
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePDMS-coated hypercrosslinked porous organic polymers modified via double postsynthetic acidifications for ammonia capture†
Dong Won
Kang
a,
Minjung
Kang
a,
Minkyu
Moon
a,
Hyojin
Kim
a,
Sunhwi
Eom
a,
Jong Hyeak
Choe
a,
Woo Ram
Lee
b and
Chang Seop
Hong
 *a
*a
aDepartment of Chemistry, Korea University, 136-713 Seoul, Republic of Korea. E-mail: cshong@korea.ac.kr
bDepartment of Chemistry, Sejong University, Seoul 05006, Republic of Korea
First published on 18th July 2018
Abstract
A hypercrosslinked porous organic polymer was modified by post-oxidation and post-sulfonation to obtain a porous platform with a high density of acidic groups. Such an acidified material exhibits record high NH3 adsorption capacity per surface area, fast adsorption rate, and recyclability at low desorption temperature. Noticeably, the coating of the polymer with PDMS represents a facile and efficient route to enable both a significant improvement of low-pressure NH3 adsorption capacity (∼40-fold enhancement; from 0.04 to 1.41 mmol g−1) with respect to the non-modified polymer at 500 ppm and hydrophobicity associated with the selective sorption of NH3 over water vapor (hydrophilic for the non-coated material). This material is easy to prepare, cost-effective, and scalable to mass production.
Introduction
Ammonia (NH3) is one of the most important chemical gases that are annually produced on a large scale, because it is utilized in fertilizer production, chemical resources, and building blocks for the synthesis of many pharmaceutical products.1–4 However, NH3 is a corrosive and harmful chemical even at low concentrations,5 and its effective and immediate removal is demanded to secure safety in industrial fields and human health.Recently, metal–organic frameworks (MOFs) and porous organic polymers (POPs) have been actively explored to remove toxic NH3 gas.6–11 Based on the basic NH3 capture, acidic sites such as open metal sites or acidic functional groups including boronic acid, carboxylic acid, phosphonic acid, and sulfonic acid are incorporated into the backbone of a framework.12–14 The acidic sites can form strong bonding with NH3 molecules, but, however, it requires high temperature (>200 °C) for the desorption of NH3, as found in some MOFs with open metal sites and POPs with large NH3 adsorption capacity.9,13 Moreover, although the selectivity of NH3 over water vapor is a key factor for practical applications, the competitive hydrogen bonding of NH3 and H2O molecules with acidic sites reduces the NH3 adsorption capacity.7 Therefore, material regeneration at low desorption temperature and protection of the acidic sites from water access are challenging yet essential for practical NH3 uptake.
The polymerization of monomers with acidic moieties has synthetic problems due to the strong reactivity of acidic functional groups during the preparation of porous frameworks.8,12 To overcome the limitation of bottom-up synthesis, acidic groups can be first protected during the formation of a framework and then deprotected to effectively remove NH3. However, this method inevitably requires complicated synthetic multi-steps and results in low total yields. Hypercrosslinked porous organic polymers (HCPs) are prepared using the Friedel–Craft reaction which entails simple synthetic procedures, fast reaction rate, and high yields.15–17 Due to these advantages, HCPs have been investigated in various research fields including CO2 conversion, dye separation, organic vapor adsorption, and gas storage.18–20 Nonetheless, to the best of our knowledge, HCPs modified by post acidifications have not been demonstrated for NH3 capture applications.
Herein, we report facile yet unique double postsynthetic acidifications of an HCP platform via post-oxidation and post-sulfonation processes. This sequential double modification protocol represents an effective route to generate a framework with significant NH3 uptake per surface area and recyclability at low desorption temperature. Surprisingly, the coating of the acidified polymer with hydroxyl-terminated poly(dimethylsiloxane) (PDMS), which has not been demonstrated before for HCP modification, proves to be a reasonable strategy to obtain a cost-effective and scalable platform with enhanced low-pressure NH3 uptake by ∼40 times compared to the non-modified polymer and exceptional hydrophobic characteristics suitable for NH3 capture selectivity over water vapor.
