
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemically stable ionic viologen-organic network: an efficient scavenger of toxic oxo-anions from water†
Partha
Samanta
 a,
Priyanshu
Chandra‡
a,
Subhajit
Dutta‡
a,
Aamod V.
Desai
a and
Sujit K.
Ghosh
*ab
a,
Priyanshu
Chandra‡
a,
Subhajit
Dutta‡
a,
Aamod V.
Desai
a and
Sujit K.
Ghosh
*ab
aDepartment of Chemistry, Indian Institute of Science Education and Research, Dr. Homi Bhabha Road, Pashan, Pune 411008, India. E-mail: sghosh@iiserpune.ac.in; Tel: +91 20 2590 8076
bCentre for Energy Science, IISER Pune, Dr. Homi Bhabha Road, Pashan, Pune 411008, India
First published on 20th August 2018
Abstract
Detoxification of water has been demonstrated with a viologen-based cationic organic network (compound-1), which was stable not only in water, but also in acidic and basic media. The presence of free exchangeable Cl− ions inside the network of compound-1 and a high physiochemical stability of the materials offered a suitable scope for the capture of hazardous anionic pollutants from water. Rapid removal of the toxic water pollutant and carcinogenic chromate (CrO42−) from water was shown with compound-1. Furthermore, the oxo-anion of the radioactive isotope of technetium (99Tc), i.e. the TcO4− ion, also counts as a toxic water pollutant and by using surrogate anions (MnO4− and ReO4−), a model capture study was performed. Notably, compound-1 showed high capacity values for each of the oxo-anions and these were comparable to some of the well-performing compounds reported in the literature. Furthermore, to check the real time aspect, removal of all of the aforementioned anions from water was demonstrated, even in the presence of other concurrent anions.
Introduction
Increasing water pollution has become a global concern in recent years, and remediation of such toxic pollutants has drawn much attention worldwide. Pollution due to metal derived oxo-anions (CrO42−, TcO4−, SeO32−, AsO43−etc.) has become a pressing challenge, as most of them are omnipresent in the environment.1 Especially, Cr(VI) based oxo-anions are found to be very carcinogenic and mutagenic to living systems.2,3 Understanding the importance of this, the EPA (Environmental Protection Agency, U.S.) has included such oxo-anions in the priority pollutant list.4,5 Chromate has been found in a wide range of applications in various industries like leather tanning, textile dyes and pigments, steel manufacturing, wood preservation, electroplating etc., where tanning industries alone discharge ∼30–35 liters of Cr(VI) contaminated water for each kilogram of leather.6,7 It also affects the vitrification of low activity radioactive waste, as it forms spinels and has resulted in weakening the integrity of waste glass.8,9 Furthermore, the Hinkley groundwater contamination is one of the well known disasters caused by dumped Cr(VI) in California.10 Apart from this case, several more incidents were found, and moreover the problems are still continuing, which has led researchers to design affordable and efficient techniques to capture Cr(VI) based oxo-anions.11,12 Apart from chromate, another oxo-anion, the pertechnetate (TcO4−) ion, has caused much concern as one isotope of technetium (99Tc) is a radioactive element with a very high half-life time (2.1 × 105 years). 99Tc has been found to be formed as a nuclear fission product of 235U or 239Pu with a high fission yield. Up to 2010 it was estimated that ∼305 metric tons of 99Tc was produced from weapons testing and nuclear reactors.13 Furthermore, Tc exists mostly as TcO4− in the environment, which is highly soluble in water with high mobility. As a consequence of this, TcO4− may also exist in low level waste. To date, many different techniques have been employed for the removal of oxo-anions, like ion exchange, chemical precipitation, adsorption, electrodialysis, photocatalysis etc.14–17 The ion exchange method has been considered as a more efficient technique over the others owing to its low cost, being comparatively simple and safe, and its efficient performance even for waste which has a low concentration of pollutants etc.18,19 Although several anion exchange resins have been reported, their poor selectivity, poor exchange kinetics and lack of stability have led researchers to render new materials for efficient oxo-anion capture from water.20 As an alternative, porous cationic framework based materials, such as layered double hydroxides (LDHs), metal–organic frameworks (MOFs) etc., have emerged in recent years.21–35 Recent reports have shown that both MOF and LDH materials can be useful for the removal of toxic and hazardous anionic pollutants from waste water.36–39Progress in the area of porous materials has skyrocketed over the past few decades due to their wide range of applications. Among them, metal–organic frameworks (MOFs) and lately evolved porous organic materials have seen much progress in recent years over congener materials, because of features like amenability in their design, high surface areas and tuneable pore surfaces as per requirements.40–47 Although MOFs have been employed to capture oxo-anions from water, a lack of sufficient physiochemical stability of most of the MOFs and difficulties in the bulk scale synthesis have hindered their applications.48,49 In contrast, porous organic materials are constructed from strong covalent bonds, which result in a high physiochemical stability. Such stability has made them one of the frontrunners in the area of porous materials, and it has been witnessed that for real time applications the stability has gained more priority over direct structure property correlations.50,51 These compounds have already acquired much attention in the field of gas adsorption, catalysis, drug delivery, fuel cell applications etc.; but are very rarely employed to capture hazardous oxo-anions from water.52–63 To execute anion capture via ion exchange, a cationic network with exchangeable anions is a primary requirement. But direct synthesis of such cationic organic frameworks has rarely been explored.64–68 In the literature very few reports are present on the direct synthesis of ionic organic frameworks, and viologen based organic frameworks are one of them, which have been synthesized via a Schiff base reaction, Sonogashira–Hagihara coupling, a Menshutkin reaction and trimerisation of –CN groups.69–73 Zincke’s reaction is an effective tool to synthesize viologen based compounds in a single step, but this reaction has not been used much to synthesize extended porous organic networks.74–76 Trabolsi & co-workers showed that the morphologies of compounds can be different depending on the polarity of solvents used during the synthesis, which can directly influence the properties of those compounds.74 Owing to such a structural diversity and chemical stability, cationic frameworks can be well suited to the capture of hazardous anionic pollutants from water. Herein, we have demonstrated the efficient capture of toxic oxo-anions with a viologen based organic network (compound-1) owing to its chemical stability and free exchangeable anions (Fig. 1).
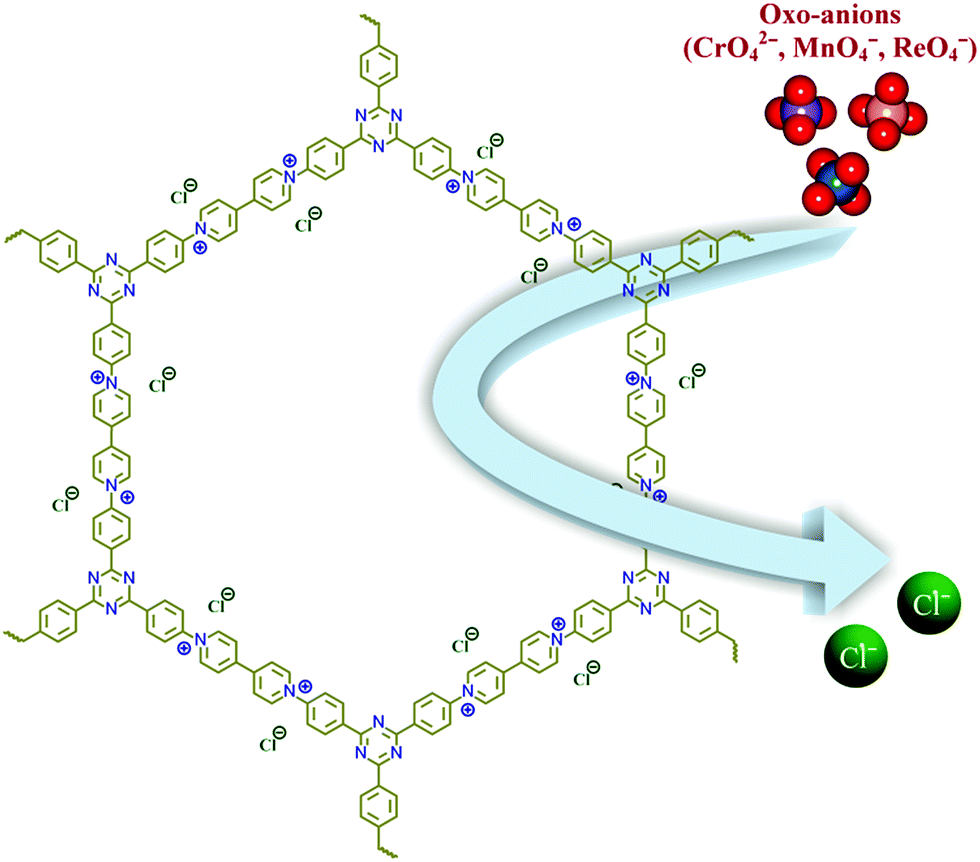 | ||
| Fig. 1 Schematic representation of the oxo-anion capture in compound-1. | ||
Experimental
Materials
4,4′-Dipyridyl, 1-chloro-2,4-dinitrobenzene and 4-aminobenzonitrile were purchased from Sigma-Aldrich, KReO4 was purchased from Alfa-Aesar; triflic acid, KMnO4 and K2CrO4 and all other solvents were purchased locally. The obtained chemicals were used without any further purification.Physical measurements
All infra-red (IR) spectra were recorded using a NICOLET 6700 FT-IR spectrophotometer, using KBr pellets in the range of 400–4000 cm−1. Thermogravimetric analysis (TGA) was recorded on a Perkin-Elmer STA 6000 TGA analyser under an N2 atmosphere with a heating rate of 10 °C min−1. Gas adsorption measurements were carried out using a BelSorp-max instrument from Bel Japan. FESEM was performed using a FEI Quanta 3D dual beam ESEM at 30 kV. UV spectra were acquired on a Shimadzu UV 2600 Spectrophotometer.Synthesis of 4,4′,4′′-(1,3,5-triazine-2,4,6-triyl)trianiline (precursor 1)
Precursor 1 was synthesised from 4-aminobenzonitrile via an acid catalyzed trimerisation reaction by following a previously reported protocol.77Synthesis of 1,1′-bis(2,4-dinitrophenyl)-4,4′-bipyridinium dichloride (precursor 2)
Precursor 2 was also synthesised by following a previously reported protocol.78Synthesis of compound-1
Compound-1 was synthesized via the Zincke reaction of precursor 1 and 2 (Scheme S1†). First, a Schlenk tube was charged with 560 mg of precursor 2 (1 mmol) and 177 mg of precursor 1 (0.5 mmol); then 20 mL of ethanol (EtOH), 20 mL of 1,4-dioxane and 20 mL of Cl-benzene were added respectively to the reaction mixture. After that, the reaction mixture was purged with N2 gas and allowed to reflux for 2 days under these conditions. After completion of the reaction, the reaction mixture was filtered and washed with different solvents like dimethylformamide (DMF), water, tetrahydrofuran (THF), MeCN, acetone, methanol (MeOH) and dichloromethane (DCM) to remove unreacted materials and small polymers (oligomers). A brown colored solid material was obtained, which was further kept in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of chloroform and THF to exchange guest molecules (with a high boiling point) with low boiling solvent molecules. After 4 days, the compound was filtered off and heated at ∼100 °C under vacuum to remove the occluded guest molecules. Yield: 240 mg.
:1 mixture of chloroform and THF to exchange guest molecules (with a high boiling point) with low boiling solvent molecules. After 4 days, the compound was filtered off and heated at ∼100 °C under vacuum to remove the occluded guest molecules. Yield: 240 mg.
Time dependent study of oxo-anion removal
In the case of the CrO42− removal study, we took 2 mL of an aqueous solution of 0.25 mM CrO42− ions in a cuvette and recorded the initial absorbance with UV-Vis spectroscopy. To the cuvette, 1 mg of desolvated compound-1 was added, and after several time intervals we recorded the corresponding absorbance spectra of the supernatant solution. In a similar way, we carried out UV-Vis spectroscopy of MnO4− and ReO4− ions. But in this case, we took an aqueous solution of 0.5 mM for each of the MnO4− and ReO4− ions. Furthermore, from this time dependent study we calculated the % removal and decrease in the concentration of the oxo-anions vs. time using the following equation,61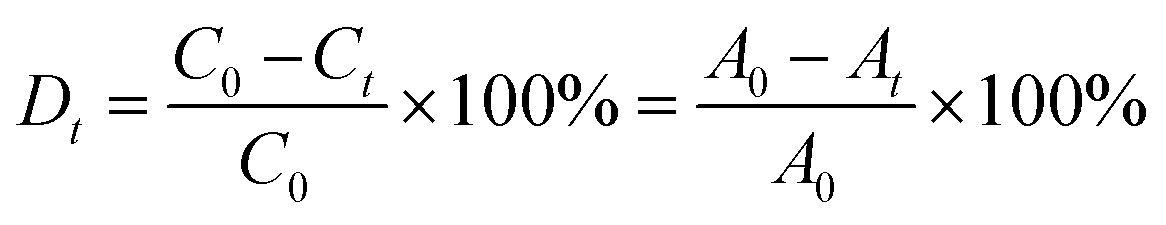
where Dt is the exchange capacity, C0 and A0 are the initial concentration and absorbance of the oxo-anion solution respectively, and Ct and At, are the concentration and absorbance of the oxo-anion solution at specific times respectively.
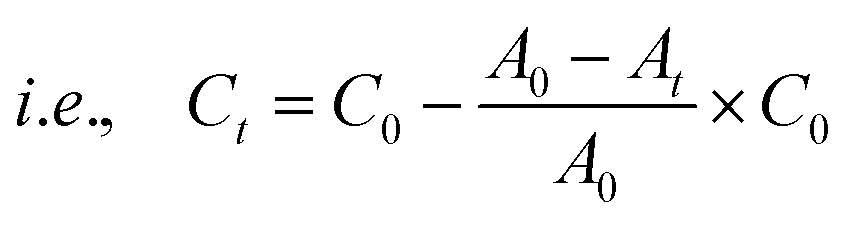
Furthermore, kinetics data for CrO42− and ReO4− ions were fitted in a pseudo second-order model with the help of the following equation,29
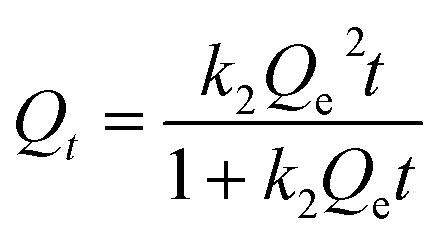
Study of oxo-anion trapping in the presence of competing anions
In this study, we took Cl−, Br−, NO3− and SO42− ions as the competing anions, which are omnipresent in common water sources and waste waters. We took an equimolar (2.5 mM; 1:1) aqueous solution of targeted oxo-anions and the competing anion. To the mixture, 2 mg of desolvated compound-1 was added and stirred for 18 hours. After 18 hours, the solution was filtered off to separate compound-1, and the solution was characterised with UV-Vis spectroscopy. The obtained solution was diluted 10 times to measure the UV-Vis spectroscopy, and furthermore the capture efficiency of compound-1 in the presence of other anions was studied by comparison with a blank (Blank: only 2.5 mM of the oxo-anion was taken instead of a mixture). Along with UV-Vis spectroscopy, this study was also affirmed by ICP-AES analyses.
Calculation of capacity
5 mg of desolvated compound-1 was kept with 2.5 mL of the 5 mM oxo-anion solution for 24 hours under stirring conditions. After 1 day, compound-1 was filtered out and the filtrate was used for further characterisation. UV-Vis measurements were carried out by diluting the solution. From the initial and final absorbance values of the oxo-anion solutions we calculated the storage capacity of compound-1 in 1 day using the following equation,61where, Qt, C0, Ct, V and m are the capacity of the adsorbent, the initial concentration of the oxo-anion solution, the concentration of the oxo-anion solution at specific times, the volume of the solution and the mass used for the adsorbent respectively. Furthermore, the capacity for compound-1 was rechecked with ICP-AES analyses.

Recyclability test of compound-1
Compound-1⊃oxo-anion (10 mg) was regenerated with a 3 M HCl solution (2 mL) by keeping it for ∼20 hours.35 Reusability of the regenerated material was checked with 5 mL of the 1 mM oxo-anion (CrO42− and ReO4−) solutions. After ∼20 hours, the concentrations of the oxo-anion solutions were measured by UV-Vis spectroscopy. These studies were repeated for up to four cycles for each of the cases (CrO42− and ReO4− ions). Furthermore, a similar method was employed for the column where 3 M HCl was passed through the column to regenerate the material. Then, the CrO42− ion solution was passed through the column to check the performance.Results and discussion
Compound-1 was synthesized from precursor 1 and 2 via Zincke’s reaction and was found to be insoluble in various solvents, which indicated the formation of an extended network (Scheme S1†). Coskun & co-workers recently reported the structure of compound-1, synthesized via the trimerisation of –CN groups in an ionothermal method.70 Very recently, Wen & co-workers reported the compound via Zincke’s reaction with the same precursors, but different reaction conditions resulted in a different morphology of the material and this was further employed for the electrosynthesis of H2O2.76 Furthermore, to remove occluded solvent molecules, compound-1 was kept in CHCl3–THF (1:1) for 4 days and then desolvated under vacuum at ∼100 °C. The desolvated phase was characterized with Fourier transform infra-red (FT-IR) spectroscopy, thermo-gravimetric analysis (TGA), solid state 13C-NMR, gas adsorption, field emission scanning electron microscopy (FE-SEM) and EDX analysis. An almost negligible change in weight up to ∼250 °C in the TGA profile revealed the desolvation of compound-1 (Fig. S1†). Replacement of the 2,4-dinitroaniline via Zincke’s reaction from precursor-2 was confirmed by FT-IR, as the peak at ∼1550 cm−1 (for –NO2 groups) disappeared in compound-1 (Fig. S2†). The amount of N2 uptake (at 77 K) was found to be 34.2 mL g−1, and such a low uptake corroborated the presence of Cl− ions in compound-1 (Fig. S3†). Furthermore, CO2 adsorption at 195 K showed strong hysteresis in the desorption profile due to interactions between the Cl− ions and the CO2 molecules (Fig. S4†). A solid state 13C-NMR study supported the presence of the triazine core along with the viologen moiety in compound-1 (Fig. S5†). The presence of Cl− ions was verified by EDX analysis (Fig. S6–S8†). Furthermore, to check the chemical stability, we kept compound-1 (25 mg) in 2 M HCl and 2 M KOH solutions for 7 days. After 1 week we found a negligible change in the weight for each, and further characterized these with FT-IR, TGA, FE-SEM and EDX analyses. No change in the FT-IR peak positions affirmed there were no changes in the functional groups (Fig. S9†). The morphologies of acid–base treated phases of compound-1 were also found to be unaltered in an FE-SEM study (Fig. S10†). The decomposition temperatures for each of the compounds were also found in a similar position to that of compound-1, which indicated that the networks remained similar (Fig. S13†).
Enthused by such physiochemical stability and the presence of free Cl− ions inside compound-1, we sought to check an anion exchange study in a water medium. Due to the ionic nature of compound-1, it was well dispersed in water which can be advantageous for anion exchange studies. Furthermore, the yellow color of the CrO42− solution turned to colorless with time when compound-1 was added to it. Encouraged by this observation, we thought to perform the CrO42− ion capture study from water with compound-1 and monitored it with UV-Visible spectroscopy. In situ titration was carried out with 0.25 mM CrO42− ions (2 mL) in water with 1 mg of compound-1 by monitoring the absorption peak at 372 nm (λmax for CrO42−) in the UV-Vis spectrum (Fig. 2a). The UV-Vis spectra of the supernatant at different time intervals revealed a continuous decrease of the peak at 372 nm due to the removal of CrO42− ions. Almost 77% removal of the CrO42− ions was observed by compound-1 in only 5 min, and reached ∼98.25% with saturation in 30 min with the decolorization of the solution (Fig. S14 and S15†). The % removal vs. time plot showed the rapid capture of CrO42− ions with compound-1 (Fig. 2d). Furthermore, the capacity of compound-1 for CrO42− ions was calculated (133 mg g−1) from the UV-Vis study and further verified with ICP-AES analysis (Fig. S16†). Notably, the capacity of compound-1 for CrO42− can be counted as one of the highest values in the area of porous materials (Table S1†). The peak at 894 cm−1 in the FT-IR spectra revealed the presence of CrO42− in compound-1⊃CrO42− (Fig. S17†). Also, the appearance of Cr in the EDX analysis affirmed the capture of CrO42− in compound-1, whereas the FE-SEM study showed no change in the morphology (Fig. S18–S20†).
 | ||
| Fig. 2 UV-Vis spectroscopy in the presence of compound-1 at different time intervals for the water solution of (a) CrO42− ions, (b) MnO4− ions and (c) ReO4− ions; removal (in %) of (d) CrO42− ions, (e) MnO4− ions and (f) ReO4− ions with compound-1 at different time intervals; the bar diagrams represent the removal efficiency of compound-1 in the presence of anions like Cl−, Br−, NO3− and SO42− for (g) CrO42− ions, (h) MnO4− ions and (i) ReO4− ions. | ||
Apart from CrO42−, TcO4− ions also count as hazardous water pollutants due to the radioactive nature of 99Tc. As handling such radioactive species is not convenient in a common laboratory, we took MnO4− and ReO4− as model ions which are non-radioactive surrogates for TcO4−. Furthermore, MnO4− and ReO4− ions are close chemical analogues of TcO4− ions (Mn, Tc and Re belong to the same group in the periodic table) and also the size of TcO4− ions is in between those of the MnO4− and ReO4− ions, which will help to mimic a real time scenario. Detailed studies of the removal of MnO4− and ReO4− ions from water were performed similarly to the CrO42− ion capture. Here, 2 mL of the 0.5 mM solutions of each of the oxo-anions were used to perform the study with 1 mg of compound-1. For the MnO4− ions, the capture study was monitored at 525 nm (λmax) while for the ReO4− ions it was monitored at 208 nm (λmax) (Fig. 2b and c). For the MnO4− ions, a rapid decrease in the absorption spectra was observed and within 5 minutes the solution was decolorized from purple (Fig. S21†). As the size of ReO4− ions is much bigger, relatively slow kinetics were observed as compared to both the MnO4− and the CrO42− ions. The presence of MnO4− and ReO4− ions in compound-1⊃MnO4− and compound-1⊃ReO4− was affirmed by FT-IR and EDX analyses. The peaks at ∼902 cm−1 and ∼918 cm−1 were observed in the FT-IR spectra for MnO4− and ReO4− ions respectively (Fig. S22 and S23†). The EDX analysis revealed the presence of Mn and Re corresponding to their oxo-anions; whereas the absence of Cl− also confirmed the anion exchange phenomenon (Fig. S24–S27†). FESEM revealed the morphologies of compound-1⊃MnO4− and compound-1⊃ReO4− remained similar to that of compound-1 (Fig. S28 and S29†). Furthermore, a time dependent decrease in the concentration as well as the % removal of oxo-anions from water were calculated from UV-Vis profiles (Fig. 2e, f, S30 and S31†). Almost 99.9% of MnO4− ions were removed within 5 min, while for bulky ReO4− ions >80% removal was achieved with 1 mg of compound-1 within only 60 minutes (Fig. 2e and f). Enthused by this, we checked the capacity of compound-1 for each of the oxo-anions from UV-Vis studies (Fig. S32 and S33†) and further verified this from the ICP-AES analyses. Notably, the high capacities of each of the oxo-anions (MnO4−: 297.3 mg g−1 ReO4−: 517 mg g−1) were again some of the highest values in the area of porous materials (Table S2†). Furthermore, it was found that removal of both the CrO42− and ReO4− ions with compound-1 follows the pseudo second order kinetics (Fig. S37 and S38†).
By virtue of such a high capacity, we thought to explore the oxo-anion capture by mimicking real time situations. Waste water contains other competing anions (Cl−, NO3−, SO42−etc.) along with the targeted oxo-anions. In this regard, we performed binary mixture studies for all of the oxo-anions with different competing anions (Cl−, Br−, NO3− and SO42−). A considerable amount of capture of the respective oxo-anions was observed even in the presence of competing anions (Fig. 2g–i). Removal of the MnO4− ions remains undisturbed even in the presence of other anions, while ≥80% ReO4− was captured by compound-1 from the mixture (Fig. 2h and i). In the case of the CrO42− ion, efficient removal was observed in the presence of Br−, NO3− and SO42− ions (Fig. 2g). Only the mixture of Cl−:CrO42− ions exhibited a relatively lower efficiency (∼70%) which may be due to the presence of Cl− ions in both compound-1 and in solution. This binary mixture study showed the efficiency of compound-1 in capturing oxo-anions even in the presence of competing anions. In addition, since the pH of waste water can vary in different ranges, we performed removal of CrO42− ions in both acidic and alkaline media (Fig. S39†). While TcO4− ions are mostly found in alkaline media, we demonstrated the removal of ReO4− ions in alkaline media (Fig. S40†). Again, the reusability of compound-1 was tested with a 3 M HCl solution. In the case of both CrO42− and ReO4− ions, compound-1 was found to maintain its efficiency without any significant changes for up to four cycles (Fig. S41 and S42†). This study showed that compound-1 was also stable even after the capture of the respective oxo-anions and regeneration with 3 M HCl, and was further useful for the capture of those anions over a period of cycles.
Moreover, a chromatographic column was prepared embedded with compound-1 and employed for the removal of oxo-anions from water (Fig. S43†). A 2.5 mM stock solution of each of the oxo-anions was passed through the column (see the video in the ESI†). For MnO4− and CrO42− ions, a distinct color change of the eluent was noticed, as a consequence of being captured by compound-1 (Fig. 3a and c). Furthermore, UV-Vis studies revealed the absence of the oxo-anions in the eluted water, which ascertained that the compound-1 based column can be useful for the removal of toxic oxo-anions (Fig. 3b, d and S44†). To validate whether the process is anion exchange or surface adsorption in the column, water was passed through the compound-1⊃oxo-anion loaded column (see the video in the ESI†). A colorless eluent affirmed the anion exchange process, discarding any option of surface adsorption. In addition, as the compound-1 can be regenerated after treatment with 3 M HCl solution, we tried to reuse the column in a similar way. When 3 M HCl was passed through the column, a yellow colored solution came out corresponding to the Cr(VI) oxo-anions (Fig. S45†). The column was used further for a second cycle of the CrO42− ion capture study (Fig. S46†).
 | ||
| Fig. 3 (a) Image and (b) UV-Vis spectra of the 2.5 mM CrO42− solution before and after passing through the compound-1 loaded packed column; (c) image and (d) UV-Vis spectra of the 2.5 mM MnO4− ion solution before and after passing through the compound-1 loaded packed column. | ||
Conclusions
In conclusion, the rapid capture of oxo-anions from water was demonstrated with an ionic viologen based covalently linked organic network (compound-1) owing to its chemical stability and the presence of free exchangeable Cl− ions inside the network. Rapid decolorization of the yellow colored CrO42− solution was observed due to the removal of CrO42− ions from water in the presence of compound-1. Furthermore, to study the capture of the radioactive 99Tc based oxo-anion (TcO4−), the close analogues MnO4− and ReO4− were used as a surrogate of TcO4−. The capacities for each of the oxo-anions were found to be high and comparable with some of the well studied materials known for their highest capacities. As waste water contains competing anions like Cl−, NO3−, SO42−etc. along with toxic oxo-anions, we demonstrated efficient capture of the oxo-anions with compound-1 even in the concurrent presence of competing anions. These results demonstrate a unique class of features where the dual capture of toxic oxo-anions has been well performed by a porous organic material which is not common in this area. We believe that this result can open up a new avenue for the capture of toxic anions based on ionic porous covalently linked organic materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. S. and P. C. acknowledge UGC (India) and INSPIRE (India) for their fellowships respectively. S. D. and A. D. are thankful to IISER Pune for fellowships. S. K. G. acknowledges funding support from the DST-SERB project (EMR/2016/000410), IISER Pune and DST Nanomission Thematic Unit, Govt. of India.Notes and references
- L. H. Keith and W. A. Teillard, Environ. Sci. Technol., 1979, 13, 416–423 CrossRef.
- Toxicological Profile for Chromium, Public Health Service Agency for Toxic Substances and Diseases Registry, U.S. Department of Health and Human Services, Atlanta, GA, 2012 Search PubMed.
- R. Vonburg and D. Liu, J. Appl. Toxicol., 1993, 13, 225–230 CrossRef.
- H. Fei, D. L. Rogow and S. R. J. Oliver, J. Am. Chem. Soc., 2010, 132, 7202–7209 CrossRef PubMed.
- R. Cao, B. D. McCarthy and S. J. Lippard, Inorg. Chem., 2011, 50, 9499–9507 CrossRef PubMed.
- L. Khezami and R. Capart, J. Hazard. Mater., 2005, 123, 223–231 CrossRef PubMed.
- M. B. Luo, Y. Y. Xiong, H. Q. Wu, X. F. Feng, J. Q. Li and F. Luo, Angew. Chem., Int. Ed., 2017, 56, 16376–16379 CrossRef PubMed.
- W. Liu, Y. Wang, Z. Bai, Y. Li, Y. Wang, L. Chen, L. Xu, J. Diwu, Z. Chai and S. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 16448–16457 CrossRef PubMed.
- S. R. J. Oliver, Chem. Soc. Rev., 2009, 38, 1868–1881 RSC.
- C. Pellerin and S. M. Booker, Environ. Health Perspect., 2000, 108, A402–A407 CrossRef PubMed.
- H. Yoshitake, T. Yokoi and T. Tatsumi, Chem. Mater., 2002, 14, 4603–4610 CrossRef.
- The World’s Worst Pollution Problems 2016: The Toxics Beneath Our Feet, Pure Earth and Green Cross Switzerland, 2016, http://www.worstpolluted.org/docs/WorldsWorst2016.pdf Search PubMed.
- S. Wang, P. Yu, B. A. Purse, M. J. Orta, J. Diwu, W. H. Casey, B. L. Phillips, E. V. Alekseev, W. Depmeier, D. T. Hobbs and T. E. Albrecht-Schmitt, Adv. Funct. Mater., 2012, 22, 2241–2250 CrossRef.
- T. H. Boyer and P. C. Singer, Environ. Sci. Technol., 2008, 42, 608–613 CrossRef PubMed.
- B. Gammelgaard, Y. P. Liao and O. Jons, Anal. Chim. Acta, 1997, 354, 107–113 CrossRef.
- B. Wen, X. Q. Shan and J. Lian, Talanta, 2002, 56, 681–687 CrossRef PubMed.
- A. Syty, R. G. Christensen and T. C. Rains, J. Anal. At. Spectrom., 1988, 3, 193–197 RSC.
- D. Sheng, L. Zhu, C. Xu, C. Xiao, Y. Wang, Y. Wang, L. Chen, J. Diwu, J. Chen, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Environ. Sci. Technol., 2017, 51, 3471–3479 CrossRef PubMed.
- D. Banerjee, D. Kim, M. J. Schweiger, A. A. Kruger and P. K. Thallapally, Chem. Soc. Rev., 2016, 45, 2724–2739 RSC.
- A. V. Desai, B. Manna, A. Karmakar, A. Sahu and S. K. Ghosh, Angew. Chem., Int. Ed., 2016, 55, 7811–7815 CrossRef PubMed.
- L. Zhu, D. Sheng, C. Xu, X. Dai, M. A. Silver, J. Li, P. Li, Y. Wang, Y. Wang, L. Chen, C. Xiao, J. Chen, R. Zhou, C. Zhang, O. K. Farha, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, J. Am. Chem. Soc., 2017, 139, 14873–14876 CrossRef PubMed.
- P.-F. Shi, B. Zhao, G. Xiong, Y.-L. Hou and P. Cheng, Chem. Commun., 2012, 48, 8231–8233 RSC.
- A. J. Howarth, Y. Liu, J. T. Hupp and O. K. Farha, CrystEngComm, 2015, 17, 7245–7253 RSC.
- H. Fei, M. R. Bresler and S. R. J. Oliver, J. Am. Chem. Soc., 2011, 133, 11110–11113 CrossRef PubMed.
- H. Fei, C. S. Han, J. C. Robins and S. R. J. Oliver, Chem. Mater., 2013, 25, 647–652 CrossRef.
- D. Banerjee, W. Xu, Z. Nie, L. E. V. Johnson, C. Coghlan, M. L. Sushko, D. Kim, M. J. Schweiger, A. A. Kruger, C. J. Doonan and P. K. Thallapally, Inorg. Chem., 2016, 55, 8241–8243 CrossRef PubMed.
- X. Li, H. Xu, F. Kong and R. Wang, Angew. Chem., Int. Ed., 2013, 52, 13769–13773 CrossRef PubMed.
- H.-R. Fu, Z.-X. Xu and J. Zhang, Chem. Mater., 2015, 27, 205–210 CrossRef.
- Q. Zhang, J. Yu, J. Cai, L. Zhang, Y. Cui, Y. Yang, B. Chen and G. Qian, Chem. Commun., 2015, 51, 14732–14734 RSC.
- H. Fei and S. R. J. Oliver, Angew. Chem., Int. Ed., 2011, 50, 9066–9070 CrossRef PubMed.
- J. J. Neeway, R. M. Asmussen, A. R. Lawter, M. E. Bowden, W. W. Lukens, D. Sarma, B. J. Riley, M. G. Kanatzidis and N. P. Qafoku, Chem. Mater., 2016, 28, 3976–3983 CrossRef.
- S. Rapti, A. Pournara, D. Sarma, I. T. Papadas, G. S. Armatas, A. C. Tsipis, T. Lazarides, M. G. Kanatzidis and M. J. Manos, Chem. Sci., 2016, 7, 2427–2436 RSC.
- C.-P. Li, B.-L. Liu, L. Wang, Y. Liu, J.-Y. Tian, C.-S. Liu and M. Du, ACS Appl. Mater. Interfaces, 2017, 9, 7202–7208 CrossRef PubMed.
- H. Yang and H. Fei, Chem. Commun., 2017, 53, 7064–7067 RSC.
- S. Rapti, D. Sarma, S. A. Diamantis, E. Skliri, G. S. Armatas, A. C. Tsipis, Y. S. Hassan, M. Alkordi, C. D. Malliakas, M. G. Kanatzidis, T. Lazarides, J. C. Plakatouras and M. J. Manos, J. Mater. Chem. A, 2017, 5, 14707–14719 RSC.
- L. Zhu, L. Zhang, J. Li, D. Zhang, L. Chen, D. Sheng, S. Yang, C. Xiao, J. Wang, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Environ. Sci. Technol., 2017, 51, 8606–8615 CrossRef PubMed.
- X. Zhao, X. Bu, T. Wu, S.-T. Zheng, L. Wang and P. Feng, Nat. Commun., 2013 DOI:10.1038/ncomms3344.
- A. V. Desai, A. Roy, P. Samanta, B. Manna and S. K. Ghosh, iScience, 2018, 3, 21–30 CrossRef.
- Y. Li, Z. Yang, Y. Wang, Z. Bai, T. Zheng, X. Dai, S. Liu, D. Gui, W. Liu, M. Chen, L. Chen, J. Diwu, L. Zhu, R. Zhou, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Nat. Commun., 2017 DOI:10.1038/s41467-017-01208-w.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- A. G. Slater and A. I. Cooper, Science, 2017, 348 DOI:10.1126/science.aaa8075.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O’Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef PubMed.
- A. Karmakar, A. V. Desai and S. K. Ghosh, Coord. Chem. Rev., 2016, 307, 313–341 CrossRef.
- S. Furukawa, J. Reboul, S. Diring, K. Sumida and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5700–5734 RSC.
- A. J. Howarth, M. J. Katz, T. C. Wang, A. E. Platero-Prats, K. W. Chapman, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2015, 137, 7488–7494 CrossRef PubMed.
- T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- W. Lu, D. Yuan, J. Sculley, D. Zhao, R. Krishna and H. –C. Zhou, J. Am. Chem. Soc., 2011, 133, 18126–18129 CrossRef PubMed.
- S. Keskin, T. M. van Heest and D. S. Sholl, ChemSusChem, 2010, 3, 879–891 CrossRef PubMed.
- W. Lu, D. Yuan, D. Zhao, C. I. Schilling, O. Plietzsch, T. Muller, S. Bräse, J. Guenther, J. Blümel, R. Krishna, Z. Li and H.-C. Zhou, Chem. Mater., 2010, 22, 5964–5972 CrossRef.
- M. G. Rabbani and H. M. El-Kaderi, Chem. Mater., 2011, 23, 1650–1653 CrossRef.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef PubMed.
- P. J. Waller, F. Gándara and O. M. Yaghi, Acc. Chem. Res., 2015, 48, 3053–3063 CrossRef PubMed.
- L. Zou, Y. Sun, S. Che, X. Yang, X. Wang, M. Bosch, Q. Wang, H. Li, M. Smith, S. Yuan, Z. Perry and H.-C. Zhou, Adv. Mater., 2017, 29, 1700229 CrossRef PubMed.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- U. Diaza and A. Corma, Coord. Chem. Rev., 2016, 311, 85–124 CrossRef.
- P. Samanta, A. V. Desai, B. Anothumakkool, M. M. Shirolkar, A. Karmakar, S. Kurungot and S. K. Ghosh, J. Mater. Chem. A, 2017, 5, 13659–13664 RSC.
- R. P. Bisbey and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 533–543 CrossRef PubMed.
- B. Aguila, Q. Sun, J. A. Perman, L. D. Earl, C. W. Abney, R. Elzein, R. Schlaf and S. Ma, Adv. Mater., 2017, 29 DOI:10.1002/adma.201700665.
- P. Samanta, P. Chandra, A. V. Desai and S. K. Ghosh, Mater. Chem. Front., 2017, 1, 1384–1388 RSC.
- Y. Su, Y. Wang, X. Li, X. Li and R. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 18904–18911 CrossRef PubMed.
- D. Banerjee, S. K. Elsaidi, B. Aguila, B. Li, D. Kim, M. J. Schweiger, A. A. Kruger, C. J. Doonan, S. Ma and P. K. Thallapally, Chem.–Eur. J., 2016, 22, 17581–17584 CrossRef PubMed.
- B. Li, Y. Zhang, D. Ma, Z. Xing, T. Ma, Z. Shi, X. Ji and S. Ma, Chem. Sci., 2016, 7, 2138–2144 RSC.
- N. Huang, P. Wang, M. A. Addicoat, T. Heine and D. Jiang, Angew. Chem., Int. Ed., 2017, 56, 4982–4986 CrossRef PubMed.
- H. Ma, B. Liu, B. Li, L. Zhang, Y.-G. Li, H.-Q. Tan, H.-Y. Zang and G. Zhu, J. Am. Chem. Soc., 2016, 138, 5897–5903 CrossRef PubMed.
- S. Fischer, A. Schimanowitz, R. Dawson, I. Senkovska, S. Kaskel and A. Thomas, J. Mater. Chem. A, 2014, 2, 11825–11829 RSC.
- S. Mitra, S. Kandambeth, B. P. Biswal, M. A. Khayum, C. K. Choudhury, M. Mehta, G. Kaur, S. Banerjee, A. Prabhune, S. Verma, S. Roy, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2016, 138, 2823–2828 CrossRef PubMed.
- Z. Li, H. Li, X. Guan, J. Tang, Y. Yusran, Z. Li, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2017, 139, 17771–17774 CrossRef PubMed.
- S.-B. Yu, H. Lyu, J. Tian, H. Wang, D.-W. Zhang, Y. Liu and Z.-T. Li, Polym. Chem., 2016, 7, 3392–3397 RSC.
- O. Buyukcakir, S. H. Je, S. N. Talapaneni, D. Kim and A. Coskun, ACS Appl. Mater. Interfaces, 2017, 9, 7209–7216 CrossRef PubMed.
- C. Hua, B. Chan, A. Rawal, F. Tuna, D. Collison, J. M. Hook and D. M. D’Alessandro, J. Mater. Chem. C, 2016, 4, 2535–2544 RSC.
- O. Buyukcakir, S. H. Je, D. S. Choi, S. N. Talapaneni, Y. Seo, Y. Jung, K. Polychronopoulou and A. Coskun, Chem. Commun., 2016, 52, 934–937 RSC.
- G. Chen, Y. Zhou, X. Wang, J. Li, S. Xue, Y. Liu, Q. Wang and J. Wang, Sci. Rep., 2015, 5, 11236-1–11236-14 Search PubMed.
- G. Das, T. Skorjanc, S. K. Sharma, F. Gándara, M. Lusi, D. S. S. Rao, S. Vimala, S. K. Prasad, J. Raya, D. S. Han, R. Jagannathan, J.-C. Olsen and A. Trabolsi, J. Am. Chem. Soc., 2017, 139, 9558–9565 CrossRef PubMed.
- G. Das, T. Prakasam, S. Nuryyeva, D. S. Han, A. Abdel-Wahab, J.-C. Olsen, K. Polychronopoulou, C. Platas-Iglesias, F. Ravaux, M. Jouiad and A. Trabolsi, J. Mater. Chem. A, 2016, 4, 15361–15369 RSC.
- L.-Z. Peng, P. Liu, Q.-Q. Cheng, W.-J. Hu, Y. A. Liu, J.-S. Li, B. Jiang, X.-S. Jia, H. Yang and K. Wen, Chem. Commun., 2018, 54, 4433–4436 RSC.
- R. Gomes, P. Bhanja and A. Bhaumik, Chem. Commun., 2015, 51, 10050–10053 RSC.
- S. Asaftei, D. Huskens and D. Schols, J. Med. Chem., 2012, 55, 10405–10413 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02456a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |