
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bright insights into palladium-triggered local chemotherapy†
Thomas L.
Bray
a,
Mark
Salji
 bc,
Alessandro
Brombin
ad,
Ana M.
Pérez-López
a,
Belén
Rubio-Ruiz
a,
Laura C. A.
Galbraith
bc,
E. Elizabeth
Patton
ad,
Hing Y.
Leung
*bc and
Asier
Unciti-Broceta
*a
bc,
Alessandro
Brombin
ad,
Ana M.
Pérez-López
a,
Belén
Rubio-Ruiz
a,
Laura C. A.
Galbraith
bc,
E. Elizabeth
Patton
ad,
Hing Y.
Leung
*bc and
Asier
Unciti-Broceta
*a
aCancer Research UK Edinburgh Centre, Institute of Genetics and Molecular Medicine, University of Edinburgh, Crewe Road South, Edinburgh EH4 2XR, UK. E-mail: Asier.Unciti-Broceta@igmm.ed.ac.uk
bInstitute of Cancer Sciences, University of Glasgow, Bearsden, Glasgow G61 1QH, UK. E-mail: Hing.Leung@glasgow.ac.uk
cCRUK Beatson Institute, Bearsden, Glasgow G61 1BD, UK
dMRC Human Genetics Unit, Institute of Genetics & Molecular Medicine, University of Edinburgh, Crewe Road South, Edinburgh EH4 2XR, UK
First published on 17th July 2018
Abstract
The incorporation of transition metal catalysts to the bioorthogonal toolbox has opened the possibility of producing supra-stoichiometric amounts of xenobiotics in living systems in a non-enzymatic fashion. For medical use, such metals could be embedded in implantable devices (i.e. heterogeneous catalyst) to “synthesize” drugs in desired locations (e.g. in a tumour) with high specificity and for extended periods of time, overcoming the useful life limitations of current local therapy modalities directed to specific organ sites (e.g. brachytherapy, controlled release systems). To translate this approach into a bona fide therapeutic option, it is essential to develop clinically-accessible implantation procedures and to understand and validate the activation process in relevant preclinical models. Herein we report the development of a novel Pd-activatable precursor of the red-fluorescent drug doxorubicin and Pd devices of optimized size and activity. Screening in state-of-the-art cancer models provided fundamental insights into the insertion protocols, safety and stability of the devices and into the prodrug distribution profile before and after activation.
Introduction
Advances in cancer diagnosis have led to major improvements in the early detection of clinically-localized prostate cancers.1 Because of the adverse effects of prostatectomy and external beam radiotherapy (impotence, incontinence, etc.), in most patients this low-to-moderate risk form of prostate cancer has been traditionally managed through active surveillance programs, which has resulted in an increment in the risk of dying from prostate malignancies.2,3 Focal therapies were introduced to facilitate localized tumour treatment with preservation of healthy prostate tissue, thus reducing the morbidity associated with whole-gland treatments without jeopardizing cancer control.1,3 Although current technologies mediating local treatment of disease (e.g. brachytherapy, external-beam focal radiotherapy, drug-eluting devices) can successfully improve a patient's quality of life, they have important limitations4 that either restrict their widespread clinical use (high cost of brachytherapy and external-beam focal radiotherapy) or the duration of their therapeutic effect (radioisotope's short half-life and limited drug cargo capacity of brachytherapy and controlled-release implants, respectively).Our capacity to visualize and modulate physiological and pathological processes in the biological milieu has expanded enormously in the last 15 years through the development of a rich diversity of bioorthogonal tools and processes.5–9 Among them, the use of abiotic transition metals (ruthenium,10–14 palladium,15–21 copper,22–24 gold25–27) has emerged to facilitate the catalytic modification or manufacture of biomolecules and xenobiotics in living systems, contributing to the advent of a new prodrug modality that is not activated by a metabolic event but through a bioorthogonal bond cleavage reaction.9 Exploration of this novel prodrug paradigm in combination with heterogeneous Pd catalysis has stimulated the development of masking strategies that efficiently suppress the bioactivity of different classes of therapeutic agents while enabling selective drug activation – via Pd-mediated N- or O-dealkylation – both in vitro and in vivo.20,28–31 It is proposed that, by surgical implantation of Pd-containing inserts in a tumour followed by systemic administration of Pd-activatable therapeutic precursors, bioorthogonal drug release could take place exclusively in the disease site and minimise adverse effects in distant organs and tissues. Since the triggering mechanism is a catalytic process, multiple doses of one or more prodrugs could be intratumourally activated by the same implant to extend and/or customize the therapeutic intervention, thereby facilitating an adaptive form of local chemotherapy that can respond to the severity and progression of the disease. Although studies in mammal models supporting the effectiveness of such an approach to treat tumours locally have not yet been reported, recent studies with Pd-based nanostructures have shown their capacity to generate tumour-controlling levels of drugs in xenografts in mice.32,33 However, the development of cancer-targeting nanotechnologies remain a significant challenge by itself.34
From a translational perspective, the use of activating devices that can be surgically implanted in or near the tumour using well-established clinical procedures3,35 would ensure that the release of cytotoxic agents occurs exclusively at the disease site. Essential questions such as appropriate implantation protocols, stability and safety of the catalyst in vivo and optimal biodistribution of the prodrug before and after activation need to be first addressed to translate this novel therapeutic modality into a medically-viable alternative to brachytherapy and drug eluting devices. In our path towards this goal, herein we report the development of Pd devices of optimal size for ultrasound-guided intratumoural insertion and Pd-activatable prodrugs of doxorubicin: a strongly fluorescent cytotoxic drug that allowed us to monitor biodistribution in prostate cancer explants. Screening in state-of-the-art cancer models provided fundamental insights into the safety and catalytic properties of the activating device, and into the prodrug activation process.
Results and discussion
Design and synthesis of bioorthogonal prodrugs of doxorubicin
Doxorubicin, 1, is an anthracycline anticancer antibiotic used alone or in combination in the treatment of a wide range of hematological and solid malignancies.36 After entering cells by diffusion, this potent cytotoxic drug engages with various cytoplasmic and nuclear targets to synergistically disrupt cellular function in cancer cells and induce apoptosis (Scheme 1).371 has a multitude of moderate to severe side effects that negatively impact on its therapeutic window, limiting its maximum tolerated dose and frequency of administration (typically administered as a single intravenous infusion in days 1 and 2 of 21 day cycles). The most serious adverse effect of 1 is cardiotoxicity, typically observed by an early onset of cardiomyopathy that may progress to myocardial infarction.38 As a result, much of the research pertaining to 1 and their anthracycline analogues focuses on suppressing the cardiotoxic effect of this class of cytotoxic drugs. A variety of prodrug strategies are currently under investigation to improve tumour targetability39–43 and have shown that blockade of the primary amino group of the daunosamine moiety of 1 significantly reduces the bioactivity of the resulting derivative. Based on this evidence, the masking of the NH2 group as a carbamate group was investigated to develop novel biochemically-stable, Pd-sensitive prodrugs of 1 (Scheme 1).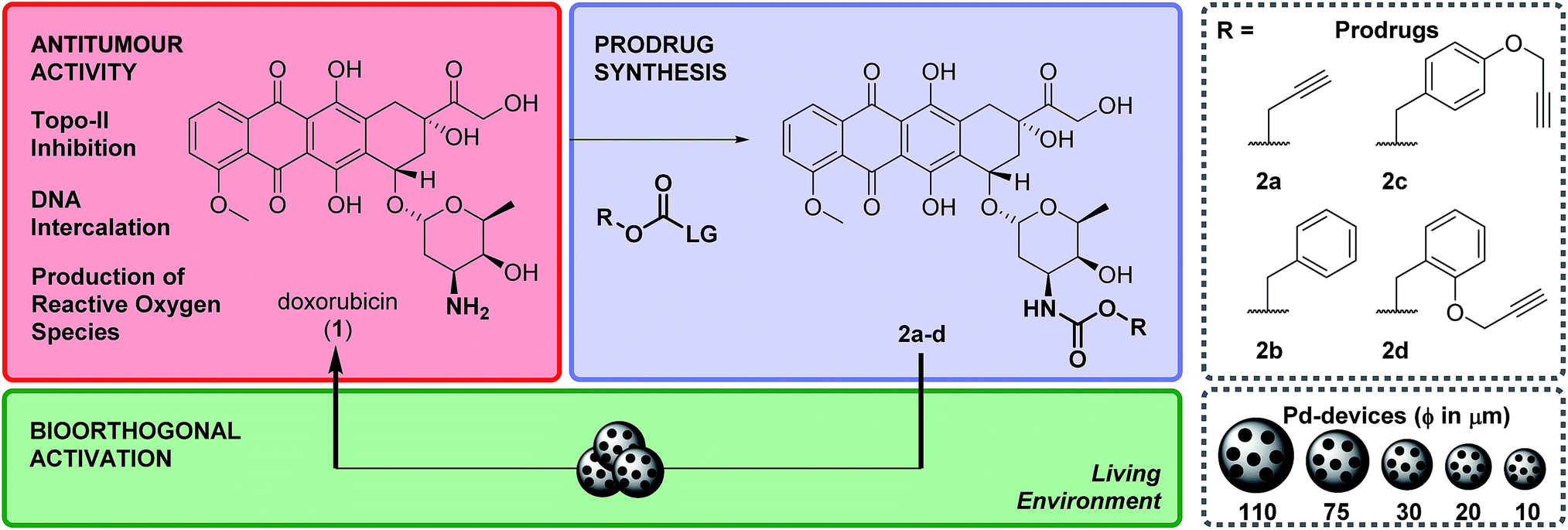 | ||
| Scheme 1 Overview of the strategy and research work. Prodrugs 2a–d were prepared by reacting 1 with their corresponding 4-nitrophenyl (LG) carbonate. Pd-devices of a range of sizes (from 110 to 10 μm in diameter) were developed to investigate prodrug activation in biocompatible conditions, in cell culture and in prostate cancer explants. | ||
Four carbamate derivatives of 1 were prepared as shown in Scheme 1 (see full experimental protocols in the ESI†). As the propargyloxycarbonyl (Poc) has been previously shown to be relatively resistant to metabolic activation and sensitive to Pd catalysis, derivative 2a was developed as a positive control, whereas the Cbz-protected analogue 2b (which is expected to be even more biochemically stable than Poc but unreactive to Pd catalysis under physiological conditions28) was used as a negative control. With the aim of increasing steric hindrance to further protect the carbamate bond from unspecific enzymatic cleavage, novel compounds containing a propargyloxybenzyl carbonyl (PBC) group were developed to enhance the metabolic stability of the derivatives whilst providing sensitivity to Pd catalysis. The para and ortho isomers 2c and 2d, which are designed to undergo 1,6- or 1,4-elimination upon O-depropargylation (ESI, Fig. S1†), were prepared to test which position provides superior protection and, consequently, a greater therapeutic window relative to the active drug (EC50 (2c, d)/EC50 (1)).
Cytotoxicity study: drug vs. prodrug
The efficacy of the deactivation strategy was evaluated by treating human prostate cancer DU145 and glioma U87 cells (models of human cancers that are clinically treated with local therapies)3,35 for 5 d with increasing concentrations of 1 and 2a–d (0.001 to 10 μM). EC50 values were calculated from the generated 10-point semilog dose–response curves (see ESI, Fig. S2†) and plotted in Table 1. All prodrugs showed a significant reduction in cytotoxicity relative to 1. A direct relationship between the reduction of bioactivity and increasing steric bulk around the carbamate group was consistently observed. Derivative 2a (containing the small Poc group) showed a relatively low difference in activity compared to 1 (30–40 fold). Notably, PBC-masked prodrugs 2c and 2d exhibited much greater reduction of cytotoxicity, with the ortho analog 2d displaying a difference in antiproliferative activity of more than two orders of magnitude relative to unmodified drug 1 (approx. 350- and 150-fold in DU145 and U87, respectively). These results indicate that the masking group of 2d provides superior protection to the carbamate bond against unspecific biological cleavage, even compared to the para analog 2c and without the need of further increment of its size.| Cell line | DU145 | U87 | ||
|---|---|---|---|---|
| Reagent | EC50 | EC50 (2)/EC50 (1) | EC50 | EC50 (2)/EC50 (1) |
| 1 | 0.02 | — | 0.08 | — |
| 2a | 0.83 | 41.5 | 2.49 | 31 |
| 2b | 2.69 | 134.5 | 3.13 | 39 |
| 2c | 5.54 | 277 | 5.27 | 66 |
| 2d | 7.13 | 356.5 | 11.8 | 147.5 |
Zebrafish cardiotoxicity study
As mentioned above, the induction of cardiotoxicity is the most severe side effect of 1. Given the greater therapeutic window afforded by 2d, an in vivo cardiotoxicity assay was designed to study its effect on the heart of developing zebrafish in comparison to 1. Dechorionated zebrafish embryos (1 dpf) were treated for 4 days with 1 or 2d at a range of concentrations (50–200 μM) and then imaged (brightfield and fluorescent microscopy). Embryos in E3 medium alone or with DMSO were used as negative controls. To determine the enlargement of the heart (=estimate of pericardial edema), the ratio between the cardiac area and the total body of the treated fish was measured and compared with the untreated controls. As shown in Fig. 1, treatment of 1 at 50 μM induced significant increment of the cardiac area, with 100% lethality observed in all populations treated at higher concentrations. In contrast, 2d did not induce lethality or cardiac edema even up to 200 μM; well above the theoretical blood concentration achieved with the maximum therapeutic dosage of clinical dosing regimens.36 Since spectrophotometry analysis of the optical properties of 1 and 2d showed similar red fluorescent properties (ESI, Fig. S3†), drug/prodrug distribution patterns were analyzed by fluorescent microscopy (Ex/Em = 520/560 nm). Image analysis showed that significant levels of 1 accumulated across the body of the zebrafish, including the heart area, while 2d did not (ESI, Fig. S4†). This observation is consistent with 2d's lack of capacity to engage with the in vivo molecular targets of 1.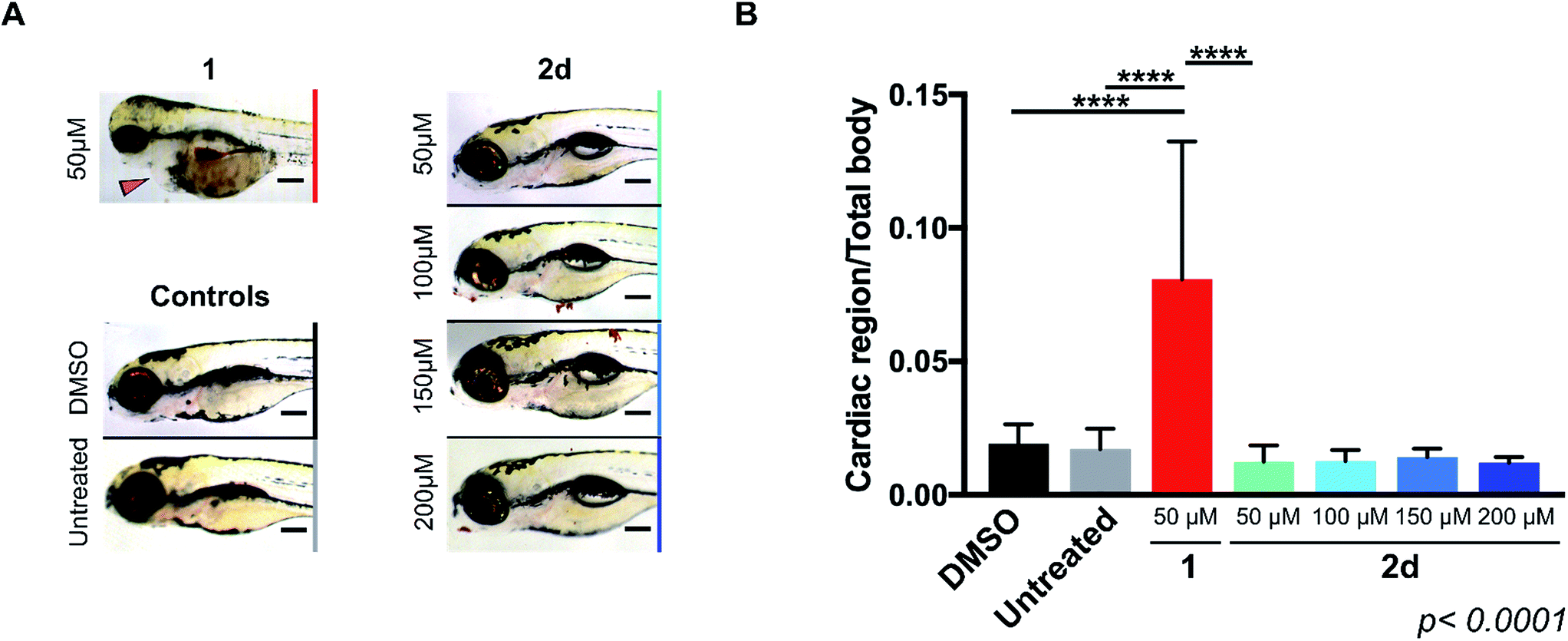 | ||
| Fig. 1 (A) Lateral view of 120 hpf zebrafish larvae after exposition to 50 μM of 1 or 50–200 μM of 2d for 4 d. Zebrafish treated with 0.1% DMSO and E3 medium only (untreated) were used as negative controls. Cardiac edema (only observed for fish treated with 1) is indicated with a red arrow. Additional toxicity in the eyes and the brain is also apparent. Scale bar = 20 μm. (B) Histogram displaying the ratio of cardiac area/total body area for zebrafish populations in 0.1% DMSO (black), E3 medium alone (untreated, grey), 1 at 50 μM (red) and 2d at 50–200 μM (light to dark blue with increasing concentrations). Zebrafish populations treated with >50 μM of 1 died during treatment and were excluded from analysis. Error bars: ±SD from n = 3 (15 fish per biological replicate); p < 0.0001, **** (ANOVA). | ||
Prodrug-into-drug conversion studies
Pd-devices were prepared from amino-functionalized TentaGel® HL resins of 110 μm in average diameter (Rapp Polymere GmbH) as previously described.20 Content in Pd metal was determined to be 4.47% w/w by inductively coupled plasma optical emission spectrometry (ICP-OES). The conversion of inactive 2a–d into cytotoxic 1 was first tested by performing a Pd-mediated activation assay in cancer cell culture at a single concentration, using cell viability measurements as a functional readout of the experiment. DU145 cells were treated with prodrugs 2a–d (0.3 μM) and Pd-devices (0.6 mg mL−1) and cell viability determined at day 5 using the PrestoBlue® reagent. Cells treated with only Pd-devices (0.6 mg mL−1) or 2a–d (0.3 μM) were used as negative controls and unmodified 1 (0.3 μM) as positive control.As expected, Cbz-protected 2b displayed no signs of toxicity either on its own or in the presence of the catalyst, which confirms its resistance to both metabolic and Pd-mediated activation (ESI, Fig. S5†). On the contrary, prodrugs 2a–2d elicited no harm on their own but significant antiproliferative effects in the presence of the catalytic devices, thus proving the selective in situ generation of 1 in response to the presence of Pd. However, the assay revealed a statistically-significant gap in cytotoxicity between 1 and the prodrug/Pd-devices combinations, particularly for prodrugs 2c and 2d. By simply mixing the prodrugs and the Pd-devices in a test tube, it was visibly noticed that the devices had a remarkable capacity to sequester 2d from the solution, an effect that was not seen for 1 (Fig. 2A). The polymeric component of these devices is made of TentaGel®, a copolymer consisting of a low crosslinked polystyrene (PS) matrix on which polyethylene glycol (PEG) is grafted.44 Based on the strong “trapping” effect observed for 2d, we reasoned that the lipophilic polystyrene core of the devices were likely responsible for capturing the prodrug, partially deterring its effective interaction with the catalyst and, in turn, resulting in a moderate reduction of the reaction rate and subsequent liberation of 1 into the cell culture.
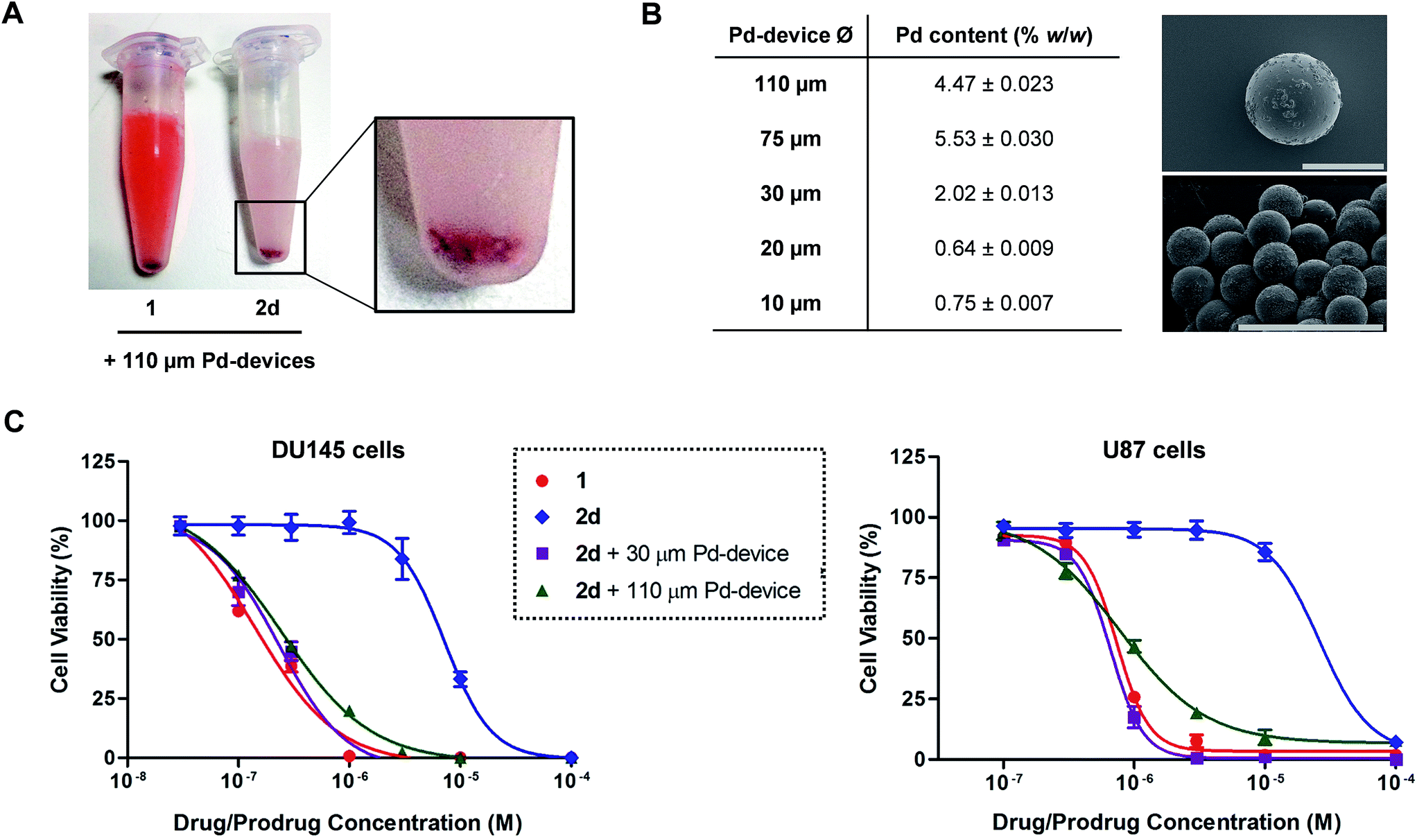 | ||
| Fig. 2 (A) Sequestration effect of prodrug 2d by Pd-devices. 100 μM of 1 or 2d and 1 mg mL−1 of 110 μm Pd-devices were mixed in PBS and stirred for 30 min. The mixtures were then frozen by immersing the tubes in liquid nitrogen and photographed. Equivalent results were observed for prodrugs 2a–c. (B) ICP-OES determination of Pd loading in Pd-devices ranging from 110 to 10 μm in diameter. SEM images of 110 μm Pd-devices (top; ×450 magnification) and 30 μm Pd-devices (bottom; ×800 magnification). Scale bar = 100 μm. (C) Study of the activation of 2d with 30 μm and 110 μm Pd-devices in DU145 (left) and U87 (right) cancer cell culture at a range of concentrations. Experiments: 1 (red), 2d (blue), and 2d + 0.6 mg mL−1 30 μm Pd-devices (purple), and 2d + 0.6 mg mL−1 110 μm Pd-devices (green). Semi-log dose–response curves were generated by measuring cell viability at day 5 using PrestoBlue® reagent. Error bars: ±SD from n = 3. | ||
Aiming to optimize the functionality of the devices, we prepared, characterized and tested a range of Pd-devices using polymer resins of reduced size and greater PEG/PS ratio (=increased hydrophilicity). Pd-functionalized devices with sizes of 10, 20, 30 and 75 μm were synthesized using commercially available TentaGel® resins (Rapp Polymere GmbH) following the same protocol used for the preparation of 110 μm Pd-devices.20 ICP-OES analysis demonstrated the presence of the metal in each of the devices, although lower Pd content was observed in the smaller ones (Fig. 2B). The catalytic capacity of the devices was tested by 24 h treatment with nonfluorescent bis-N,N′-(propargyloxycarbonyl)rhodamine 110 (3) in cell culture conditions (PBS + 10% serum, pH = 7.4, 37 °C). This assay is based on the fluorogenic release of rhodamine 110 (4) upon Pd-mediated O-depropargylation of 3. Fluorescence intensity was measured with a spectrophotometer. As shown in Fig. S5C (ESI),† the 30 μm Pd-devices displayed slightly superior catalytic activity then the rest of the devices. The catalytic properties of the devices remained stable after storage at 4 °C for 20 months (ESI, Fig. S6†). Study of the tolerability of DU145 and U87 cells to the presence of 30 μm Pd-devices showed full biocompatibility at the concentrations tested (ESI, Fig. S7†).
Prodrug candidate 2d was then incubated with 30 μm Pd-devices in serum-free media at physiological conditions (pH = 7.4, isotonicity, 37 °C) and analyzed by UPLC. Remarkably, whereas the analysis indicated that the 30 μm devices also sequestered the prodrug very rapidly (note the immediate disappearance of the prodrug signal from the chromatogram in Fig. S8 of the ESI†), it showed efficient synthesis and release of 1, an optimal situation for the therapeutic goal of the strategy. In principle, the superior affinity of the prodrug for the devices could potentially serve to pull the prodrug molecules out of circulation, whereby they would be uncaged and released as an active drug at the intended site of treatment. As shown in Fig. 2C, dose–response studies in cancer cell culture using 30 μm Pd-devices provided equivalent curves and EC50 values for the prodrug 2d/Pd combinations and the unmodified drug 1 in both cell lines (see complete studies in ESI, Fig. S9 and S10†). Based on the safety, catalytic performance and convenient size of the 30 μm Pd-devices, they were then chosen for the next phase of the investigation.
In vivo/ex vivo assays: implantation, safety and stability of Pd-devices
Given the medical interest of our team in developing improved prostate cancer treatments, a state-of-the-art orthotopic murine model of human prostate cancer45,46 was used to test the in vivo biocompatibility and functional stability of the Pd-devices. DU145 prostate cancer cells were carefully inserted in the prostate of anesthetized male mice (n = 4) and tumours allowed to grow for 4 weeks (see full details in the ESI†). One of the clinical logistics of implant-based focal therapy modalities is to insert the devices in the tumour with high precision. In brachytherapy, this procedure is facilitated by the echogenic nature of the radioactive inserts (which are made of metal), as it enables to control the correct placement of the radioactive pellets using ultrasound imaging.47 Since the Pd-devices contain a significant amount of metallic material, we envisioned that ultrasound-guided insertion would be an optimal method to place such small devices with millimeter range precision in the prostate tumour (Fig. 3A). A standard 1 mL syringe fitted with a 9-gauge needle was used to deliver the devices. The size of the 30 μm Pd-devices was found optimal to perform this procedure with ease, whereas partial blockage of the needle was observed with the larger devices. Hence, 1 mg of 30 μm Pd-devices suspended in 25 μL of sterile PBS was injected into the prostate tumour of anaesthetized mice under real time monitoring with ultrasound imaging. Fig. 3B and Movie S1† shows the highly echogenic Pd-devices being precisely inserted into a hypoechogenic area of the tumour. The mice were then transferred for recovery. The injected region was imaged 30 min after the procedure (Movie S2†) and monitored for 3 days (ESI, Fig. S11†) to ensure that Pd-devices did not leak out the tumour. Tumour growth was not affected by the presence of the devices. Apart from the presence of the tumour, animals remained healthy and behaved normally during the time of the study. After 21 days, the animals were sacrificed. Necropsy of animals and gross observation of major organs showed no macroscopic signs of toxicity. Tumours were removed and sliced in half to study the distribution of the Pd-devices (ESI, Fig. S11†). Examination of the tissue surrounding the injection site further confirmed that the devices had not been displaced from the cancerous tissue.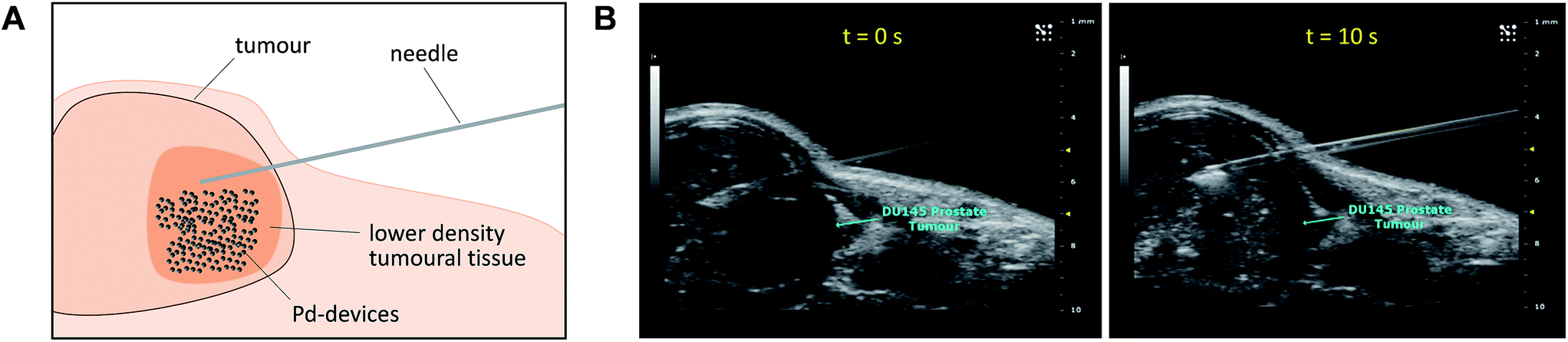 | ||
| Fig. 3 (A) Schematic of the in vivo intratumoural insertion of 30 μm Pd-devices (1 mg in 25 μL of PBS) into a murine prostate tumour. (B) Ultrasound image captions of the prostate tumour area of an anaesthetised mouse taken during the implantation procedure at t = 0 s (left) and 10 s (right). | ||
The catalytic capacity of Pd-devices hosted in the prostate tumour of live mice for 3 weeks (21 day-in-tumour Pd-devices) was then tested ex vivo. Explants bearing Pd-devices were sliced longitudinally using a rat brain slicer instrument into 1 mm sections, soaked in media containing fluorogenic probe 3 and visualized by live-cell imaging confocal microscopy for 24 h (using the green fluorescent channel with 488 nm laser excitation). The catalytic activity of 21 day-in-tumor Pd-devices was compared with devices directly implanted in the explants (0 day-in-tumour Pd-devices). Incubation of Pd-free explants with 3, 4 or DMSO were used as controls. As shown in Fig. 4A, a rapid increase in fluorescent intensity (=release of fluorescent dye 4) was achieved by treating Pd-bearing explants with 3. Importantly, equivalent fluorescence generation was observed with both 0- and 21 day-in-tumour Pd-devices, evidence that maintaining the devices in living tissue for extended periods of time does not have a negative impact in the underlying turnover rate of the devices. In contrast, in the absence of the activation devices, tumour explants treated with 3 showed minimal background signal (Fig. 4A, right). The good biocompatibility and catalytic stability of the catalyst in living tissue, which are herein shown for the first time for a bioorthogonal catalyst that has been hosted for weeks in a tumour in vivo, are optimal features that support the use of heterogeneous Pd devices for site-specific prodrug activation in precision medicine.
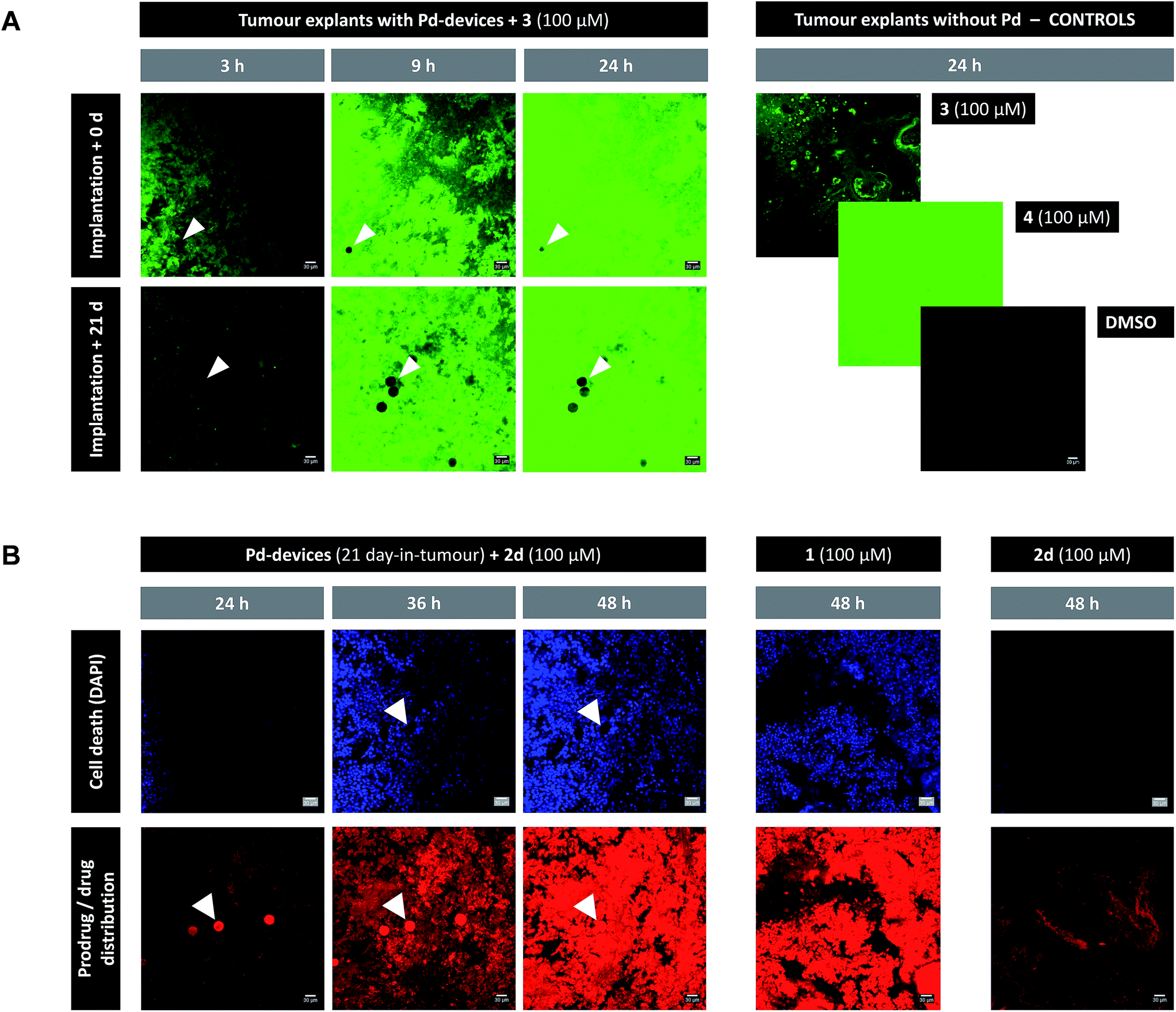 | ||
| Fig. 4 (A) Live-cell imaging study of the catalytic activity of tumour-implanted Pd-devices: 0 d vs. 21 d after implantation. Pd-bearing explants (n = 2) were incubated with 3 (100 μM) and imaged by live-cell confocal microscopy (488 laser excitation) at 3, 9 and 24 h. The presence of the Pd-devices is indicated with white arrows. Note that Pd-devices are identified due to their capacity to quench green fluorescence.27 Images of the controls (3 alone (100 μM); 4 alone (100 μM); and DMSO) are shown at 24 h. Pictures were generated using ImageJ software. Scale bar = 30 μm. (B) Live-cell imaging study of the Pd-mediated conversion of inactive 2d into cytotoxic 1 in a human prostate tumour explant model. Images of a representative tumour explant (n = 2) bearing 21 day-in-tumor Pd-devices after treatment with 2d (100 μM) in the presence of DAPI for 24, 36 and 48 h (left panel). Tissue samples were imaged under laser excitation at 405 nm (for DAPI staining, in blue) and 543 nm (for 2d/1 distribution, in red). The presence of the Pd-devices, indicated with white arrows, is identified by a bright fluorescence signal in the red channel (as previously reported,48 red fluorescence is not quenched by Pd). Images of the Pd-free controls 1 (100 μM, mid panel) and 2d (100 μM, right panel) are shown at 48 h. Pictures were generated using ImageJ software. Scale bar = 30 μm. | ||
Ex vivo prodrug-into-drug conversion study
Next, the capacity of the devices to generate therapeutic (=cytotoxic) levels of 1 from an inactive dose of 2d was tested in tumour explants bearing 21 day-in-tumour Pd-devices. Tissue samples were treated for 48 h in media containing 100 μM of 2d in the presence of DAPI (a nuclei stain not permeable in viable cells). Treatment of Pd-free explants (in the presence of DAPI) with either 2d (100 μM) or 1 (100 μM) were used as a negative and positive control, respectively. Cancer cell viability and prodrug/drug distribution was monitored by live-cell imaging confocal microscopy using two laser excitations: 405 nm to image nuclei staining by DAPI (=dying/dead cells) and 543 nm to visualize 2d and/or 1 (the spectroscopic properties of both compounds are indistinguishable; see Fig. S3†). In the absence of Pd-devices (Fig. 4B, right panel), 48 h treatment with 2d did not elicit any sign of cell death in the cancer tissue. Analysis of the red fluorescent channel shows that 2d was not retained inside the cancer cells, which is consistent with its expected inability to bind with the intracellular targets of doxorubicin. A drastically different distribution profile was observed upon treatment with 1, with bright red fluorescence being accumulated in the cells (Fig. 4B, middle panel; and ESI, Fig. S12†). Remarkably, the distribution pattern of the red fluorescence signal from explants bearing Pd-devices and treated with 2d became identical to that of the explants directly treated with 1 after 48 h, indicating that 2d has been chemically converted into 1. It is also noteworthy that compound 2d rapidly entered the devices during the first hours of treatment, making the Pd-devices patently visible under the 543 nm laser excitation (Fig. 4B, see panels at 24 and 36 h). This observation agrees with the high capacity of the devices to “pull” the prodrug from the environment (Fig. 2A). Importantly, the 2d/Pd-devices treatment combination induced a strong cytotoxic effect (DAPI staining), equivalent to that of 1 treatment, demonstrating the in situ bioorthogonal generation of bioactive levels of drug. Ex vivo studies with the 0 day-in-tumour Pd-devices provided equivalent results (ESI, Fig. S12†).Last, samples of an explant containing 21 day-in-tumour Pd-devices and previously treated with fluorogenic compound 3 were washed and subsequently incubated with either 1, prodrug 2d or DMSO for 48 h. Tissue samples were then fixed and analysed by immunohistochemical staining with caspase 3 (apoptosis biomarker) or γ-H2AX (biomarker for DNA double-strand breaks) antibodies (ESI, Fig. S13†). As expected, damaged cells (indicated by brown staining) were only observed in the Pd-containing samples treated with 1 and 2d. This test agrees with the inert nature of the devices and supports their capacity to activate multiple doses of prodrug.
Conclusions
In conclusion, fundamental insights into the validity, reliability and clinical feasibility of a novel approach to accurately target chemotherapy spatially within a tumour have been reported. This therapeutic approach, which is conceived for the treatment of localised cancers, consists of two components: (i) an inactive derivative of a cytotoxic drug designed to be specifically activated by palladium chemistry; and (ii) an inert implantable polymer-based device functionalised with Pd nanoparticles to catalyse drug conversion and release in a spatially controlled manner. As part of the investigation, a novel caged doxorubicin was developed by blocking the NH2 group of the drug's sugar moiety with a Pd-labile PBC group. The derivative having the propargyloxy moiety in ortho was found to offer superior reduction of bioactivity than the one in para and high sensitivity to Pd catalysis. Studies in zebrafish proved that the cardiotoxic effect induced by doxorubicin was abolished in the caged precursor; a clinically-relevant result that to the best of our knowledge is shown in this animal model for the first time for a doxorubicin prodrug. Pd-devices of optimised size and activity were also developed and tested in a state-of-the-art orthotopic murine model of human prostate cancer. The devices showed high echogenicity, enabling precise injection into the tumour using ultrasound scanning, an image-guided microsurgical technique widely used in the clinic. The study provided evidence of the non-perturbing effect of the devices to the mice and of the resilience and biocompatibility of the catalyst to the tumour microenvironment. Devices hosted in a tumour of mice for 21 d elicited catalytic release of a fluorogenic probe and a caged doxorubicin at an equivalent rate than freshly injected devices. Ex vivo time-lapse imaging enabled to monitor the distinct distribution profiles of the drug and the inactive precursor, and the capacity of the devices to “sequester” circulating prodrug molecules and generate cytotoxic levels of drug in the tumour explant. This investigation provides compelling evidence that heterogeneous palladium catalysts can be applied as bioorthogonal tools to enable local treatment of disease and reports a standardized preclinical methodology to perform and study local bioorthogonal release of bioactive substances in preclinical disease models.Conflicts of interest
The authors declare that compounds 2c, d are protected under patent application PCT/GB2017/051379.Acknowledgements
We thank EPSRC (Healthcare Technology Challenge Award EP/N021134/1) and CRUK (CRT Project Development Fund) for funding the project. T. L. B. thanks the CMVM of the University of Edinburgh (Principal's scholarship). M. S. and H. Y. L. are grateful to MRC (Clinical Research Fellowship 70128) and CRUK (Beatson Institute A15151), respectively. A. T. and E. E. P. thanks MRC (MC_PC_U127585840), ERC (ZF-MEL-CHEMBIO-648489) and the Melanoma Research Alliance – L'Oreal USA (401181). B. R.-R. thanks the EC (H2020-MSCA-IF-2014-658833, ChemoBOOM).Notes and references
- M. Perera, N. Krishnananthan, U. Lindner and N. Lawrentschuk, Nat. Rev. Urol., 2016, 13, 641 CrossRef PubMed.
- G. W. Chodak, R. A. Thisted, G. S. Gerber, J. E. Johansson, J. Adolfsson, G. W. Jones, G. D. Chisholm, B. Moskovitz, P. M. Livne and J. Warner, N. Engl. J. Med., 1994, 330, 242 CrossRef PubMed.
- H. U. Ahmed, R. G. Hindley, L. Dickinson, A. Freeman, A. P. Kirkham, M. Sahu, R. Scott, C. Allen, J. Van der Meulen and M. Emberton, Lancet Oncol., 2012, 13, 622 CrossRef PubMed.
- G. Giannarini, G. Gandaglia, F. Montorsi and A. Briganti, J. Clin. Oncol., 2014, 32, 1299 CrossRef PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef PubMed.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592 CrossRef PubMed.
- P. Shieh and C. R. Bertozzi, Org. Biomol. Chem., 2014, 12, 9307 RSC.
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129 CrossRef PubMed.
- W. R. Algar, P. E. Dawson and I. L. Medintz, Chemoselective and Bioorthogonal Ligation Reactions: Concepts and Applications, Wiley-VCH, Weinheim, 2017 Search PubMed.
- C. Streu and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 5645 CrossRef PubMed.
- J. L. Mascareñas, M. I. Sánchez, C. Penas and M. E. Vázquez, Chem. Sci., 2014, 5, 1901 RSC.
- T. Völker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536 CrossRef PubMed.
- M. Tomás-Gamasa, M. Martínez-Calvo, J. C. Couceiro and J. L. Mascareñas, Nat. Commun., 2016, 7, 12538 CrossRef PubMed.
- P. Destito, J. R. Couceiro, H. Faustino, F. López and J. L. Mascareñas, Angew. Chem., Int. Ed., 2017, 56, 10766 CrossRef PubMed.
- R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239 CrossRef PubMed.
- A. Unciti-Broceta, E. M. V. Johansson, R. M. Yusop, R. M. Sánchez-Martín and M. Bradley, Nat. Protoc., 2012, 7, 1207 CrossRef PubMed.
- N. Li, R. K. Lim, S. Edwardraja and Q. Lin, J. Am. Chem. Soc., 2011, 133, 15316 CrossRef PubMed.
- C. D. Spicer, T. Triemer and B. G. Davis, J. Am. Chem. Soc., 2012, 134, 800 CrossRef PubMed.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y. C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597 CrossRef PubMed.
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sánchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 3277 CrossRef PubMed.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. Chen, Nat. Chem., 2014, 6, 352 CrossRef PubMed.
- J. Clavadetscher, S. Hoffmann, A. Lilienkampf, L. Mackay, R. M. Yusop, S. A. Rider, J. J Mullins and M. Bradley, Angew. Chem., Int. Ed., 2016, 55, 15662 CrossRef PubMed.
- J. Miguel-Ávila, M. Tomás-Gamasa, A. Olmos, P. J. Pérez and J. L. Mascareñas, Chem. Sci., 2018, 9, 1947 RSC.
- Y. Liu, S. Pujals, P. J. M. Stals, T. Paulöhrl, S. I. Presolski, E. W. Meijer, L. Albertazzi and A. R. A. Palmans, J. Am. Chem. Soc., 2018, 140, 3423 CrossRef PubMed.
- K. Tsubokura, K. K. Vong, A. R. Pradipta, A. Ogura, S. Urano, T. Tahara, S. Nozaki, H. Onoe, Y. Nakao, R. Sibgatullina, A. Kurbangalieva, Y. Watanabe and K. Tanaka, Angew. Chem., Int. Ed., 2017, 56, 3579 CrossRef PubMed.
- A. M. Pérez-López, B. Rubio-Ruiz, V. Sebastián, L. Hamilton, C. Adam, T. L. Bray, S. Irusta, P. M. Brennan, G. Lloyd-Jones, D. Sieger, J. Santamaría and A. Unciti-Broceta, Angew. Chem., Int. Ed., 2017, 56, 12548 CrossRef PubMed.
- C. Vidal, M. Tomás-Gamasa, P. Destito, F. López and J. L. Mascareñas, Nat. Commun., 2018, 9, 1913 CrossRef PubMed.
- J. T. Weiss, J. C. Dawson, C. Fraser, W. Rybski, C. Torres-Sánchez, M. Bradley, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, J. Med. Chem., 2014, 57, 5395 CrossRef PubMed.
- J. T. Weiss, N. O. Carragher and A. Unciti-Broceta, Sci. Rep., 2015, 5, 9329 CrossRef PubMed.
- B. Rubio-Ruiz, J. T. Weiss and A. Unciti-Broceta, J. Med. Chem., 2016, 59, 9974 CrossRef PubMed.
- B. Li, P. Liu, H. Wu, X. Xie, Z. Chen, F. Zeng and S. Wu, Biomaterials, 2017, 138, 57 CrossRef PubMed.
- M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nat. Commun., 2017, 8, 15906 CrossRef PubMed.
- M. Hoop, A. S. Ribeiro, D. Rösch, P. Weinand, N. Mendes, F. Mushtaq, X. -Z. Chen, Y. Shen, C. F. Pujante, J. Puigmartí-Luis, J. Paredes, B. J. Nelson, A. P. Pêgo and S. Pané, Adv. Funct. Mater., 2018, 28, 1705920 CrossRef.
- D. Bobo, K. Robinson, J. Islam, K. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373 CrossRef PubMed.
- J. Perry, A. Chambers, K. Spithoff and N. Laperriere, Curr. Oncol., 2007, 14, 189 CrossRef PubMed.
- Cancer Drug Information: Doxorubicin HCl, National Cancer Institute, http://www.cancer.gov/cancertopics/druginfo/doxorubicinhydrochloride. Date accessed: 10/12/17.
- J. Nitiss, Nat. Rev. Cancer, 2009, 9, 338 CrossRef PubMed.
- Y. Octavia, C. G. Tocchetti, K. L. Gabrielson, S. Janssens, H. J. Crijns and A. L. Moens, J. Mol. Cell. Cardiol., 2012, 52, 1213 CrossRef PubMed.
- R. Mahato, W. Tai and K. Cheng, Adv. Drug Delivery Rev., 2011, 63, 659 CrossRef PubMed.
- B. Rubio-Ruiz, T. L. Bray, A. López-Pérez and A. Unciti-Broceta, Masking strategies for the bioorthogonal release of anticancer glycosides, in Coupling and decoupling of diverse molecular units in glycosciences, ed. Z. J. Witczak and R. Bielski, Springer International Publishing, Cham, 1st edn, 2018, pp. 269–298 Search PubMed.
- R. M. Versteegen, R. Rossin, W. ten Hoeve, H. M. Janssen and M. S. Robillard, Angew. Chem., Int. Ed., 2013, 52, 14112 CrossRef PubMed.
- J. M. M. Oneto, I. Khan, L. Seebald and M. Royzen, ACS Cent. Sci., 2016, 2, 476 CrossRef PubMed.
- B. J. Stenton, B. L. Oliveira, M. J. Matos, L. Sinatra and G. J. L. Bernardes, Chem. Sci., 2018, 9, 4185 RSC.
- R. Quarrell, T. D. W. Claridge, G. W. Weaver and G. Lowe, Mol. Diversity, 1995, 1, 223 CrossRef.
- I. Ahmad, O. J. Sansom and H. Y. Leung, Expert Rev. Mol. Med., 2008, 10, e16 CrossRef PubMed.
- R. Patel, J. Fleming, E. Mui, C. Loveridge, P. Repiscak, A. Blomme, V. Harle, M. Salji, I. Ahmad, K. Teo, F. C. Hamdy, A. Hedley, N. van den Broek, G. Mackay, J. Edwards, O. J. Sansom and H. Y. Leung, EMBO Mol. Med., 2018, e8347 CrossRef PubMed.
- M. A. L. Bell, N. P. Kuo, D. Y. Song, J. U. Kang and E. M. Boctor, J. Biomed. Opt., 2014, 19, 126011 CrossRef PubMed.
- A. Delgado-Gonzalez, E. Garcia-Fernandez, T. Valero, M. V. Cano-Cortes, M. J. Ruedas-Rama, A. Unciti-Broceta, R. M. Sánchez-Martin, J. J. Diaz-Mochon and A. Orte, ACS Omega, 2018, 3, 144 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of materials, biological methods, in vivo/ex vivo protocols and Fig. S1–S13. See DOI: 10.1039/c8sc02291g |
| This journal is © The Royal Society of Chemistry 2018 |