
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalent organic nanosheets for bioimaging†
Gobinda
Das‡
a,
Farah
Benyettou‡
a,
Sudhir Kumar
Sharama
b,
Thirumurugan
Prakasam
a,
Felipe
Gándara
 c,
Victor A.
de la Peña-O’Shea
d,
Na'il
Saleh
e,
Renu
Pasricha
a,
Ramesh
Jagannathan
b,
Mark A.
Olson
f and
Ali
Trabolsi
*a
c,
Victor A.
de la Peña-O’Shea
d,
Na'il
Saleh
e,
Renu
Pasricha
a,
Ramesh
Jagannathan
b,
Mark A.
Olson
f and
Ali
Trabolsi
*a
aChemistry Program, New York University Abu Dhabi (NYUAD), United Arab Emirates. E-mail: at105@nyu.edu
bEngineering Division, New York University Abu Dhabi (NYUAD), United Arab Emirates
cThe Materials Science Factory, Instituto de Ciencia de Materiales de Madrid - CSIC, Sor Juana Inés de la Cruz 3, 28049 Madrid, Spain
dPhotoactivated Processes Unit, IMDEA Energy, Avd. Juan de la Sagra 3, Mostoles 28935, Madrid, Spain
eUnited Arab Emirates University, College of Science, Department of Chemistry, Al-Ain, United Arab Emirates
fSchool of Pharmaceutical Science and Technology, Health Science Platform, Tianjin University, 92 Weijin Rd. Nankai, District, Tianjin 300072, P. R. China
First published on 3rd September 2018
Abstract
Covalent organic nanosheets (CONs) have attracted much attention because of their excellent physical, chemical, electronic, and optical properties. Although covalent organic nanosheets have widely been used in many applications, there are only a few CONs that have been tested for bio-medical applications. Nanometer sized triazine-based nanosheets were obtained by exfoliating their bulk counterparts in water. The obtained nanosheets were dispersible and stable in water with enhanced photoluminescence properties compared to the bulk material. The nanosheets were biocompatible and non-toxic and showed ability to stain HeLa cell nuclei without additional assistance of an external targeting agent.
Introduction
Covalent organic nanosheets (CONs) obtained by exfoliation of their counterparts, such as covalent organic frameworks (COFs),1 have emerged as a new class of porous, thin two-dimensional (2D) nanostructures with distinctive dimension related properties that differ from their corresponding bulk materials.2–6 Exfoliation of COFs into CONs reduces the size of the material while also imparting novel physical and chemical properties, which for example, allows for the circumvention of a few serious COF drawbacks, namely their dispersibility and bioavailability within the cells. In this regard, CONs are a promising nanomaterial for applications in biomedicine including drug delivery and bioimaging.2,3,7,8Non-toxic CONs with high photoluminescence quantum yields can be of great interest for bioimaging as they are stable in solution, possess tunable optical characteristics, and exhibit limited photobleaching. Reported materials for bioimaging are mainly (i) inorganic in nature with the presence of heavy metals,9 (ii) carbon nitride,10 or (iii) graphene-based nanomaterials,11–13 all with considerably challenging syntheses.14 While several examples in the literature have employed CONs in drug delivery,2,15 they have yet to be investigated for bioimaging.
We describe here the synthesis of a crystalline, porous, and luminescent triazine-based COF (denoted as TTA–DFP COF) from the condensation of 2,6-diformylpyridine (DFP) with 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA) under 30 min of microwave irradiation (Fig. 1). As synthesized, the bulk material is constituted of nanosheets and is weakly fluorescent with micrometer lateral dimensions. Exfoliation of the bulk material in pure water drastically reduces the lateral dimensions to the nanometer scale and enhances the photoluminescence properties by a factor of 6. The TTA–DFP CONs exhibit a high luminescence efficiency with a maximum peak centred at 435 nm when excited at λmax = 375 nm.
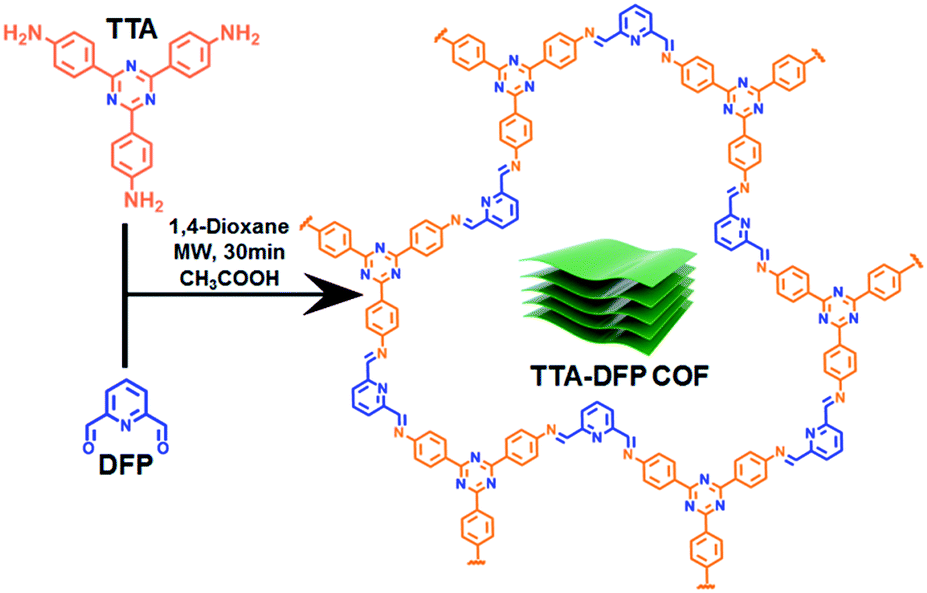 | ||
| Fig. 1 Chemical structure and synthetic route of the TTA–DFP COF under microwave irradiation. Cartoon representation (green block) illustrates the stacked nanosheets of the TTA–DFP COF. | ||
Cellular labelling is of great interest in biology, in particular the nucleus, which is regarded as one of the main targets for cancer therapeutics.16 Nuclear staining dyes (organic or peptide-based) are limited by poor photo-sensitivity, challenging syntheses, and cost.11 Incubation experiments with HeLa cells demonstrated that TTA–DFP CONs could be used for label-free selective intracellular localization within the cellular nucleus without hampering the cell architecture and viability, thus making this intrinsically luminescent CON material a promising nuclear probe for bioimaging.
Results and discussion
The bulk material, denoted as TTA–DFP COF, was synthesized by co-condensation of DFP (12 mg, 0.09 mmol, 3 equiv.) and TTA (21.24 mg, 0.06 mmol, 2 equiv.) in 3 mL of anhydrous 1,4-dioxane in the presence of a catalytic amount of acetic acid, sealed in a 10 mL glass microwave tube and heated under microwave irradiation at 110 °C for 30 min (Fig. 1). The formed yellowish precipitate was collected by filtration, washed with 1,4-dioxane and ethanol, and dried at 110 °C for 12 h. After centrifugation and multiple washings with ethanol, the product was completely insoluble in water and all organic solvents tested, including THF, acetone, CHCl3, DMF and DMA, indicating the formation of a stable polymeric material.The formation of imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) linkages for the as-synthesized material was first confirmed by Fourier-transform infrared (FT-IR) spectroscopy. The material displayed stretching modes at 1625 cm−1 as well as the disappearance of the TTA primary amine stretches at ∼3420 cm−1 and the DFP aldehyde stretch at 1700 cm−1 (Fig. S1†). Thermogravimetric analyses (TGA) revealed that the TTA–DFP COF exhibits excellent thermal stability, maintaining more than 60% of its weight up to 500 °C (Fig. S2†).
N) linkages for the as-synthesized material was first confirmed by Fourier-transform infrared (FT-IR) spectroscopy. The material displayed stretching modes at 1625 cm−1 as well as the disappearance of the TTA primary amine stretches at ∼3420 cm−1 and the DFP aldehyde stretch at 1700 cm−1 (Fig. S1†). Thermogravimetric analyses (TGA) revealed that the TTA–DFP COF exhibits excellent thermal stability, maintaining more than 60% of its weight up to 500 °C (Fig. S2†).
The crystallinity of the as-synthesized material was demonstrated by powder X-ray diffraction (PXRD), which shows a strong peak at 2θ = 5° that can be assigned to the (110) plane of the regularly ordered lattice, as well as a broad peak centred at 2θ = 25.6°, corresponding to the reflection from the (003) plane (Fig. 2a).
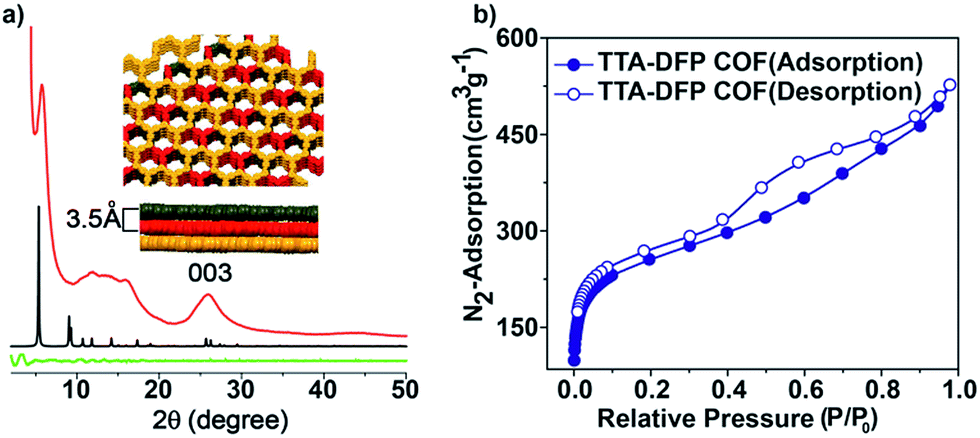 | ||
| Fig. 2 (a) PXRD patterns and simulated structures of the TTA–DFP COF: experimental (red line), simulated (black line) and difference (green line). TOP view: space-filling representation of the stacked layers of the TTA–DFP COF. The simulated unit cell parameters are a = b = 33.0 Å and c = 10.39 Å. (b) Nitrogen adsorption/desorption isotherms at 77 K of the TTA–DFP COF. | ||
The spacing between the π–π stacking planes was calculated and found to be ∼3.5 Å, which matches the simulated structure. Following reticular chemistry principles, crystal structure models were built (Fig. 2a) based on the geometry of the building blocks. Models based on distorted hcb layered structures were generated in the trigonal P3 space group and geometrically optimized with universal force field energy minimizations. The best agreements between experimental and simulated PXRD patterns were obtained for layer stacked structures with an ABC sequence, resulting in unit cell parameters of a = b = 33.0 Å and c = 10.39 Å (Rwp = 2.07% and Rp = 0.76% after Pawley refinement). The structure is distorted compared to regular hcb layers because the meta disposition of the aldehyde groups in the pyridine linker imposes a non-linear connection between the triangular nodes, which is, indeed, not common in reported COFs. On account of the sheet-like layered morphology of TTA–DFP COFs having an imperfect long-range order, broadening of the wide-angle diffraction peaks was observed. PXRD patterns of this sort are commonly reported for other layered triazine-based covalent organic networks.17–20
The porosity of the TTA–DFP COF was evaluated by measuring nitrogen gas adsorption/desorption isotherms at 77 K (Fig. 2b). The samples were activated at 85 °C for 24 hours prior to the measurements in order to remove solvent and trapped gas. The TTA–DFP COF exhibits a sorption isotherm, which is a characteristic of microporous materials.21 The Brunauer–Emmett–Teller (BET) surface area of the TTA–DFP COF was found to be 900 m2 g−1, while the pore size distribution of the COF was calculated with a DFT fitting of the adsorption branch, based on a model with cylindrical pores. A narrow distribution of pores with an average width of 1.7 nm (Fig. S3†) was obtained which is in agreement with the proposed structural models, where hexagonal channels are formed upon stacking of the layers. The calculated pore volume for the TTA–DFP COF is 0.76 cm3 g−1.
The morphology of the TTA–DFP COF was investigated by high resolution transmission electron microscopy (HRTEM) and atomic force microscopy (AFM). HRTEM images showed transparent nanosheets, an indication of the thickness of TTA–DFP COFs (Fig. 3a). AFM analysis of TTA–DFP COFs revealed a 2D sheet-like morphology (Fig. 3b). The topological height profile for the 2D sheets was found to be 1.5 nm. PXRD analysis indicates an interlayer stacking distance of 0.35 nm, which is consistent with the well-defined lattice fringes obtained from HRTEM analysis (0.38 nm, Fig. 3a). Based on these microscopy data, we determined that TTA–DFP COFs are comprised of about four stacked layers.
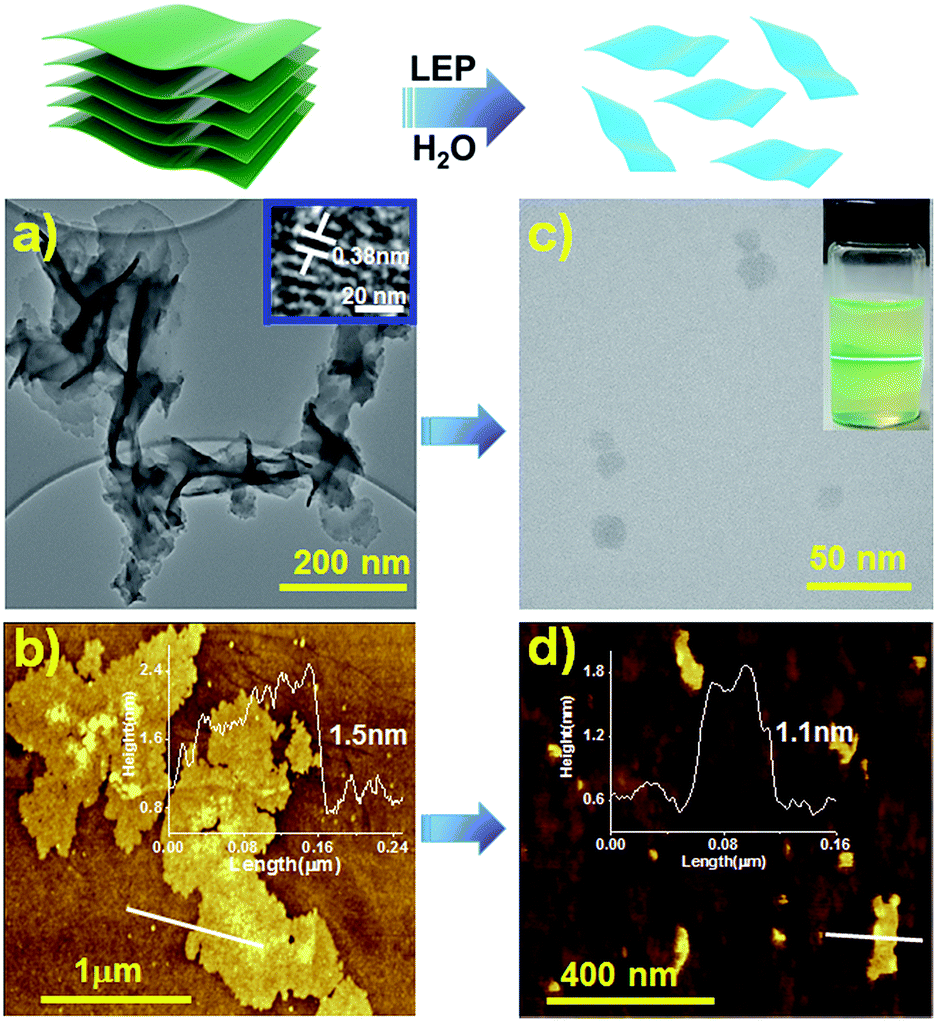 | ||
| Fig. 3 Schematic representation of the liquid exfoliation process (LEP) in water (5 hours). HR-TEM (a and c) and AFM (b and d) images with line profiles of the TTA–DFP COF and TTA–DFP CONs, respectively. Inset of (c) shows the Tyndall effect of TTA–DFP CONs in water. | ||
Prior to their use for biomedical applications, it is important to process these materials to nanometer sizes while also imparting water solubility and stability.22 For these reasons, liquid exfoliation processing (LEP)23–27 from bulk to submicron-sized TTA–DFP CONs in water was performed using ultra-sonication for 5 hours (Fig. 3, top). Exfoliation was confirmed by TEM imaging of the sample which was comprised of large quantities of 2D-nanosheets with an average size of ≈22 nm (Fig. 3c, S4 and S5†). The clear distinct Tyndall effect of TTA–DFP CON solution (inset Fig. 3c) also indicates the presence of highly monodisperse ultrathin nanosheets in water.10 AFM confirmed the formation of ultrathin 2D-nanosheets with smaller lateral diameters (Fig. 3d). Upon liquid phase exfoliation, the sheet thickness decreased from 1.5 to 1.0–1.3 nm (Fig. 3d). Not only did the diameter of the nanosheets decrease but also sheets of uniform thickness and shape were produced. PXRD analysis showed that the intensity of the (110) peak gets reduced upon exfoliation and the (003) peak gets broader (Fig. S7†), which is consistent with the smaller number of vertically stacked layers. Such peak broadening is also noted for previously reported CONs.28 Aqueous solutions of exfoliated nanosheets were found to be stable with excellent dispersibility with no precipitation observed over time. At pH 7.4 in water, the hydrodynamic diameter of TTA–DFP CONs was found to be 37.8 nm (Fig. S8†). The stability of TTA–DFP CONs was then investigated in foetal bovine serum (FBS) to simulate the physiological conditions in cells (Fig. S9†). The hydrodynamic diameter of the TTA–DFP CONs was found to be 23.55 nm, which is close to the size observed by TEM (22 nm). Additionally, when the mixture was stirred for twenty-four hours at room temperature, the size remained unchanged (22.80 nm). Together, these results confirm the stability and minimal aggregation of TTA–DFP CONs under simulated physiological conditions.
The electronic properties of the bulk material and the exfoliated nanosheets were studied by absorption and fluorescence spectroscopies. As shown in Fig. 4a, the UV-vis absorption maximum for an aqueous dispersion of TTA–DFP CONs is blue shifted compared to the bulk material (λmax = 387 nm versus 438 nm, respectively). In water, the TTA–DFP COF exhibited a broad emission band centred at λmax of 538 nm when excited at 375 nm (Fig. 4b). After the exfoliation of the bulk material into TTA–DFP CONs, the fluorescence emission spectrum is wider with a maximum that is blue shifted from 538 nm to 435 nm, with an intensity enhancement by a factor of ∼6 (Fig. 4b), which is the result of reduced interlayer π–π interactions in the nanosheets. Such a change in the π–π interactions induces a sharp increase of the optical band gap, from 2.45 eV in the bulk sample to 2.7 eV in the nanosheets and explains the observed increase in intensity.4,5,10,29–31 The photoluminescent nature of both the TTA–DFP COF and TTA–DFP CONs can be explained by intramolecular donor/acceptor charge transfer from the phenyl groups (donors) to the central triazine moieties (acceptors). A control experiment, in which the luminescence of the TTA monomer condensed with pyridine-monoaldehyde was measured, revealed that the monomer was not luminescent in water (Fig. S10†). The less emissive character of the bulk material could be explained by the aggregation-induced quenching mechanism resulting from the π–π layered structure of the COF.21
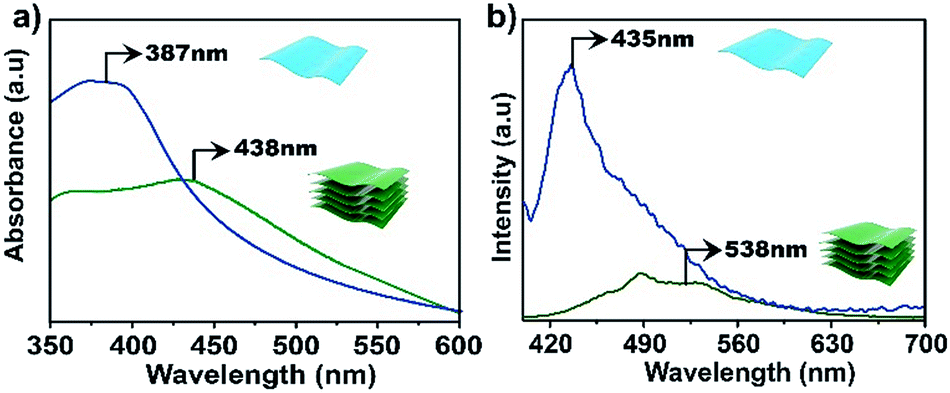 | ||
| Fig. 4 UV-vis (a) and emission (b) spectra of bulk TTA–DFP COFs (green line) and exfoliated TTA–DFP CONs (blue line) dispersed in water at room temperature, λex = 375 nm. | ||
To gain more insight into the band structure of the TTA–DFP COF, plane wave density functional theory (PW-DFT) calculations were performed for bulk and single layers following optimization of the calculated crystal structures to determine their electronic and optical properties, and differences between bulk and single layers. The density of states (DOS) shows (Fig. 5) an increase of states at the edge of the conduction band in the bulk sample, indicating a blue shift in both the wavelengths of light absorbed and subsequent emission. As expected, the TTA–DFP COF displays semiconducting behaviour, with a Valence Band (VB) and a Conduction Band (CB) formed by hybridized C 2p and N 2p states as depicted in the charge density plots (Fig. 5). In addition, the energy gaps obtained from HSE06 calculations are 2.45 eV for the bulk sample and 2.7 eV for a single layer nanosheet (Fig. S11†). These results are in agreement with the optical measurements obtained from UV-vis diffuse reflectance spectroscopy (UV-vis DRS) which reveal a broad absorption band with an indirect band gap of 2.55 eV (Fig. S12†). Time dependent density functional theory (TD-DFT) calculations were used to determine the optoelectronic properties of the materials and indicate that the electronic transitions mainly occur with UV excitation and are associated with intramolecular charge-transfer processes from several highest occupied molecular orbital (HOMO) degenerate orbitals centred in the imine moieties to the lowest unoccupied molecular orbital (LUMO) degenerate orbitals (Fig. S13†). Finally, we calculated the surface energy of the monolayer from DFT energy calculations. The calculated surface energy is 98.8 mJ m−2, which is comparable to the value reported for other two-dimensional structures10 and also similar to the surface energy of water (∼102 mJ m−2) suggesting a potentially high dispersibility of the TTA–DFP COF in this solvent.
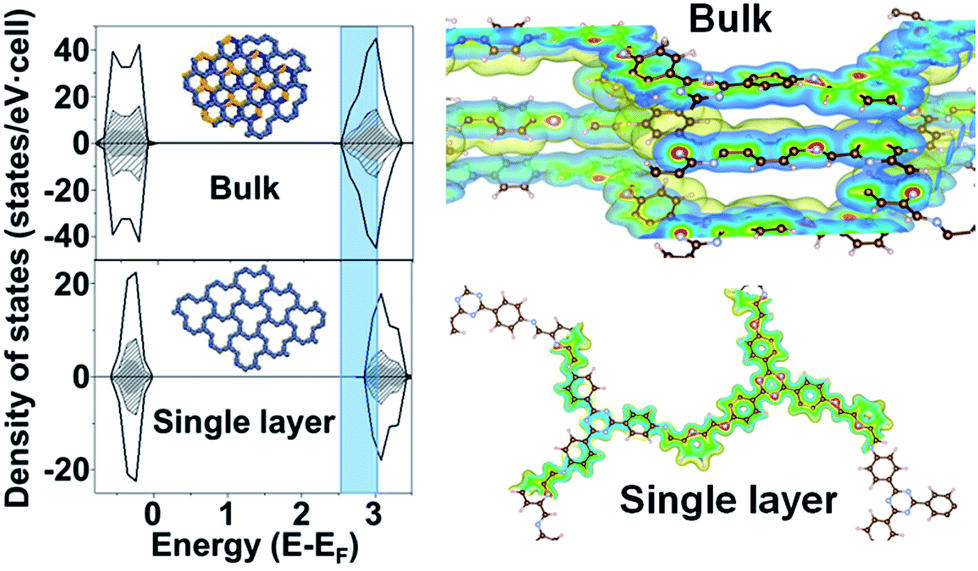 | ||
| Fig. 5 Total density of states (DOS) (black) and atom-projected density of states (PDOS) for C (black dash) and N (grey) and schematic representation of the isosurfaces of a section of charge density for: bulk and single layer nanosheets. Atom colors: brown for C, blue for N, and white for H. Shadow part clearly indicates an increase of states at the edge of the conduction band in the bulk sample. | ||
The enhanced luminescence properties of the exfoliated TTA–DFP CONs, their nanometer size and their high dispersibility in biological media compelled us to use them as a fluorescent probe to perform cellular labelling on HeLa cells. Cells treated with the exfoliated nanosheets (100 μg mL−1) for 30 minutes up to 24 hours exhibited a strong blue signal (λex = 360 nm) which increases in intensity with time demonstrating the ability of the material to internalize and accumulate into cells. Time-dependent incubation experiments (Fig. 6 a–c and S14–S17†) revealed that TTA–DFP CONs could enter the cell nucleus after 30 min. It is worth noting that TTA–DFP CONs are stable under acidic conditions with no hydrolysis of the imine bond observed. Also, the monomers are not fluorescent. We conclude that the fluorescence observed cannot come from fragmentation of the TTA–DFP CONs but from their presence in the nucleus. Few aggregates can be seen outside cells but are not fluorescent and therefore cannot be considered as aggregated nanosheets but are actually due to a small fraction of non-exfoliated material which is not fluorescent at this excitation wavelength.
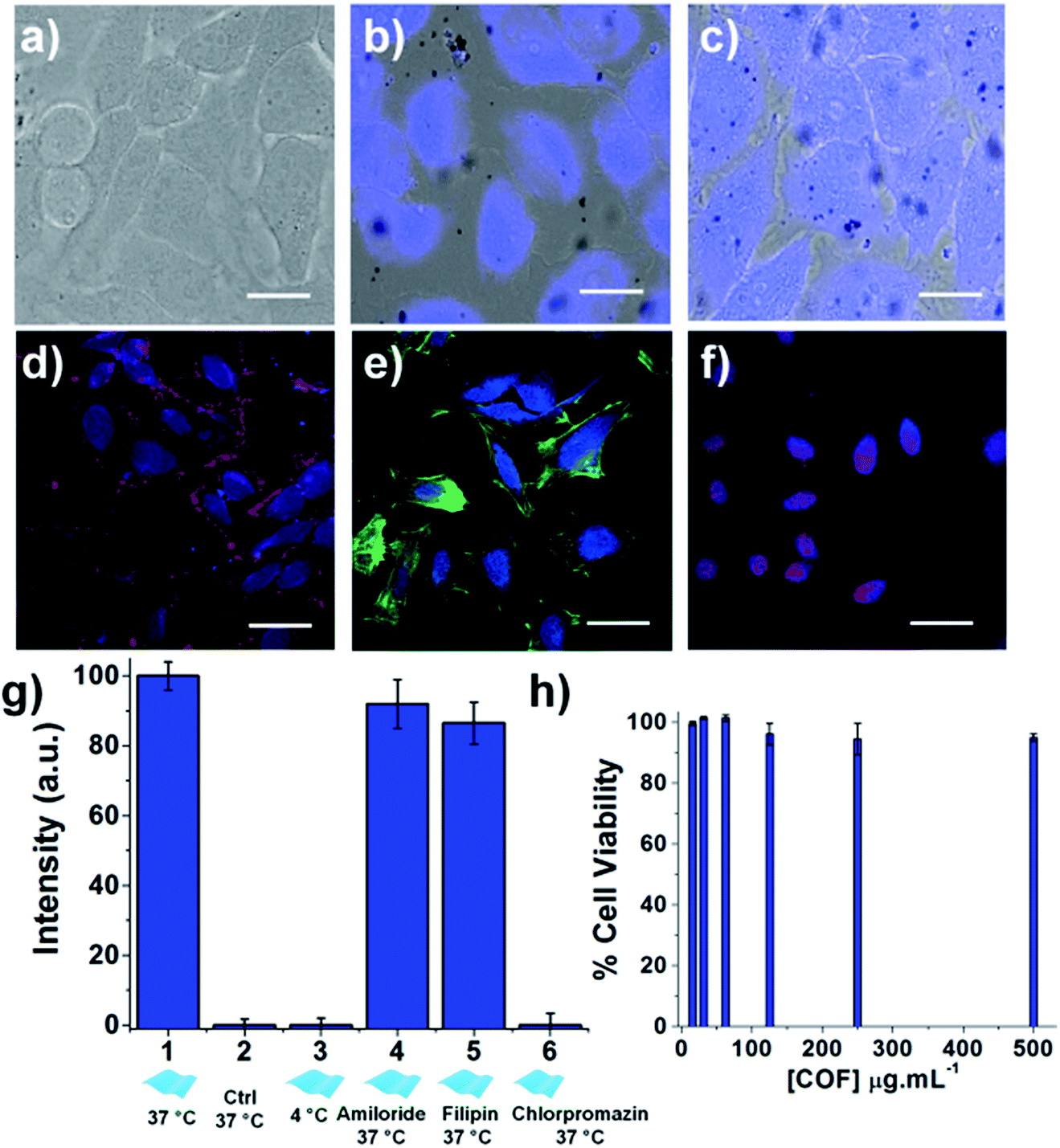 | ||
| Fig. 6 In vitro study. Overlay of bright field and fluorescence images of HeLa cells incubated with the TTA–DFP CONs (100 μg mL−1) for (a) 0, (b) 60 and (c) 180 minutes of incubation, showing that accumulation of the TTA–DFP CONs in the nucleus does not hamper the cell integrity (λex = 360 nm; scale bars = 10 μm). Confocal images of HeLa cells incubated for 4 hours with the exfoliated TTA–DFP CONs (50 μg mL−1) and (d) a plasmic membrane marker, (e) an actin marker and (f) a nucleus marker, showing that after internalization in cells, the exfoliated TTA–DFP CONs specifically target the nucleus (blue channel (λex = 405 nm), red channel (λex = 633 nm), green channel (λex = 561 nm), and scale bars = 20 μm). (g) Quantitative comparison of fluorescence intensity in HeLa cells incubated with TTA–DFP CONs (50 μg mL−1) at 4 °C versus 37 °C as well as in the presence of endocytotic inhibitors. (h) Viability assessment of HeLa cells after 48 h incubation with increasing concentrations of exfoliated TTA–DFP CONs up to 500 μg mL−1. | ||
To further confirm the localization of the nanosheets in the cells, the nucleus, the membrane, and the cytoplasm of the cells were specifically labelled using organelle markers and co-localized with the exfoliated TTA–DFP CON signal (Fig. 6d–f). The blue emission in the cells arising from the nanosheets does not overlap with the emission of either the membrane (Fig. 6d) or the actin in the cytoplasm (Fig. 6e). However, the blue emission does overlap with the fluorescence staining from the red labelled nucleus (Fig. 6f), with also accumulation in the endoplasmic reticulum (ER). The accumulation of the TTA–DFP CONs in the ER is particularly interesting because the lumen of the ER is contiguous with the space between the inner and outer nuclear membranes.32 Recently, Mal et al.6 reported the interactions of self-exfoliated ionic covalent organic nanosheets with DNA, which might explain the accumulation of the TTA–DFP CONs into the nucleus.
In order to determine the relative amounts of active versus passive mechanisms involved in TTA–DFP CON internalization, we compared the relative intensity of fluorescence in cells incubated with TTA–DFP CONs for 1 h at either 4 °C or 37 °C (Fig. S21†). The fluorescence intensity in the cells incubated at 4 °C is considerably diminished compared to the cells' fluorescence intensity at 37 °C, indicating that the active mechanisms of internalization, such as endocytosis, are predominant. To understand the TTA–DFP CON cellular uptake mechanism, HeLa cells were pre-treated with different inhibitors of various endocytotic mechanisms (Fig. S21†). For amiloride and filipin-treated cells, the fluorescence intensity did not change much after 1 hour of incubation, suggesting that macropinocytosis and caveolae-mediated endocytosis are not involved in the internalization process.33 In contrast, the fluorescence intensity of the chlorpromazine-treated cells was significantly less pronounced, implying that clathrin-mediated endocytosis may be the major pathway for TTA–DFP CON internalization (Fig. 6g).33 Clathrin-mediated endocytosis is one of the pathways that sorts molecules to organelles such as the Golgi, the ER and the nucleus.34,35 This result is consistent with the observation of TTA–DFP CON accumulation in the ER and nucleus. In order to assess if the intensity of the fluorescence arising from TTA–DFP CONs diminishes over time inside the cells, HeLa cells were incubated with TTA–DFP CONs for 4 h and then subsequently incubated for 24 h with fresh media without TTA–DFP CONs (Fig. S18†). Fluorescence images showed that the cells' fluorescence intensity does not fade and remains strong and localized within the nucleus. It is worth noting that after 24 hours of incubation with TTA–DFP CONs, cells were present in the ER and nucleus demonstrating that even after prolonged exposure of cells to TTA–DFP CONs up to 24 hours, the sheets continue accumulating into the cells without hampering the cell integrity. We conclude that after internalization through clathrin-mediated endocytosis within the HeLa cells, the exfoliated TTA–DFP CONs specifically target and label the nucleus in a relatively short time of 30 minutes. A similar phenomenon was observed for graphene quantum dots with a diameter below 100 nm.13
The exfoliated TTA–DFP CONs did not weaken cell activity and the cells maintained their normal morphology up to 24 hours of incubation (Fig. S15†), which encouraged us to assess the biocompatibility of this material. Overall the exfoliated TTA–DFP CONs elicit no cytotoxic effects on HeLa cells after 48 hours of incubation with concentrations up to 500 μg mL−1 (Fig. 6h).
Conclusions
In summary, we have successfully synthesized a crystalline, porous and luminescent triazine-based COF (denoted as TTA–DFP COF) using a facile microwave-assisted method. Liquid-exfoliation of the bulk TTA–DFP COF in water gave rise to nanometer-sized 2D-nanosheets whose luminescence emission was considerably blue shifted and enhanced compared to the bulk material. Our in vitro results following the incubation of the nanosheets with HeLa cells confirmed that the exfoliated materials did not affect the cell architecture and viability, which constitutes a preliminary demonstration of biocompatibility and nontoxicity. Two commercially available blue-emitting dyes are commonly used in biology to stain DNA. One of these two, DAPI, is used to stain dead cells or cells with compromised membranes and has high toxicity. The second is Hoechst 33258, which is less toxic and suitable for staining both live and dead cells. Hoechst enters the cell by an unmediated diffusion transport mechanism through the cell membrane prior to DNA binding.36,37 The exfoliated TTA–DFP CONs function as nuclear probes for bioimaging without the additional assistance of external capping or targeting agents, penetrating the cell nucleus through clathrin-mediated endocytosis in less than 30 minutes. Considering that very few nuclear labelling commercial dyes are available for cellular imaging, TTA–DFP CONs are a novel and cost-effective alternative. Research on further biomedical applications such as targeted delivery of drugs to the nucleus is ongoing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research described here was sponsored by New York University Abu Dhabi (NYUAD), UAE, and we are thankful for its generous support of the research program. The research was carried out using the Core Technology Platform resources at NYUAD. We would like to acknowledge the Al Jalila Foundation (AJF 201646). The authors also thank Miss Khulood Alawadi for 3D cartoons. F. G. acknowledges the Spanish Ministry of Economy and Competitiveness for funding through the “Ramón y Cajal” and “Retos de la Sociedad (CTQ2017-87262-R)” programs.References
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef PubMed.
- S. Mitra, H. S. Sasmal, T. Kundu, S. Kandambeth, K. Illath, D. Díaz Díaz and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 4513–4520 CrossRef PubMed.
- Y. Peng, Y. Huang, Y. Zhu, B. Chen, L. Wang, Z. Lai, Z. Zhang, M. Zhao, C. Tan, N. Yang, F. Shao, Y. Han and H. Zhang, J. Am. Chem. Soc., 2017, 139, 8698–8704 CrossRef PubMed.
- J. Dong, K. Zhang, X. Li, Y. Qian, H. Zhu, D. Yuan, Q.-H. Xu, J. Jiang and D. Zhao, Nat. Commun., 2017, 8, 1142 CrossRef PubMed.
- G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931–3939 RSC.
- A. Mal, R. K. Mishra, V. K. Praveen, M. A. Khayum, R. Banerjee and A. Ajayaghosh, Angew. Chem., Int. Ed., 2018, 57, 8443–8447 CrossRef PubMed.
- H. Wang, W. Zhu, L. Feng, Q. Chen, Y. Chao, Z. Dong and Z. Liu, Nano Res., 2018, 11, 3244–3257 CrossRef.
- H. Wang, W. Zhu, J. Liu, Z. Dong and Z. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 14475–14482 CrossRef PubMed.
- H. Soo Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165 CrossRef PubMed.
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CrossRef PubMed.
- M. K. Kumawat, M. Thakur, R. B. Gurung and R. Srivastava, Sci. Rep., 2017, 7, 15858 CrossRef PubMed.
- G. Yang, C. Zhu, D. Du, J. Zhu and Y. Lin, Nanoscale, 2015, 7, 14217–14231 RSC.
- W. Congyu, W. Chong, H. Ting, Z. Xuejiao, G. Shouwu and Z. Jingyan, Adv. Healthcare Mater., 2013, 2, 1613–1619 CrossRef PubMed.
- R. Liu, D. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2011, 133, 15221–15223 CrossRef PubMed.
- V. S. Vyas, M. Vishwakarma, I. Moudrakovski, F. Haase, G. Savasci, C. Ochsenfeld, J. P. Spatz and B. V. Lotsch, Adv. Mater., 2016, 28, 8749–8754 CrossRef PubMed.
- P. Ding, H. Wang, B. Song, X. Ji, Y. Su and Y. He, Anal. Chem., 2017, 89, 7861–7868 CrossRef PubMed.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef PubMed.
- J. Guo, Y. Xu, S. Jin, L. Chen, T. Kaji, Y. Honsho, M. A. Addicoat, J. Kim, A. Saeki, H. Ihee, S. Seki, S. Irle, M. Hiramoto, J. Gao and D. Jiang, Nat. Commun., 2013, 4, 2736 CrossRef PubMed.
- M. J. Bojdys, S. A. Wohlgemuth, A. Thomas and M. Antonietti, Macromolecules, 2010, 43, 6639–6645 CrossRef.
- M. J. Bojdys, J. Jeromenok, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 2202–2205 CrossRef PubMed.
- S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. Jiang, J. Am. Chem. Soc., 2016, 138, 5797–5800 CrossRef PubMed.
- S. K. Nune, P. Gunda, P. K. Thallapally, Y.-Y. Lin, M. Laird Forrest and C. J. Berkland, Expert Opin. Drug Delivery, 2009, 6, 1175–1194 CrossRef PubMed.
- J. Shen, Y. He, J. Wu, C. Gao, K. Keyshar, X. Zhang, Y. Yang, M. Ye, R. Vajtai, J. Lou and P. M. Ajayan, Nano Lett., 2015, 15, 5449–5454 CrossRef PubMed.
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O'Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O'Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624 CrossRef PubMed.
- C. Backes, N. C. Berner, X. Chen, P. Lafargue, P. LaPlace, M. Freeley, G. S. Duesberg, J. N. Coleman and A. R. McDonald, Angew. Chem., 2015, 127, 2676–2680 CrossRef.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563 CrossRef PubMed.
- S. D. Bergin, Z. Sun, D. Rickard, P. V. Streich, J. P. Hamilton and J. N. Coleman, ACS Nano, 2009, 3, 2340–2350 CrossRef PubMed.
- S. Mitra, S. Kandambeth, B. P. Biswal, A. Khayum M, C. K. Choudhury, M. Mehta, G. Kaur, S. Banerjee, A. Prabhune, S. Verma, S. Roy, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2016, 138, 2823–2828 CrossRef PubMed.
- G. Tai, T. Hu, Y. Zhou, X. Wang, J. Kong, T. Zeng, Y. You and Q. Wang, Angew. Chem., Int. Ed., 2015, 54, 15473–15477 CrossRef PubMed.
- X. Li, H. Ren, Z. Zou, J. Sun, J. Wang and Z. Liu, Chem. Commun., 2016, 52, 453–456 RSC.
- J. Dong, X. Li, K. Zhang, Y. Di Yuan, Y. Wang, L. Zhai, G. Liu, D. Yuan, J. Jiang and D. Zhao, J. Am. Chem. Soc., 2018, 140, 4035–4046 CrossRef PubMed.
- T. Levine and C. Rabouille, Curr. Opin. Cell Biol., 2005, 17, 362–368 CrossRef PubMed.
- K. Bin, C. Shuquan, D. Yaodong, Y. Decai and C. Da, Small, 2010, 6, 2362–2366 CrossRef PubMed.
- J. G. R. Elferink, Biochem. Pharmacol., 1979, 28, 965–968 CrossRef PubMed.
- D. Devos, S. Dokudovskaya, F. Alber, R. Williams, B. T. Chait, A. Sali and M. P. Rout, PLoS Biol., 2004, 2, e380 CrossRef PubMed.
- R. B. Weisenfeld, Bioorganic Med. Chem., 2007, 15, 6361–6366 CrossRef PubMed.
- M. E. Lalande, V. Ling and R. G. Miller, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 363–367 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02842g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |