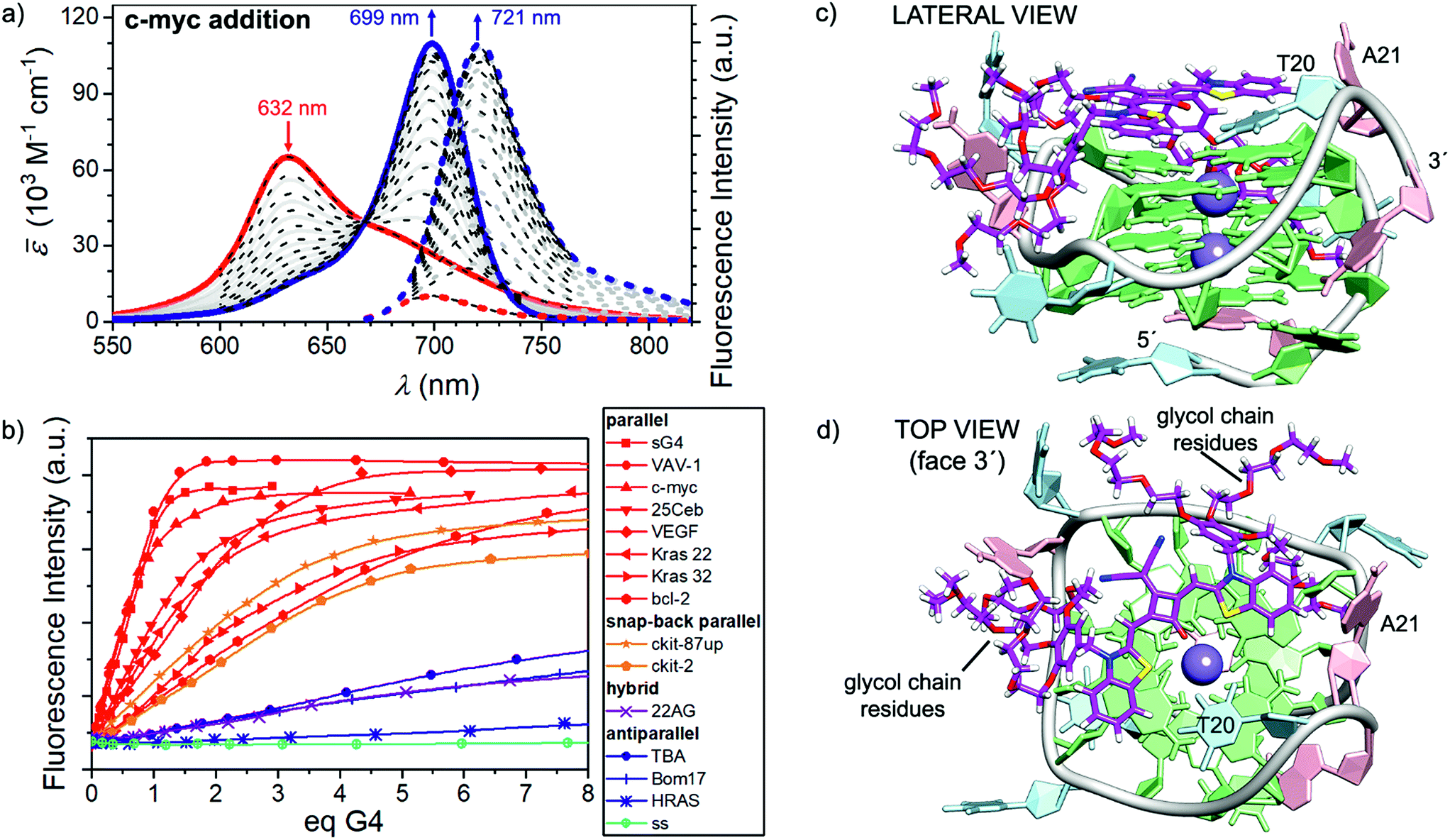 | ||
| Fig. 3 (a) Visible/NIR absorption (solid lines) and fluorescence spectra (dotted lines, excitation on the isosbestic point at 660 nm) of a CAS-C1 buffered solution in water (1.66 × 10−6 M, TRIS buffer c = 10 mM, pH = 7.2, KCl c = 100 mM) upon addition of c-myc G4 at 25 °C (red and blue lines correspond to the spectra at 0 eq. and 3.4 eq. respectively. Superimposed black dotted line results from a global fitting with a 1:1 binding model (Kb = 9.7 × 106 M−1). (b) Fluorescence titrations of the CAS-C1 buffered solution in water upon addition of various G4s sequences at 25 °C: the plots show a clear-cut preference of the squaraine for the parallel G4s (in red). (c and d) Docking lowest energy binding models for CAS-C1 with c-myc (score function ΔE =−5.9 kcal mol−1). | ||
It turned out that probe accommodation within the hydrophobic G4 scaffolds exert changes in the polarity of the surrounding that well-match a ETN value of 0.6 (Fig. S2†). The optical changes arising in the absorption and emission profiles of the CAS-C1-parallel G4 systems result in excellent one-photon brightness (defined as the product ε × ΦF) in the NIR region with values comprised between 56 and 84 × 103 M−1 cm−1 (Table 1).