Results and discussion
The dark-brownish solid 1T was obtained from a microwave-assisted solvothermal reaction of toluene, formaldehyde dimethyl acetal (FDA), 1,2-dichloroethane (DCE), and anhydrous FeCl3 as the catalyst. The application of the microwave method to the reaction system significantly reduced the reaction time (5 h) compared with conventional reflux reactions (18–24 h), which can be ascribed to the fast nucleation and crystal growth under microwave irradiation.21–23 In order to oxidize methyl groups on the benzene rings to carboxylic acid groups, 1T was suspended over a mixture of water and ethanol, and refluxed for 2 d after the addition of KMnO4 and 2 N NaOH. The resultant bright-brown solid 1TC was then treated with chlorosulfonic acid to give sulfonated polymer 1TCS with –SO3H groups (Fig. 1). | ||
| Fig. 1 (a) Synthetic scheme for preparing 1T and its post-modified products, 1TC and 1TCS. (b) 1TCS@PDMS coating synthetic scheme. | ||
Powder X-ray diffraction (PXRD) data of all the samples showed no distinct peaks, indicating the formation of amorphous phases (Fig. S1†). The incorporation of the functional groups was inspected by infrared (IR) spectra (Fig. S2†). After oxidation, the distinct peak of C–H stretching at 2980 cm−1 in 1T was suppressed and a new peak assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of carboxylic acid at 1675 cm−1 appeared, indicative of the conversion from 1T to 1TC.23
O stretching of carboxylic acid at 1675 cm−1 appeared, indicative of the conversion from 1T to 1TC.23
Further post-acidification of 1TC with ClSO3H afforded 1TCS, as evidenced by the presence of the S–OH bending mode at 888 cm−1 and the SO2 stretching at 1166 and 1031 cm−1.12,24 The elemental analysis also supported the existence of sulfur in 1TCS (Table S1†). These results specify that the –SO3H groups are incorporated into the framework backbone of 1TC. From the elemental analysis data, we estimated that 45.45% of the methyl groups of toluenes in 1T were oxidized to the carboxylic acid groups in 1TC and then 30.84% of benzene rings in 1TC were sulfonated in 1TCS. After the double post-acidifications, the surface morphologies of 1T, 1TC, and 1TCS remained almost identical, as shown in the scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. S3 and S4†).
To examine the compositional feature, X-ray photoelectron spectroscopy (XPS) was performed for 1T, 1TC, and 1TCS (Fig. S5 and S6†). In the survey scan of 1T, the O1s and Cl2p peaks indicated the presence of a trace of unreacted reagents. The intensity of the O1s peak in 1TC became stronger than that in 1T, suggesting that the methyl group in 1T was oxidized to the –CO2H group in 1TC. In particular, the S2s and S2p peaks were visible in the survey scan of 1TCS, while these peaks were absent in 1TC. This observation verified the successful sulfonation in 1TCS by treating 1TC with ClSO3H. The S2p band was deconvoluted into two peaks of S2p1/2 (169.52 eV) and S2p3/2 (168.11 eV). The binding energies are attributed to –SO3H in good agreement with the IR data.25,26 The same results were also obtained using energy dispersive X-ray spectroscopy-scanning electron microscopy (EDX-SEM) data (Fig. S7†). We performed solid state 13C NMR spectroscopy to gain insight into the structures of the three polymers (Fig. S8 and Table S2†). The spectrum of 1T showed characteristic peaks assignable to methyl and aromatic carbons. In particular, the peak at 34 ppm corresponded to the carbons in the methylene link resulting from the Friedel–Craft alkylation reaction. The band observed in the range of 128–140 ppm was assignable to the various chemical environments of the aromatic carbons upon sequential post-modifications.23,27
The thermogravimetric analysis (TGA) revealed that the frameworks were thermally robust in the range of 25–170 °C (Fig. S9†). From the elemental analysis and IR data before and after regeneration at 120 °C, the elemental contents in 1TC and 1TCS remained invariant, together with almost no variation in IR spectra (Table S3 and Fig. S10†). These results indicate that there is no decay of the functional groups during the regeneration process. To evaluate the porosity of the polymers, nitrogen isotherms at 77 K were collected (Fig. S11†). The Brunauer–Emmett–Teller (BET) surface areas were determined to be 915 ± 2 m2 g−1 for 1T, 552 ± 1 m2 g−1 for 1TC, and 72.5 ± 0.1 m2 g−1 for 1TCS. The sequential modifications gradually reduced the surface area, which are induced from the incorporation of –CO2H and –SO3H groups into the pores.
We collected the NH3 isotherms of three samples at several temperatures, as depicted in Fig. 2 after degassing the pores at a relatively low temperature of 120 °C. This low desorption temperature is beneficial for the reusability of the material, compared with the desorption temperature (>200 °C) observed in some MOF and COF systems with high NH3 adsorption capacity.9,13 While the NH3 adsorbed amount of 1T was below 3.8 mmol g−1 at 298 K and 1 bar, the adsorption capacities of 1TC and 1TCS significantly increased to 6.41 and 8.52 mmol g−1, respectively, despite their reduced BET surface areas (Fig. 2a–c). At a low pressure of interest (500 ppm), the NH3 adsorption of 1TC with carboxylic acid groups was 0.33 mmol g−1, which was 10 times greater than that of the non-modified 1T. The uptake of 1TCS with the carboxylic and sulfonic acid groups was even greater, reaching 0.92 mmol g−1. This capacity of 1TCS was 20 times greater than that of 1T. The acid-functionalization generates a high density of acidic sites in the pores, which accounts for the enhanced affinity and uptake for NH3 especially at low pressure (Fig. S12†). Thus, the low-pressure adsorption trend clearly correlates with the density of acidic functional groups in the pores. Using the Clausius–Clapeyron formula, the enthalpy of NH3 adsorption (−Qst) for the solids was estimated to give the magnitude of −Qst in the order of 1TCS > 1TC > 1T, which is associated with the density of the acidic groups in the systems (Fig. S13†). To check recyclability, we measured the NH3 isotherm of 1TCS again, showing no capacity change (Fig. S14†).
 | ||
| Fig. 2 NH3 isotherms of (a) 1T, (b) 1TC and (c) 1TCS at several temperatures (298 K – black, 323 K – red, 348 K – blue, and 373 K – magenta). Filled and open symbols indicate adsorption and desorption, respectively. (d) NH3 adsorption capacity of 1TCS under humid conditions and gas capacity changes after regeneration. | ||
To investigate NH3 uptake under humid conditions, we conducted titration experiments (Fig. 2d and S15†). The activated 1TCS was exposed to 4% NH3 solution for 30 min (Fig. S16a†).28 The sample was then located in the apparatus and degassed at 120 °C and for 10 h under He gas. After the degassing process, the outgas containing desorbed NH3 was connected to 0.5 N H2SO4 solution and the resulting solution was then titrated with 0.0250 M NaOH solution (Fig. S16b†). After 30 min, the titration result demonstrated that the adsorbed NH3 amount of 1TCS was rapidly saturated at 2.94 mmol g−1 (Fig. S15†). This uptake amount under the given conditions was significant and more than three times higher than (0.88 mmol g−1) that observed in an isonicotinate-bridged Zn–MOF.29 We performed adsorption-regeneration cycles of 1TCS over 10 times and the adsorption capacity was maintained during the repeated runs (Fig. 2d). We collected the IR spectroscopic data of 1TCS before and after the co-vapor exposure (Fig. S17†). Upon the exposure, the asymmetric vibration modes of carboxylate (–CO2−) and sulfonate (–SO3) groups in the IR spectrum were visible at 1590 and 1207 cm−1, respectively.12 Moreover, the IR pattern of the regenerated 1TCS was almost identical to that of the pristine 1TCS. This result suggests that the adsorption–desorption cycles are reversible, thereby indicating the potential of 1TCS as a promising NH3 adsorbent.
To carefully examine the affinity of the pore surface of the framework depending on the density of acidic sites, we converted the NH3 adsorption amounts at 298 K to an adsorbed amount of NH3 per surface area at a low pressure of 0.5 mbar (Fig. 3a and Table S4†).30 The NH3 adsorption per surface area of 1TCS was calculated to be 1.278 × 10−2 mmol m−2. Notably, this value is the highest recorded among the porous NH3 capture materials in the low pressure range up to 1 mbar NH3 (Table S4†). Such a substantial density of acidic functional groups in the pores originates from the double postsynthetic acidifications first demonstrated in this study. The highly acidic surface of 1TCS could contribute to the rapid adsorption rate of NH3 (Fig. S15†).
 | ||
| Fig. 3 (a) Correlation diagram of NH3 adsorbed amounts per surface area around 0.5 mbar. (1: 1TCS, 2: P2-SO3H,12 3: P2-CO2H,12 4: P1-PO3H2,12 5: Fe-MIL-101-SO3H,8 6: P1-SO3H,12 7: UiO-66-NH2,8 8: P1-NH3Cl,12 and 9: P2-NH3Cl12). (b) NH3 isotherms of coated samples at low pressure and 298 K. | ||
Postsynthetic acidifications of polymer 1T promote the affinity toward water vapor. As the acidic modifications of the surface progressed, the framework represented enhanced hydrophilic nature, as shown in the water vapor isotherms of Fig. S18.†25,311TCS captured 12 mmol g−1 around P/Po ∼ 0.9 and its adsorption amount was three times greater than that of 1T at the same relative pressure. Impedance spectroscopic data also supported the hydrophilic affinity of the samples through proton conductivity changes (Fig. S19†). The conductivity of 1T was 2.47 × 10−7 S cm−1 at 30 °C and 90% relative humidity (RH). After post-oxidation of the methyl group, the carboxylic groups were decorated on the pore surface of 1TC, increasing the conductivity to 2.85 × 10−6 S cm−1 under the same conditions. Further acidification of the solid with sulfonic groups allowed for a 3 orders of magnitude conductivity enhancement of 1TCS, reaching 2.09 × 10−3 S cm−1 at 30 °C and 90% RH. At 80 °C and 90% RH, 1TCS showed an even higher conductivity of 6.27 × 10−3 S cm−1 with inductance error corrections, which means that 1TCS belongs to the range of high-performance POP conductors.32–34 In particular, no semicircle was observed in the Nyquist plots of 1TCS, which is due to the reduction of the time constant related to the product of the resistance and capacitance values.35 The sequential post-acidification process engenders a gradual increase of the conductivity, which demonstrates that the density of acidic functional groups increases upon post-modifications.
To enhance the selectivity of NH3 over water vapor, we coated the surface of 1TCS using a hydrophobic polymer PDMS.36 The successful introduction of PDMS on 1TCS was confirmed by IR, XPS, and EDX-SEM analyses (Fig. S20 and S21†). The new peaks were observed at 2965 and 1257 cm−1, assigned to the antisymmetric C–H stretching and symmetric bending in the methyl group, respectively, and their intensities were proportional to the loading of PDMS (Fig. S20a†).37 The existence of Si atoms was verified by distinct peaks related to Si2s and Si2p in the XPS and EDX-SEM spectra, corroborating the reliable surface coating of 1TCS (Fig. S20b and S21†).
For PDMS-coated samples with different loadings, N2 isotherms at 77 K and NH3 isotherms at 298 K were collected (Fig. 3b, S22 and S23†). While no N2 adsorption was observed for all samples, NH3 adsorption was significant. The NH3 adsorption in the second cycle remained identical to the adsorption in the first cycle (Fig. S24†). Remarkably, the NH3 uptake of PDMS-coated materials increased in the entire pressure range. Hydroxyl groups in the terminal of the PDMS chain can create the preferred adsorption sites for NH3, which could be responsible for the superior uptake.38,39 In particular, at 500 ppm, the adsorption capacity of 1TCS@PDMS10 increased to 1.41 mmol g−1, which is ∼40 times greater than that of the non-modified polymer 1T (Fig. 3b). Considering the low pressure level of NH3 required for human safety, the fact that such a considerable enhancement of low-pressure NH3 adsorption capacity is achievable by simply modifying the porous polymer with the proper coating protocol is a promising and encouraging result for real applications. Furthermore, to examine the hydrophobic character against water molecules, we conducted a water droplet test (Fig. 4a and S25†). While the water droplet was rapidly absorbed by the non-coated 1TCS due to the hydrophilicity of the solid surface, PDMS-coated samples showed large contact angles (121–134°), implying hydrophobic properties on the surface.36,40 The impedance data demonstrated that the activation energy of 1TCS@PDMS10 was 0.31 eV, which is higher than that of the non-coated solid (0.23 eV), suggesting that the PDMS coating diminishes conduction pathways via occlusion of some water channels and promotes hydrophobicity (Fig. S26†).
 | ||
| Fig. 4 (a) Water droplet test of 1TCS@PDMS10. (b) Ammonia adsorption capacity of 1TCS@PDMS10 under humid conditions and gas capacity changes after regeneration. | ||
To investigate the co-adsorption of NH3 and water vapor, 1TCS@PDMS10 was placed in a 4% NH3 solution atmosphere at 25 °C for 30 min and the outlet gas was titrated. In the IR data, characteristic peaks of –CO2− and –SO3− were observed after NH3 adsorption (Fig. S27†). The adsorption capacity of 2.1 mmol g−1 in the first run was almost maintained over 4 cycles, suggesting that the PDMS-coated sample was recyclable without capacity loss (Fig. 4b and S28†).
Thus, the double postsynthetic acidification method provides an efficient route to maximize the density of the acidic functional groups which improves the NH3 adsorption capacity and rate at low pressures. More importantly, the post-coating process of acidified solids with PDMS not only increases NH3 adsorption capacity but also reinforces hydrophobicity linked to water inaccessibility. Given that the NH3 adsorption property and hydrophobicity are mutually antagonistic, the PDMS coating method represents a remarkable strategy to achieve both desirable characteristics optimal to NH3 uptake applications.
Conclusions
In summary, we prepared a hypercrosslinked porous organic polymer which was then modified via sequential post-oxidation and post-sulfonation to achieve a superior NH3 capture material. The unique double postsynthetic acidifications offer the incorporation of a high density of acidic functional groups into the porous polymer framework, enabling high NH3 adsorption per surface area at low pressures and excellent recyclability at low desorption temperature. More remarkably, the PDMS coating of the acidified polymer platform provides a successful approach to achieve the desirable NH3 adsorption properties of superb low-pressure NH3 adsorption capacity and hydrophobicity. This polymer material with significant low-pressure NH3 adsorption capacity, rapid adsorption rate, recyclability, and hydrophobicity is cost-effective and scalable to mass production.Experimental
Preparation
All starting materials and solvents including formaldehyde dimethyl acetal, anhydrous 1,2-dichloroethane, and anhydrous FeCl3 employed in the synthesis were purchased from commercial suppliers and utilized without further purification except for toluene which was used after distillation. Hydroxyl-terminated poly(dimethylsiloxane) (PDMS) having viscosity 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–22000 cST was bought from Sigma-Aldrich.
000–22000 cST was bought from Sigma-Aldrich.
Synthesis of 1T
After anhydrous FeCl3 (4.5 g) was added to a 35 mL Pyrex cell for microwave reaction, the cell was sealed with a septum and treated in a vacuum. Then, formaldehyde dimethyl acetal (FDA) and 1,2-dichloroethane (DCE) were added to the cell using a cannula and Schlenk line and the cell was sonicated for 30 minutes. Finally, the mixture was sealed with a PTTE cap and irradiated in a CEM Discover microwave reactor (100 W, 150 psi, 40 °C for 1.5 h, 80 °C for 1.5 h, 100 °C for 1 h, and 120 °C for 1 h) after distilled toluene was added to the mixture. Then, the dark-brownish precipitate was transferred into a mixture of 1 M HCl 100 mL and methanol and stirred for 1 h. The precipitate was collected by filtration and washed several times with 1 M HCl, deionized water, tetrahydrofuran, ethanol, acetone, and methanol. 1T was dried overnight in an oven at 100 °C and degassed at 120 °C in a vacuum for 12 h.Synthesis of 1TC
1TC was synthesized by a slight modification of the reported procedure.23 After H2O (100 mL) and ethanol (50 mL) were poured into a 250 mL-round-bottom flask containing 1T (1 g), the mixture was stirred for 1 h. Then, KMnO4 (1.40 g) and 2 N NaOH 8 mL were added into the flask and the mixture was refluxed at 90 °C for 60 h. The product was collected by filtration and washed with 1 M HCl solution, acetone, water, tetrahydrofuran, and methanol several times. 1TC was dried overnight in an oven at 100 °C and degassed at 120 °C in a vacuum for 12 hours.Synthesis of 1TCS
After methylene chloride (150 mL) was added to a 250 mL-round-bottom flask containing ∼3.00 g of 1TC, the mixture was stirred for 30 min. Chlorosulfonic acid (36 mL) was then added dropwise to the dispersed mixture in an ice-water bath. After stirring at room temperature for 5 d, the mixture was poured into a beaker containing 1 L of ice-water and stirred for 12 h. The solid was collected by filtration and neutralized to pH 7 by washing several times with water and methanol. The product was dried overnight in an oven at 100 °C and degassed at 120 °C in a vacuum for 12 h.Synthesis of 1TCS@PDMSX (X = 10, 20, and 50)
After X mg of PDMS was dissolved in hexane (3 mL), 1TCS (100 mg) was dispersed in the solution and sonicated for 10 min. The dispersed solution was dried under N2 flowing conditions and desiccated overnight in an oven at 100 °C.Physical measurements
Infrared spectra were obtained with an ATR module using a Nicolet iS10 FT-IR spectrometer. Thermogravimetric analysis (TGA) was carried out in an Ar (99.999%) atmosphere (flow rate = 30 mL min−1) in the temperature range 30–800 °C (heating rate = 10 °C min−1) using a Scinco TGA-N 1000. Powder XRD patterns were recorded using Cu Kα (λ = 1.5406 Å) on a Rigaku Ultima III diffractometer with a scan speed of 2° min−1 and a step size of 0.02°. Deionized water in the experiment was purified using an aqua MAX™ Basic 360 series. Elemental analyses for C, H, and S were performed at the Elemental Analysis Service Center of Sogang University. SEM images were acquired using a JSM-7001F. XPS was performed at the Semiconductor & Display Green Manufacturing Research Center (GMRC) at Korea University. NMR data were obtained from a 500 MHz Avance II + Bruker Solid-state NMR at the National Center for Inter-University Research Facilities (NCIRF) at Seoul National University. EDX-SEM analysis was performed at the KBSI Seoul Center using a Hitachi SU-70 and other EDX-SEM data were obtained from a Quanta 250 FEG at GMRC. Transmission Electron Microscopy (TEM) was carried out using a Tecnai G2 20 S-Twin microscope. The contact angle was measured using a Phoenix-MT(T).Gas sorption measurements
Before sorption analysis, the samples except for coated samples were degassed at 120 °C in a vacuum for 10 h. Coated samples were degassed at 160 °C in a vacuum for 10 h. All gases used in measurements were highly pure (99.999%) except for water vapor and NH3 (99.9995%). N2 gas sorption measurements at 77 K were carried out on a Micromeritics ASAP2020 instrument up to 1 atm of gas pressure. Water vapor isotherms at 298 K were obtained using a Micrometrics ASAP2020 from a vapor source after additional purification using an ASAP2020 (freezing-vacuum). Isotherms of NH3 at several temperatures were collected using a Micromeritics 3-Flex up to 1 atm of gas pressure. Filled and open symbols indicate adsorption and desorption, respectively.Impedance analyses
All pellets for measuring AC impedance data were prepared by homogeneously grinding the powder samples with a mortar and pestle and pressing at 2500–4200 kg for a few minutes. The thickness of the pellets ranged from 0.01 to 0.05 cm and their diameter was 0.5 cm. When all samples were measured, the pellet was placed in a home-made sample holder composed of Pt-based electrodes and the holder was located in a temperature and humidity controlled chamber of ESPEC SH-222/Bench-top type. To equilibrate temperature and humidity in the chamber, the sample was maintained over 6–24 h before AC measurements. To obtain more accurate data, impedance data were collected repeatedly until the value-convergence. AC measurements were carried out using a Solartron SI 1260 Impedance/Gain-Phase Analyzer and Dielectric Interface with Pt-pressed electrodes and an applied AC voltage amplitude of 100 mV and a frequency range of 3 MHz to 0.1 Hz. ZView and ZPlot software were used to analyze and fit impedance plot to acquire the resistance value through a designed equivalent circuit. Especially, to correct inductance error generated from wires, etc. in a high frequency range, we re-designed an equivalent circuit containing a series inductance and accurately measured the resistance from the spectra of 1TCS and 1TCS@PDMS10. Then, the accurate conduction values and activation energies were calculated using equations below.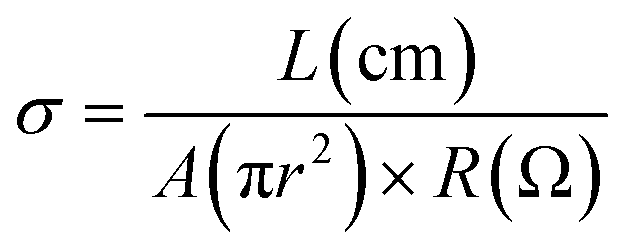

Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by a Korea CCS R&D Center (KCRC) grant funded by the Korean government (the Ministry of Science, ICT, & Future Planning (MSIP)) (NRF-2014M1A8A1049253), by the Basic Science Research Program (NRF-2015R1A2A1A10055658 and NRF-2017R1C1B5018060), and by a Korea University Grant. We appreciate Prof. Kwangyeol Lee for the TEM measurements.Notes and references
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef.
- H. Doh, H. Y. Kim, G. S. Kim, J. Cha, H. S. Park, H. C. Ham, S. P. Yoon, J. Han, S. W. Nam, K. H. Song and C. W. Yoon, ACS Sustainable Chem. Eng., 2017, 5, 9370–9379 CrossRef.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- F. Schüth, R. Palkovits, R. Schlögl and D. S. Su, Energy Environ. Sci., 2012, 5, 6278–6289 RSC.
- Permissible Exposure Limits for Chemical Contaminants, Cal/OSHA, Oakland, CA, http://www.dir.ca.gov/title8/5155table_ac1.html, accessed on June 15, 2018.
- N. S. Bobbitt, M. L. Mendonca, A. J. Howarth, T. Islamoglu, J. T. Hupp, O. K. Farha and R. Q. Snurr, Chem. Soc. Rev., 2017, 46, 3357–3385 RSC.
- K. Vikrant, V. Kumar, K. Kim and D. Kukkar, J. Mater. Chem. A, 2017, 5, 22877–22896 RSC.
- J. F. Van Humbeck, T. M. McDonald, X. Jing, B. M. Wiers, G. Zhu and J. R. Long, J. Am. Chem. Soc., 2014, 136, 2432–2440 CrossRef PubMed.
- C. J. Doonan, D. J. Tranchemontagne, T. G. Glover, J. R. Hunt and O. M. Yaghi, Nat. Chem., 2010, 2, 235–238 CrossRef PubMed.
- J. W. Lee, G. Barin, G. W. Peterson, J. Xu, K. A. Colwell and J. R. Long, ACS Appl. Mater. Interfaces, 2017, 9, 33504–33510 CrossRef PubMed.
- A. J. Rieth, Y. Tulchinsky and M. Dinca, J. Am. Chem. Soc., 2016, 138, 9401–9404 CrossRef PubMed.
- G. Barin, W. G. Peterson, V. Crocellà, J. Xu, A. K. Colwell, A. Nandy, A. J. Reimer, S. Bordiga and R. J. Long, Chem. Sci., 2017, 8, 4399–4409 RSC.
- G. T. Grant, G. W. Peterson, B. J. Schindler, D. Britt and O. M. Yaghi, Chem. Eng. Sci., 2011, 66, 163–170 CrossRef.
- A. J. Rieth and M. Dinca, J. Am. Chem. Soc., 2018, 140, 3461–3466 CrossRef PubMed.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef PubMed.
- B. Tan and L. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- K. J. Msayib and N. B. McKeown, J. Mater. Chem. A, 2016, 4, 10110–10113 RSC.
- Z. Yang, H. Wang, G. Ji, X. Yu, Y. Chen, X. Liu and Z. Liu, New J. Chem., 2017, 41, 2869–2872 RSC.
- P. Samanta, P. Chandra, A. V. Desai and S. K. Ghosh, Mater. Chem. Front., 2017, 1, 1384–1388 RSC.
- H. Gao, L. Ding, H. Bai and L. Li, ChemSusChem, 2017, 10, 618–623 CrossRef PubMed.
- B. Li, R. Gong, W. Wang, X. Huang, W. Zhang, H. Li, C. Hu and B. Tan, Macromolecules, 2011, 44, 2410–2414 CrossRef.
- C. Wilson, M. J. Main, N. J. Cooper, M. E. Briggs, A. I. Cooper and D. J. Adams, Polym. Chem., 2017, 8, 1914–1922 RSC.
- Y. He, Q. Liu, J. Hu, C. Zhao, C. Peng, Q. Yang, H. Wang and H. Liu, Sep. Purif. Technol., 2017, 180, 142–148 CrossRef.
- R. Gomes, P. Bhanja and A. Bhaumik, J. Mol. Catal. A: Chem., 2016, 411, 110–116 CrossRef.
- D. W. Kang, K. S. Lim, K. J. Lee, J. H. Lee, W. R. Lee, J. H. Song, K. H. Yeom, J. Y. Kim and C. S. Hong, Angew. Chem., Int. Ed., 2016, 55, 16123–16126 CrossRef PubMed.
- D. W. Kang, J. H. Song, K. J. Lee, H. G. Lee, J. E. Kim, H. Y. Lee, J. Y. Km and C. S. Hong, J. Mater. Chem. A, 2017, 5, 17492–17498 RSC.
- S. Mane, Z. Gao, Y. Li, D. Xue, X. Liu and L. Sun, J. Mater. Chem. A, 2017, 5, 23310–23318 RSC.
- Y. Chen, Y. Wang, C. Yang, S. Wang, J. Yang and J. Li, ACS Sustainable Chem. Eng., 2017, 5, 5082–5089 CrossRef.
- Y. Chen, C. Yang, X. Wang, J. Yang, K. Ouyang and J. Li, J. Mater. Chem. A, 2016, 4, 10345–10351 RSC.
- S. J. Lee, S. Kim, E. J. Kim, M. Kim and Y. S. Bae, Chem. Eng. J., 2018, 335, 345–351 CrossRef.
- W. J. Phang, H. Jo, W. R. Lee, J. H. Song, K. Yoo, B. Kim and C. S. Hong, Angew. Chem., Int. Ed., 2015, 54, 5142–5146 CrossRef PubMed.
- T. Soboleva, Z. Xie, Z. Shi, E. Tsang, T. Navessin and S. Holdcroft, J. Electroanal. Chem., 2008, 622, 145–152 CrossRef.
- H. Xu, S. Tao and D. Jiang, Nat. Mater., 2016, 15, 722–726 CrossRef PubMed.
- D. B. Shinde, H. B. Aiyappa, M. Bhadra, B. P. Biswal, P. Wadge, S. Kandambeth, B. Garai, T. Kundu, S. Kurungot and R. Banerjee, J. Mater. Chem. A, 2016, 4, 2682–2690 RSC.
- D. Umeyama, S. Horike, M. Inukai, Y. Hijikata and S. Kitagawa, Angew. Chem., Int. Ed., 2011, 50, 11706–11709 CrossRef PubMed.
- G. Huang, Q. Yang, Q. Xu, S. Yu and H. Jiang, Angew. Chem., Int. Ed., 2016, 55, 7379–7383 CrossRef PubMed.
- C. Kapridaki and P. Maravelaki-Kalaitzaki, Prog. Org. Coat., 2013, 76, 400–410 CrossRef.
- G. A. Blomfield and L. H. Little, Can. J. Chem., 1973, 51, 1771–1781 CrossRef.
- S. Brandenberger, O. Kröcher, A. Tissler and R. Althoff, Catal. Rev.: Sci. Eng., 2008, 50, 492–531 CrossRef.
- S. Tao, L. Xu and J. C. Fanguy, Sens. Actuators, B, 2006, 115, 158–163 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02640h |
| This journal is © The Royal Society of Chemistry 2018 |