| G-quadruplex | λ max (nm) | ε max (103 M−1 cm−1) | λ em (nm) | K 11 (M−1) | K 12 (M−1) | τ (ns) | Φ F | ε max × ΦF (103 M−1 cm−1) |
|---|---|---|---|---|---|---|---|---|
|
a Data fitting with 1:1 and 1:2 models was performed according to ref. 28 and the software provided at the website http://supramolecular.org. We like to note that the credibility of the derived values for the 1:2 case is lower than for the 1:1 case. This is due to the presence of three species and unknown fluorescence quantum yields of the 1:1 and the 1:2 complexes, and changing amounts of these complexes during the titration experiment. To give the same weight to UV/Vis and fluorescence experiments we evaluated both titrations independently and took the average values.
|
||||||||
| sG4 | 699 | 114 | 722 | 8.1 × 106 | — | 5.8 | 0.74 ± 0.07 | 84 |
| VAV-1 | 699 | 118 | 722 | 1.2 × 107 | — | 5.6 | 0.67 ± 0.02 | 79 |
| c-myc | 699 | 108 | 721 | 1.0 × 107 | — | 5.5 | 0.52 ± 0.03 | 56 |
| 25Ceb | 698 | 108 | 719 | 9.0 × 105 | — | 6.0 | 0.66 ± 0.01 | 71 |
| VEGF | 699 | 113 | 721 | 1.6 × 102 | 3.7 × 109 | 5.7 | 0.74 ± 0.08 | 84 |
| Kras 22 | 699 | 116 | 721 | 1.5 × 107 | 1.7 × 106 | 5.4 | — | — |
| Kras 32 | 699 | 103 | 721 | 5.9 × 106 | 4.1 × 105 | 5.6 | — | — |
| bcl-2 | 699 | 113 | 721 | 36 | 6.0 × 108 | — | — | — |
| ckit-87up | 699 | 103 | 721 | 3.1 × 102 | 4.3 × 108 | 5.5 | — | — |
| ckit-2 | 699 | 92 | 720 | 5.0 × 102 | 4.0 × 108 | — | — | — |
As suggested by recent publications,27–29 the issue of the binding stoichiometry of the complexes in solution was assessed by Job's plot titration assays (Fig. S13–S15†), the tangent method and a global fitting based on both the 2:1, 1:1 and 1:2 binding models (Fig. S16–S18†). If compared with the previously reported SQgl, CAS-C1 also showed multiple binding with the G4s, except for sG4, VAV-1 and c-myc.
A detailed quantitative binding data analysis that takes into account 1:1 and 1:2 CAS-C1:G4 stoichiometries with associated (anti-)cooperative effects indicates that CAS-C1 exhibits higher binding constants for parallel topologies such as c-myc (Fig. 3c and d) with respect to antiparallel and non-G4 topologies with values that exceed by two-orders of magnitude those for the previously reported squaraine SQgI (see ESI† for details concerning the quantitative binding data analysis).18 These results are ascribed to the highly accessible parallel G-quartets which, being devoid of adjacent lateral or diagonal loops, provide better π-stacking platforms for the accommodation of the squaraine core, thereby resulting in a better planarity and thus in a more efficient rigidification as desired for a fluorescence increase. On the other hand, the negative effect of the external quartets surrounded by loops in antiparallel G4s such as 22AG and c-kit hampers to some extent the end-stacking ability of CAS-C1 inducing a lower overall optical response. The aforementioned rationale is confirmed by using the closely related c-myc G4 analogue sG4, featuring no flanking overhang residues at both 5′ and 3′ ends that limit the steric hindrance, to enhance the light-up ability of CAS-C1. Indeed, complexation of the squaraine with sG4 provides a ΦF of 0.74 which is among the highest values reported for nucleic acid complexes within comparable spectral range (Table 1).30,31 Structural insights into the binding mode of CAS-C1 with G4s were provided by docking binding analysis on the G4s whose structures in solution had been derived by NMR studies under similar experimental conditions. The elucidated structures of G4s are referenced in Table S1†. In the case of c-myc, the squaraine chromophore is the recognition unit leading to a perfect complementary match with the 3′-quartet (Fig. 3c). However, the substitution of side groups at the squaraine chromophore has surely an impact in the binding. On one side, the massive number of glycol chains are responsible for the enhanced interaction compared to SQgl by establishing strong electrostatic interactions with the polar backbone of the G4. Presumably, the glycol chains displace water molecules from the grooves and thereby provide an entropic contribution to the stability of the G4-CAS-C1 complex, especially for the parallel ones,32 which explains the strengthening of the binding affinity compared with SQgl. On the other hand, it is reasonable to assume that the steric hindrance of the benzyloxy unit, which is accommodated in the hydrophobic pockets in the side of the quartet (Fig. 3c and d), largely contribute to the selectivity of the interaction.
Two-photon absorption
A fundamental optical limitation is encountered at the nanoscale level owing to Abbe's law of diffraction, which limits the maximum spatial resolution to approximately half the wavelength of light used to image the specimen.33 One way to overcome this limitation involves the use of nonlinear optical procedures based on multi-photon absorption processes. In this approach, the excitation of a chromophore is performed by the simultaneous absorption of two-photons with half the energy required for the corresponding one-photon absorption process. As a result, the excitation wavelength is confined in the first and second NIR biological transparency windows where the diffusion and absorption of light by inherently biological matrices is minimized, and thus allowing deep tissue penetration. Moreover, the low-energy excitation prevents photobleaching and cellular damages in biological samples. Importantly, the TPA is limited to a small femtoliter volume and occurs only at the focal point of a focused laser beam resulting in 3D image resolution. Despite the lower energy of the IR light compared to visible and UV light, this excitation mode has been proven to induce damage to the samples when using classical fluorophore that possess low TPA cross-section (σ2 < 50 GM).34 It turns out that, besides having significant photoluminescence properties in the NIR, a chromophore should also possess high σ2 values to be considered as a suitable probe for two-photon microscopy procedures in NIR-to-NIR configurations. The TPA spectra of CAS-C1 complexed with parallel G4s including VAV1, VEGF and c-myc were measured by two-photon excited fluorescence (TPEF) method in the range 1000–1550 nm and are reported in Fig. 4 as σ2 × ΦF.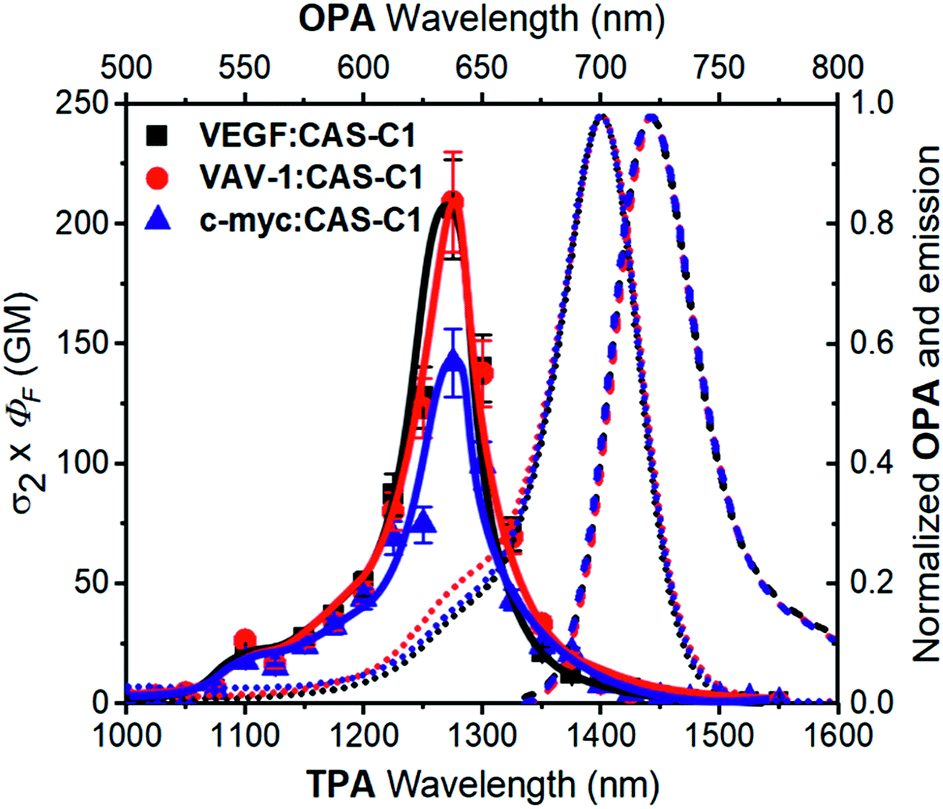 | ||
| Fig. 4 Two-photon molecular brightness (σ2 × ΦF) plot of CAS-C1 in the presence of VEGF, VAV-1 and c-myc (10 mM Tris, 100 mM K+, pH 7.2) at molar ratio (r) = 7. The values of the ΦF used are tabulated in Table 1. As a comparison, the OPA corresponding to the double of the energy of the TPA is shown (top and right axis): the absorption (dotted lines) and emission (dashed line) spectra corresponding to the CAS-C1 complexes with the G4s are shown. | ||
In general, all the complexes show similar TPA spectra composed of an intense high energy TPA band blue shifted compared to the one-photon transition energy and a weak low energy TPA band that matches well the wavelength-doubled 1PA spectra. These spectral features are usually associated with centrosymmetric quadrupolar molecules, in which selection rules for 1PA and 2PA electronic transitions are mutually exclusive. CAS-C1 presents a non-centrosymmetric character with a quasi-C2v symmetry and can be treated as bent quadrupolar chromophore, of which the quadrupolar conformation gives rise to an intense hypsochromically shifted TPA while the residual dipolar character results in a weak TPA transition overlapped with the 1PA maximum. Similar conclusions were recently reported for extended Michler's ketone, heptamethine, diarylboryl and anthracenyl derivatives.35–38 The TPA cross-section (σ2) values for the CAS-C1:G4 systems follow the order: CAS-C1:VAV1 σ2 = 312 GM, CAS-C1:VEGF σ2 = 278 GM and CAS-C1:c-myc σ2 = 273 GM at 1275 nm (Fig. S25†). Interestingly, the CAS-C1 aggregate also shows TPA absorption close to half energy of the corresponding OPA, in agreement with a centrosymmetric aggregate (Fig. S26†).
The high values of σ2 combined with the associated 2PA maximum are an optimal combination for NIR-to-NIR configurations since excitation of CAS-C1:G4 systems can be performed within the NIR-II window by TPA (λex(2PA) = 1275 nm) with emissions centered within the NIR-I spectral region (λem ≈ 720 nm). In line with the trend depicted for σ2 also the two-photon molecular brightness (σ2 × ΦF), as a result of the strong fluorescence enhancement of CAS-C1 bound to parallel G4s, follow the same order (i.e.CAS-C1:VAV1 σ2 × ΦF = 209 GM, CAS-C1:VEGF σ2 × ΦF = 206 GM and CAS-C1:c-myc σ2 × ΦF = 142 GM at 1275 nm) indicating the suitability of CAS-C1 to be used as a nonlinear biomarker for parallel G4 detection by using ultrasensitive fluorescence microscopy techniques. These values are nevertheless lower than those observed for monomeric CAS-C1 in organic solvent (σ2 × ΦF = 386 GM in isopropanol, Fig. S24†). This result is in agreement with recent results from Goodson and co-workers39 and some of us40,41 that relate the changes in the TPA cross-section to the different DNA binding modes. In particular, groove binders exhibit an enhanced TPA cross-section due to the DNA electric field induced enhancement of the transition dipole moment, while intercalators or end-stackers show a decrease in the TPA cross-section when complexed with DNA. Accordingly, this finding also supports our proposal that CAS-C1 binds to parallel G4s mainly via end-stacking mode in agreement with the docking results. The end-stacking ability of CAS-C1 was further confirmed through electronic circular dichroism (ECD) measurements (Fig S27†).42 Indeed, the native conformation of the parallel G4s was almost not affected by the presence of the squaraine dye indicating no or very weak perturbation on the quadruplex morphologies. By analogy with previously reported data for a G4 intercalator,41 the absence of conformational changes observed for CAS-C1-parallel G4 systems constitutes a clear evidence of the ability of CAS-C1 to coordinate the external G-tetrads, ruling out the possible intercalative binding behavior of our probe known to give rise to pronounced structural changes associated with a partially disruption of the stacking interactions that govern G4 formation.
Conclusions
In conclusion, the herein reported results on a new squaraine dye CAS-C1 with outstanding binding strength and selectivity towards parallel G4 motif open up a new research line for this fascinating class of NIR fluorophores and functional dyes17,43 beyond previous applications in organic electronics,44 photovoltaics45 and exciton transport.46 The CAS-C1 probe once complexed with parallel G4s exhibits a NIR light-up fluorescence response with quantum yields that go beyond 0.7. As a result, close-to-optimal one-photon molecular brightness in the far red-NIR region can be obtained. The parallel G4 preference of CAS-C1 is accompanied by binding constants that exceed by two-orders of magnitude those calculated for the previously reported squaraine SQgI. The selectivity of the probe is tested toward a large panel of anti-parallel G4s and single- and double-stranded morphologies of any size and composition making CAS-C1 a highly discriminatory parallel G4-binder. The docking results showed that the squaraine chromophore bestows the necessary selectivity for the interactions with the G4s. The molecular design of CAS-C1 allowed us, on the one hand, to increase the selectivity of the interaction by exploiting the more easily accessible G-quartet surface of parallel G4s and, on the other hand, the massive presence of glycol chains in the hydrophobic pocket of the G4 imparted strong affinity. The complexes of CAS-C1 with parallel G4s also possess strong two-photon absorption with maxima centered at 1275 nm, which make them ideally suited for NIR-to-NIR imaging procedures. Ultimately, the enhanced fluorescence quantum yields and the two-photon absorption cross-section enable one to obtain exceptionally high two-photon molecular brightness values within the NIR-II window that are desired for virtually all ultrasensitive fluorescence spectroscopy and microscopy techniques. These overall findings award CAS-C1 as the best two-photon parallel G4-specific probe currently available.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Pall Thordarson (Sydney, Australia) for his helpful advice for the analysis of the titration experiments. The research at the University of Würzburg was financially supported by the State of Bavaria with the establishment of the KeyLab for Supramolecular Polymers of the Bavarian Polymer Institute (BPI). At the Wroclaw University of Science and Technology, financial support from a statutory activity subsidy from the Polish Ministry of Science and Higher Education for the Faculty of Chemistry of WUST, KNOW – center of excellence in biotechnology project, and Harmonia grant DEC-2016/22/M/ST4/00275 are highly acknowledged.Notes and references
- P. Murat and S. Balasubramanian, Curr. Opin. Genet. Dev., 2014, 25, 22–29 CrossRef PubMed.
- R. Hänsel-Hertsch, M. Di Antonio and S. Balasubramanian, Nat. Rev. Mol. Cell Biol., 2017, 18, 279–284 CrossRef PubMed.
- A. M. Fleming, J. Zhu, Y. Ding, J. A. Visser, J. Zhu and C. J. Burrows, Biochemistry, 2018, 57, 991–1002 CrossRef PubMed.
- A. Bedrat, L. Lacroix and J.-L. Mergny, Nucleic Acids Res., 2016, 44, 1746–1759 CrossRef PubMed.
- P. Williams, L. Li, X. Dong and Y. Wang, J. Am. Chem. Soc., 2017, 139, 12426–12429 CrossRef PubMed.
- S. Zhang, Y. Wu and W. Zhang, ChemMedChem, 2014, 9, 899–911 CrossRef PubMed.
- T. M. Ou, Y. J. Lu, J. H. Tan, Z. S. Huang, K. Y. Wong and L. Q. Gu, ChemMedChem, 2008, 3, 690–713 CrossRef PubMed.
- S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261–275 CrossRef PubMed.
- Q. Cao, Y. Li, E. Freisinger, P. Z. Qin, R. K. O. Sigel and Z.-W. Mao, Inorg. Chem. Front., 2017, 4, 10–32 RSC.
- L. Tuong Vy Thi, H. Sujin, C. Junghyun and P. Hyun-Ju, Curr. Pharm. Des., 2012, 18, 1948–1972 CrossRef.
- A. Garci, K. J. Castor, J. Fakhoury, J.-L. Do, J. Di Trani, P. Chidchob, R. S. Stein, A. K. Mittermaier, T. Friščić and H. Sleiman, J. Am. Chem. Soc., 2017, 139, 16913–16922 CrossRef PubMed.
- M. Wang, Z. Mao, T.-S. Kang, C.-Y. Wong, J.-L. Mergny, C.-H. Leung and D.-L. Ma, Chem. Sci., 2016, 7, 2516–2523 RSC.
- K.-H. Leung, H.-Z. He, B. He, H.-J. Zhong, S. Lin, Y.-T. Wang, D.-L. Ma and C.-H. Leung, Chem. Sci., 2015, 6, 2166–2171 RSC.
- A. C. Bhasikuttan and J. Mohanty, Chem. Commun., 2015, 51, 7581–7597 RSC.
- B. R. Vummidi, J. Alzeer and N. W. Luedtke, ChemBioChem, 2013, 14, 540–558 CrossRef PubMed.
- Z. Li and K. B. Grant, RSC Adv., 2016, 6, 24617–24634 RSC.
- J. Zhao, D. Zhong and S. Zhou, J. Mater. Chem. B, 2018, 6, 349–365 RSC.
- V. Grande, F. Doria, M. Freccero and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 7520–7524 CrossRef PubMed.
- B. Jin, X. Zhang, W. Zheng, X. J. Liu, J. Zhou, N. Zhang, F. Y. Wang and D. H. Shangguan, Anal. Chem., 2014, 86, 7063–7070 CrossRef PubMed.
- X. Zhang, Y. Wei, T. Bing, X. Liu, N. Zhang, J. Wang, J. He, B. Jin and D. Shangguan, Sci. Rep., 2017, 7, 4766 CrossRef PubMed.
- M. Zuffo, A. Guédin, E.-D. Leriche, F. Doria, V. Pirota, V. Gabelica, J.-L. Mergny and M. Freccero, Nucleic Acids Res., 2018 DOI:10.1093/nar/gky607.
- A. Laguerre, Y. Chang, M. Pirrotta, N. Desbois, C. P. Gros, E. Lesniewska and D. Monchaud, Org. Biomol. Chem., 2015, 13, 7034–7039 RSC.
- F. Cuenca, O. Greciano, M. Gunaratnam, S. Haider, D. Munnur, R. Nanjunda, W. D. Wilson and S. Neidle, Bioorg. Med. Chem. Lett., 2008, 18, 1668–1673 CrossRef PubMed.
- U. Mayerhöffer, M. Gsänger, M. Stolte, B. Fimmel and F. Würthner, Chem.–Eur. J., 2013, 19, 218–232 CrossRef PubMed.
- T. Brixner, R. Hildner, J. Köhler, C. Lambert and F. Würthner, Adv. Energy Mater., 2017, 7, 1700236 CrossRef.
- F. Würthner, Acc. Chem. Res., 2016, 49, 868–876 CrossRef PubMed.
- L. K. S. von Krbek, C. A. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622–2637 RSC.
- D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- W. Xu, K. M. Chan and E. T. Kool, Nat. Chem., 2017, 9, 1043–1055 CrossRef PubMed.
- Y. V. Suseela, N. Narayanaswamy, S. Pratihar and T. Govindaraju, Chem. Soc. Rev., 2018, 47, 1098–1131 RSC.
- D. Miyoshi, H. Karimata and N. Sugimoto, J. Am. Chem. Soc., 2006, 128, 7957–7963 CrossRef PubMed.
- J. Moreno, M. Gerecke, L. Grubert, S. A. Kovalenko and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1544–1547 CrossRef PubMed.
- B. Dumat, G. Bordeau, E. Faurel-Paul, F. Mahuteau-Betzer, N. Saettel, G. Metge, C. Fiorini-Debuisschert, F. Charra and M.-P. Teulade-Fichou, J. Am. Chem. Soc., 2013, 135, 12697–12706 CrossRef PubMed.
- N. S. Makarov, S. Mukhopadhyay, K. Yesudas, J. L. Brédas, J. W. Perry, A. Pron, M. Kivala and K. Müllen, J. Phys. Chem. A, 2012, 116, 3781–3793 CrossRef PubMed.
- Q. Zou, Y. Zhao, N. S. Makarov, J. Campo, H. Yuan, D. C. Fang, J. W. Perry and F. Wu, Phys. Chem. Chem. Phys., 2012, 14, 11743–11752 RSC.
- S. Pascal, S. Denis-Quanquin, F. Appaix, A. Duperray, A. Grichine, B. L. Guennic, D. Jacquemin, J. Cuny, S. H. Chi, J. W. Perry, B. V. D. Sanden, C. Monnereau, C. Andraud and O. Maury, Chem. Sci., 2017, 8, 381–394 RSC.
- B. Mettra, F. Appaix, J. Olesiak-Banska, T. Le Bahers, A. Leung, K. Matczyszyn, M. Samoc, B. van der Sanden, C. Monnereau and C. Andraud, ACS Appl. Mater. Interfaces, 2016, 8, 17047–17059 CrossRef PubMed.
- P. H. Doan, D. R. G. Pitter, A. Kocher, J. N. Wilson and T. Goodson, J. Am. Chem. Soc., 2015, 137, 9198–9201 CrossRef PubMed.
- M. Deiana, B. Mettra, K. Matczyszyn, D. Pitrat, J. Olesiak-Banska, C. Monnereau, C. Andraud and M. Samoc, Biomacromolecules, 2016, 17, 3609–3618 CrossRef PubMed.
- M. Deiana, B. Mettra, L. Martinez-Fernandez, L. M. Mazur, K. Pawlik, C. Andraud, M. Samoc, R. Improta, C. Monnereau and K. Matczyszyn, J. Phys. Chem. Lett., 2017, 8, 5915–5920 CrossRef PubMed.
- R. del Villar-Guerra, J. O. Trent and J. Chaires, Angew. Chem., Int. Ed., 2018, 57, 7171–7175 CrossRef PubMed.
- A. Ajayaghosh, Acc. Chem. Res., 2005, 38, 449–459 CrossRef PubMed.
- M. Gsänger, E. Kirchner, M. Stolte, C. Burschka, V. Stepanenko, J. Pflaum and F. Würthner, J. Am. Chem. Soc., 2014, 136, 2351–2362 CrossRef PubMed.
- U. Mayerhöffer, K. Deing, K. Gruß, H. Braunschweig, K. Meerholz and F. Würthner, Angew. Chem., Int. Ed., 2009, 48, 8776–8779 CrossRef PubMed.
- C. Lambert, F. Koch, S. F. Völker, A. Schmiedel, M. Holzapfel, A. Humeniuk, M. I. S. Röhr, R. Mitric and T. Brixner, J. Am. Chem. Soc., 2015, 137, 7851–7861 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, titration experiments, optical spectra, NMR and HRMS spectra. See DOI: 10.1039/c8sc02882f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |