DOI:
10.1039/C8SC02290A
(Edge Article)
Chem. Sci., 2018,
9, 6361-6367
Enantioselective aza-Friedel–Crafts reaction of furan with α-ketimino esters induced by a conjugated double hydrogen bond network of chiral bis(phosphoric acid) catalysts†
Received
24th May 2018
, Accepted 23rd June 2018
First published on 25th June 2018
Abstract
Chiral C2- and C1-symmetric BINOL-derived bis(phosphoric acid) catalysts, which have OP(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)(OH)2/OP(O)(OH)(OR) moieties at the 2,2′-positions, were developed and used for the enantioselective aza-Friedel–Crafts reaction of 2-methoxyfuran with α-ketimino esters for the first time. The intramolecular conjugated double hydrogen bond network is a key to increasing the Brønsted acidity and preventing deactivation of the catalysts. Highly functionalized α-amino acid derivatives with a chiral quaternary carbon center could be transformed into versatile optically active N- and O-heterocycles and an α-aryl-substituted serine.
O)(OH)2/OP(O)(OH)(OR) moieties at the 2,2′-positions, were developed and used for the enantioselective aza-Friedel–Crafts reaction of 2-methoxyfuran with α-ketimino esters for the first time. The intramolecular conjugated double hydrogen bond network is a key to increasing the Brønsted acidity and preventing deactivation of the catalysts. Highly functionalized α-amino acid derivatives with a chiral quaternary carbon center could be transformed into versatile optically active N- and O-heterocycles and an α-aryl-substituted serine.
Introduction
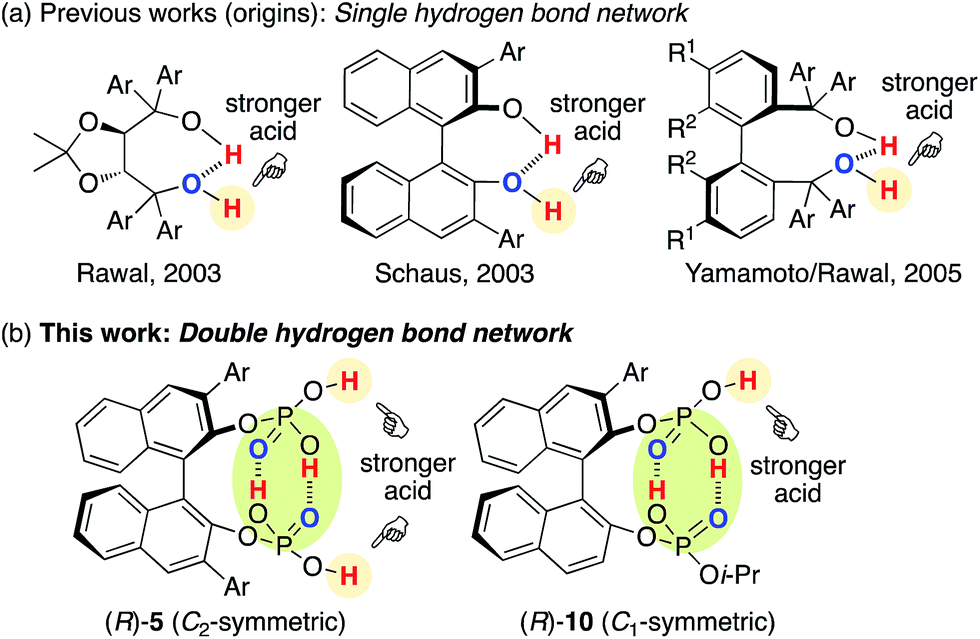
The hydrogen bond network of chiral multiprotic acid catalysts plays an important role in activating Brønsted acidity, controlling conformational flexibility, and producing high enantioselectivity.1,2 According to the general classification of combined acid catalysts described by Yamamoto,3 some chiral Brønsted acid catalysts R*(XH)2 with a hydrogen bond network could be considered part of a Brønsted acid-assisted Brønsted acid (BBA) catalyst system. In such a BBA system, one hydrogen atom of an XH group might participate in an intramolecular hydrogen bond with the other XH group, which might be activated and thus used for activation of the substrate. Since 2003, when Rawal reported the first example of an intramolecular single hydrogen bonding network in chiral TADDOLs (α,α,α′,α′-tetraaryl-1,3-dioxolan-4,5-dimethanols)4a,c for the enantioselective hetero-Diels–Alder reaction, and after Schaus developed chiral 3,3′-diaryl-BINOLs (1,1′-bi-2-naphthol)4b for the enantioselective Morita–Baylis–Hillman reaction, great effort has been devoted to this research area (Fig. 1a). Chiral 1,1′-biaryl-2,2′-dimethanol (Yamamoto/Rawal),4d chiral glycolic acid (Yamamoto),5 chiral 2-bis(triflyl)methyl-2′-hydroxy-1,1′-binaphthyl (Ishihara/Yamamoto),6 chiral binaphthyl di-carboxylic acids (Maruoka),7 3,3-linked-bis(BINOL)-derived bis(phosphoric acid)s (Gong),8 chiral binaphthyl disulfonic acids (BINSA) (our group),9 and chiral 3,3′-di(2-hydroxy-3-arylphenyl)-BINOL-derived bis(phosphoric acid)s and chiral carboxylic acid–phosphoric acid combined catalysts (Momiyama/Terada)10,11 have been developed for a variety of asymmetric catalyses. These outstanding chiral BBA catalysts might have an intramolecular single hydrogen bond network with the use of bis(monoprotic acid)s R*(XH)2. In sharp contrast, we envisioned that an intramolecular double hydrogen bond network may represent a new strategy for the design of chiral Brønsted acid catalysts. However, a simple and closed double hydrogen bond network, as seen in a dimeric structure of two molecules of carboxylic acids, would lose both the Brønsted acid- and base-functions upon neutralization. Therefore, here we developed chiral C2-symmetric BINOL-derived bis(phosphoric acid) catalysts (R)-5 as bis(diprotic acid)s R*(XH2)2, which have two OP(O)(OH)2 moieties at the 2,2′-positions of the chiral binaphthyl backbone (Fig. 1b). Based on the essence of (R)-5, chiral C1-symmetric catalysts (R)-10, which have OP(O)(OH)2/OP(O)(OH)(Oi-Pr) moieties, were also developed. Remarkably, outside of the conjugated intramolecular double hydrogen bond network, the Brønsted acid moiety would still exist and work as an active center by the BBA methodology.
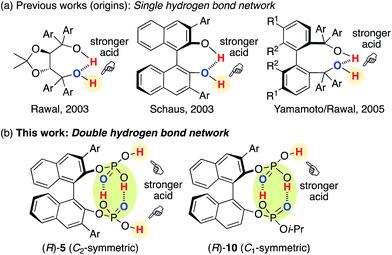 |
| | Fig. 1 Design of chiral BINOL-derived bis(phosphoric acid) catalysts. | |
Results and discussion
Experimental investigation with chiral C2-symmetric catalysts
We initially examined the aza-Friedel–Crafts (FC) reaction of 2-methoxyfuran 2 (ref. 12 and 13) with β,γ-alkynyl-α-imino esters 1a14 through the use of achiral Brønsted acid catalysts (5 mol%) in dichloromethane at −78 °C (Table 1 and also see the ESI†). As a result, suitable Brønsted acidity would be required for the reaction to proceed smoothly; catalysts that were too weakly acidic gave poor catalytic activity (entries 1 and 2) and catalysts that were too strongly acidic gave undesired byproducts due to the instability of 1a and particularly 2 under acidic conditions (entries 4–6). Fortunately, phosphoric acids could be used without the serious generation of byproducts due to their suitable Brønsted acidity for this reaction, although the yields of product 3a were moderate (entries 7 and 8). Next, we examined conventional chiral phosphoric acid (R)-4a (entry 9), which would be less aggregatable than the less bulky achiral phosphoric acids in entries 7 and 8. As a result, although the pKa of (R)-4a would be similar to those of the achiral phosphoric acids in entries 7 and 8, the catalytic activity was greatly improved (see the ESI† for details). Particularly, when (R)-4b with electron-withdrawing CF3 groups in its 3,3′-diaryl moieties was used, the reaction was accelerated, although the enantioselectivity was still low (40% ee) (entry 10). Moreover, well-acknowledged bulky (R)-4c was much less active than (R)-4a and (R)-4b (entry 11). In contrast, chiral C2-symmetric bis(phosphoric acid) (R)-5a15 was much more effective than (R)-4a–c, and 3a was obtained in 82% yield with 70% ee within 5 h (entry 12).16 Slightly modified catalyst (R)-5b derived from (R)-3,3′-(3,5-(o-Tol)2C6H3)2-BINOL improved the enantioselectivity (76% ee) of 3a (entry 13). (R)-5c with a 5,5′,6,6′,7,7′,8,8′-H8-binaphthyl backbone showed lower catalytic activity than (R)-5a and (R)-5b, and a prolonged reaction time (24 h) was needed (entry 14). Moreover, we optimized β,γ-alkynyl-α-imino esters 1 with the use of (R)-5b (see the ESI† for details).17 To avoid the effect of adventitious water, which might react with 1 and 2 to give undesired products, powdered MS 5 Å was used as a drying agent. As a result, when we used 1b with bulky i-Pr3Si protection for the alkyne moiety, (S)-3b was obtained in 83% yield with 91% ee (eqn (1)). Remarkably, we could perform a 1.89 g-scale synthesis of (S)-3b (77% yield with 91% ee), and 99% of (R)-5b could be recovered.| | 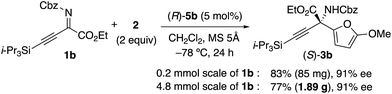 | (1) |
Table 1 Screening of Brønsted acid catalystsa
We now turn our attention to mechanistic aspects. It is important to identify the intramolecular double hydrogen bond network in the catalysts. Fortunately, (R)-5c·(pyridine)2 was crystallized, and the results of an X-ray analysis are shown in Fig. 2. As a result, two P(O)(OH)2 moieties at the 2,2′-positions of the H8-binaphthyl backbone coordinate with each other, and the conjugated double hydrogen bond network is unambiguously formed at the center of the monomeric molecules. Pyridines are coordinated BBA-activated protons, which would be considered to activate the substrate in a similar way. In this regard, the role of the two outside protons was next examined with the use of monomethyl-protected (R)-6a and dimethyl-protected (R)-6b (Scheme 1). As a result, (R)-6a showed almost the same catalytic activity (81% yield and 91% ee of 3b) as (R)-5b, whereas (R)-6b gave a poor result (22% yield and 34% ee of 3b) in the reaction of 2 with 1b. This result strongly suggests that two activated protons in (R)-5b should be independent of each other, although the absence of both protons results in a loss of catalytic activity. In contrast, the protons in the tight structure of a double hydrogen bond network might not be directly involved in promoting the reaction as possible Brønsted acids.
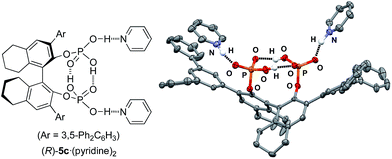 |
| | Fig. 2 X-ray analysis of (R)-5c·(pyridine)2. Hydrogen atoms are partially omitted for clarity. | |
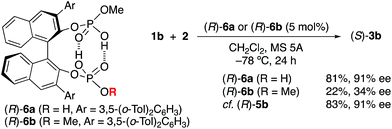 |
| | Scheme 1 Role of active H+-centers in chiral C2-symmetric catalysts (R)-5b. | |
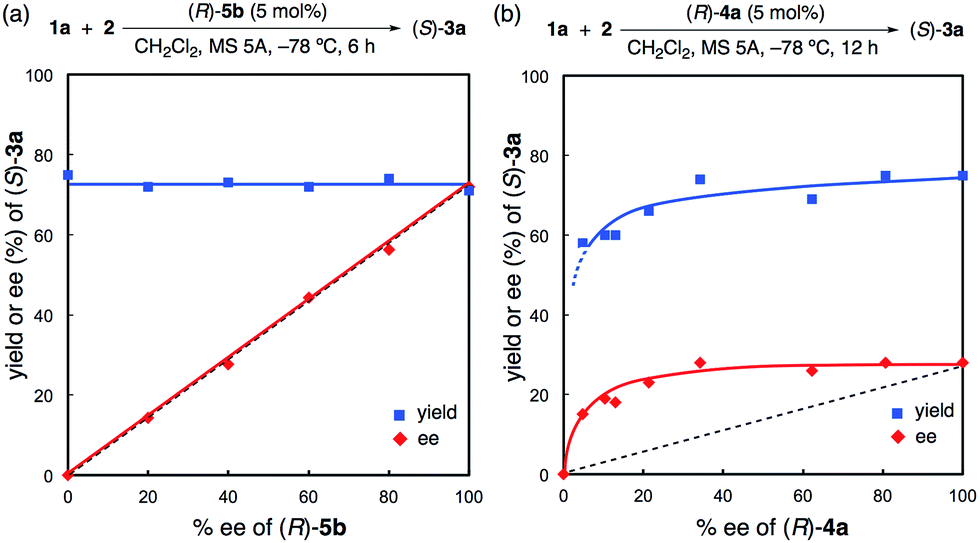
Moreover, a non-linear effect was examined in the reaction of 2 with 1a with the use of (R)-5b or (R)-4a (Fig. 3 and also see the ESI†). As expected from the X-ray structure of monomeric (R)-5c·(pyridine)2, a linear relationship was observed for (R)-5b, and the yields were almost constant (71–75%) (Fig. 3a). In contrast, a positive non-linear effect was observed for (R)-4a (Fig. 3b).18 This non-linear relationship strongly suggests that inactive dimeric species19 might be involved under the reaction conditions for (R)-4a unlike (R)-5b. Indeed, although both (R)-5b and (R)-4a have almost the same sterically hindered 3,3′-diaryl moieties, the strongly Brønsted basic PO moiety is still free in (R)-4a. It is quite unlike the situation in (R)-5b, which has a core hydrogen bond network through the “intramolecular dimerization” of two P(O)(OH)2 moieties. Thus, the much better catalytic activity of (R)-5b compared to (R)-4a might be attributed to not only the stronger acidity of (R)-5b due to a BBA system but also the monomeric active species of (R)-5b due to the closed PO moieties.20
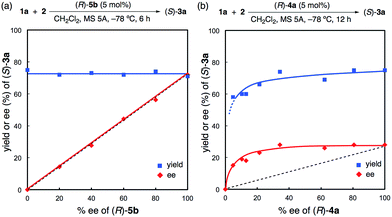 |
| | Fig. 3 Non-linear effects of (R)-5b and (R)-4a. | |
Experimental investigation with chiral C1-symmetric catalysts
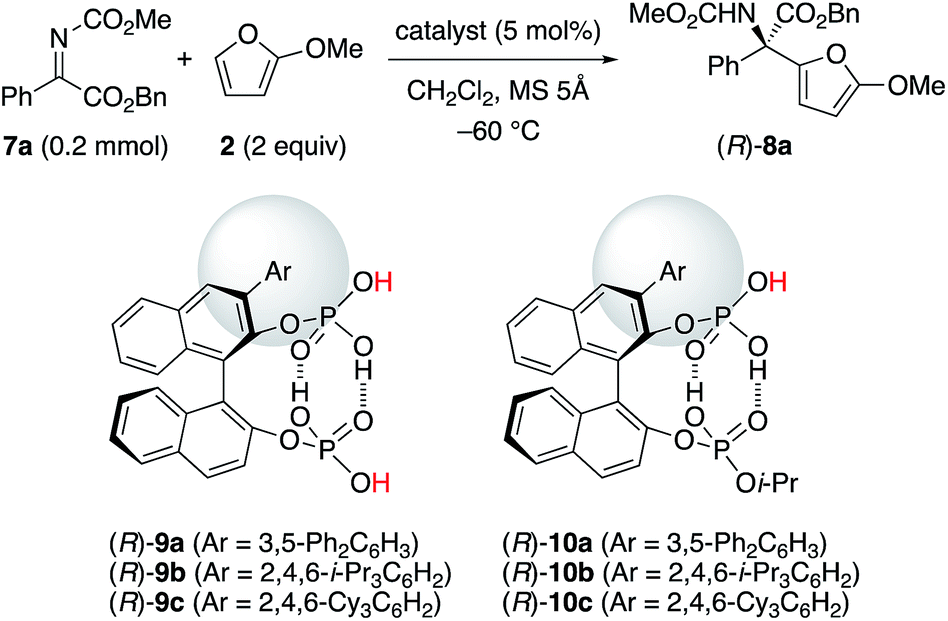
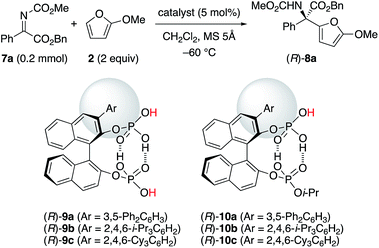
With the above understanding of the present catalyst system, we should expand the substrate scope of α-ketimino esters beyond the useful but specific substrates 1a and 1b. Due to their synthetic importance, we chose aryl α-ketimino esters, such as 7a (Table 2). Unfortunately, however, (R)-5b was not effective for the reaction of less reactive 7a, unlike more reactive 1a and 1b, and (R)-8a was barely obtained with 15% ee at slightly higher temperature (−60 °C) (entry 3). To improve the enantioselectivity, stereocontrol by more bulky substituents at the 3,3′-positions of the C2-symmetric catalysts (R)-5 should be needed. However, we could not introduce phosphoric acid moieties at the 2,2′-positions of the bulky 3,3′-Ar2-BINOL-skeleton (e.g., Ar = 2,4,6-i-Pr3C6H2). Instead, an extremely bulky C1-symmetric catalyst (R)-9c with 2,4,6-Cy3C6H2 at the 3-position could be readily prepared as well as (R)-9a (Ar = 3,5-Ph2C6H3) and (R)-9b (Ar = 2,4,6-i-Pr3C6H2). However, since two different active Brønsted acid centers would compete, the enantioselectivity was low (11–18% ee), as expected (entries 4–6). Based on the above consideration of C2-symmetric catalysts (R)-5, we designed the site-selectively mono-i-Pr-capped catalysts (R)-10.21 As a result, when we use (R)-10c with extremely bulky 2,4,6-Cy3C6H2 at the 3-position, the reaction proceeded smoothly, and (R)-8a was obtained in 93% yield with 95% ee (entry 9). The bulkiness of the aryl moiety at the 3-position is important, since slightly less hindered (R)-10b with 2,4,6-i-Pr3C6H2 was less effective (entry 8), and much less hindered (R)-10a with 3,5-Ph2C6H3 was entirely ineffective (entry 7). Conventional chiral phosphoric acids, such as (R)-4b and (R)-4c, were not effective (also see the ESI† for details): a prolonged reaction time (24 h) was needed and the enantioselectivity was low (entries 1 and 2).
Table 2 Optimization of catalysts for the reaction of α-ketimino ester 7aa
|
|
| Entry |
Catalyst |
Reaction time (h) |
Yield (%) |
ee (%) |
|
The reaction was carried out with catalyst (5 mol%), 7a (0.20 mmol, 1 equiv.), 2 (2 equiv.), and MS 5 Å in dichloromethane (0.1 M based on 7a) at −60 °C.
|
| 1 |
(R)-4b (Ar = 3,5-(CF3)2C6H3) |
24 |
83 |
14 |
| 2 |
(R)-4c (Ar = 2,4,6-i-Pr3C6H2) |
24 |
79 |
0 |
| 3 |
(R)-5b (Ar = 3,5-(o-Tol)2C6H3) |
12 |
89 |
15 |
| 4 |
(R)-9a (Ar = 3,5-Ph2C6H3) |
3 |
51 |
11 |
| 5 |
(R)-9b (Ar = 2,4,6-i-Pr3C6H2) |
3 |
74 |
13 |
| 6 |
(R)-9c (Ar = 2,4,6-Cy3C6H2) |
3 |
82 |
18 |
| 7 |
(R)-10a (Ar = 3,5-Ph2C6H3) |
3 |
86 |
36 |
| 8 |
(R)-10b (Ar = 2,4,6-i-Pr3C6H2) |
3 |
96 |
91 |
| 9 |
(R)-10c (Ar = 2,4,6-Cy3C6H2) |
3 |
93 |
95 |
With the optimized catalyst in hand, we next examined the scope of aryl α-ketimino esters 7 (Scheme 2). As a result, not only electron-withdrawing group- but also electron-donating group-substituted aryl substrates could be generally used (see 8b–j, 83–98% yield with 82–97% ee). When we investigated simple o-, m-, and p-tolyl-substituted substrates (7i–k), p-tolyl 8i and m-tolyl 8j were obtained in high yields with high enantioselectivities (94% and 95% ee, respectively), whereas o-tolyl 8k could not be obtained probably due to steric hindrance. Indeed, less sterically hindered o-F-C6H48c and 1-naphthyl 8m were obtained with high enantioselectivities (97% and 90% ee, respectively). 2-Naphthyl 8l and heteroaryl 8n were also obtained successfully (95% and 87% ee, respectively). Unfortunately, however, a low-reactive aliphatic substrate 7o could not be used, and no reaction proceeded. Moreover, instead of 2-methoxyfuran 2, 2-ethoxyfuran22 could be used, and the corresponding product 8p was obtained in 94% yield with 94% ee.
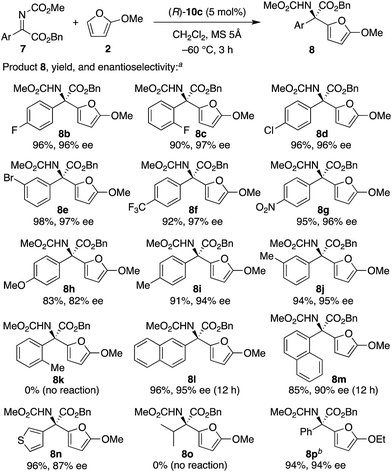 |
| | Scheme 2 Substrate scope in the enantioselective aza-FC reaction of 2-methoxyfuran 2 with aryl α-ketimino esters 7. (a) The reaction was carried out with (R)-10c (5 mol%), 7 (0.20 mmol, 1 equiv.), and 2 (2 equiv.) in dichloromethane (0.1 M based on 7) at −60 °C for 3 h. (b) 2-Ethoxyfuran was used instead of 2. | |
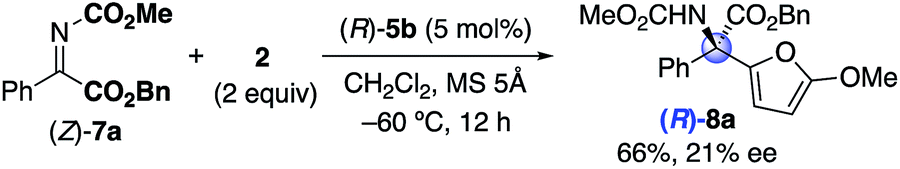
To date, it has been difficult to consider the difference between the optimized C2-symmetric (R)-5b and C1-symmetric (R)-10c. As seen above, (R)-5b-catalysis of 1b and 2 provided (S)-3b, and (R)-10c-catalysis of 7a and 2 provided (R)-8a. Therefore, the observed absolute stereochemistries in 5b and 8a were opposite each other. This changeover might be caused by the geometry of α-ketimino esters (E)-1b14 and (Z)-7a23 (see the ESI† for details). Moreover, (S)-3b was obtained in 14% yield with 6% ee by using (R)-10c (eqn (2)), whereas (R)-8a was obtained in 66% yield with 21% ee by using (R)-5b (eqn (3)). Overall, chiral C2- and C1-symmetric bis(phosphoric acid) catalysts were complementary, and either catalyst that was suitable for one reaction would not be suitable for the other reaction from the viewpoint of yield and enantioselectivity. Although preliminary possible transition states are considered as a working model (see the ESI† for details), further investigations of the catalysts are still needed.16,20
| |  | (2) |
| | 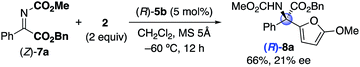 | (3) |
Transformation of products
Since optically active 3b has four versatile and transferable functional groups, including acetylene, ester, carbamate, and furyl groups, at the chiral quaternary carbon, we chose to explore the transformations of these functional groups (Scheme 3a). First, the i-Pr3Si moiety of 3b was removed by tetrabutylammonium fluoride (TBAF) to give 11 quantitatively. Next, 11 was reduced under typical reaction conditions. Wilkinson's catalyst completely reduced the acetylene moiety of 11, and 12 was obtained quantitatively. Lindlar's catalyst facilitated the selective hydrogenation of acetylene at 0 °C to give vinyl compound 13 in 94% yield, whereas both acetylene and the Cbz moieties of 11 were reduced at room temperature and 14 was obtained quantitatively. Furthermore, the NH2 moiety of 14 was protected by di-tert-butyl dicarbonate (Boc2O) in 83% yield, and the corresponding product was consequently treated with N-bromosuccinimide (NBS) to give 1,4-dicarbonyl compound 15 in 68% yield via furan cleavage. Finally, chemoselective reduction of the keto moiety of 15 with the use of CeCl3/NaBH4 gave γ-butenolide 16 in 84% yield with a diastereomeric ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20.12
:20.12
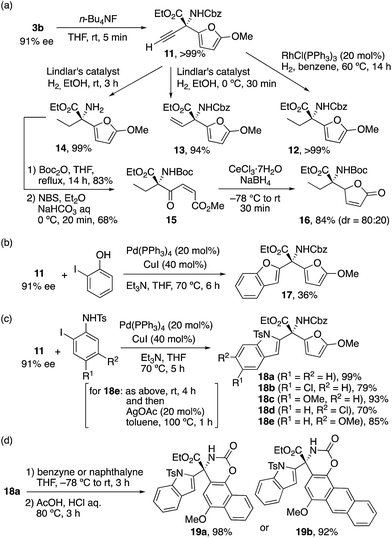 |
| | Scheme 3 Transformation of alkyne and furan moieties. | |
We further explored the transformation of 11 to some optically active N- and O-heterocycles. Sonogashira coupling of 11 with 2-iodophenol proceeded in the presence of Pd(PPh3)4/CuI catalysts, and novel 2-substituted benzofuran 17, which has a chiral quaternary carbon center with substitutions of furan, ester, and carbonate moieties, was obtained in 36% yield (Scheme 3b). A similar transformation of 11 with N-tosyl-2-iodoaniline proceeded, and novel 2-substituted indol 18a with those functional groups was obtained quantitatively (Scheme 3c). Cl- or MeO-substituted N-tosyl-2-iodoanilines were also tolerable, and 18b–e were obtained in high yields (70–93%). Moreover, a Diels–Alder reaction of the furan moiety of 18a with benzyne gave the corresponding adduct, which, without purification, was treated with HCl/acetic acid to give 19a in 98% yield (Scheme 3d).24 Naphthalyne in place of benzyne also gave the corresponding product 19b in 92% yield. These indole-derived α-amino acid derivatives 18 and 19 with extraordinary structural diversities would facilitate the process of drug discovery.25
Since optically active α-aryl-substituted serines are synthetically useful,26 we finally transformed the obtained product 8e (Scheme 4). Before the transformation, we performed a >2 g-scale synthesis of 8e (95% ee) with a catalyst loading as low as 0.2 mol%. The obtained 8e was then reduced by NaBH4, and compound 20 was obtained in 84% yield. Compound 20 was highly crystalline, and a single recrystallization increased the enantiopurity up to >99% ee. Finally, the 2-methoxyfuran moiety of 20 was oxidized by RuCl3-catalyzed NaIO4-oxidation,27 and the corresponding desired optically active α-aryl-substituted serine-derivative 21 (0.89 g) was obtained in 89% yield.
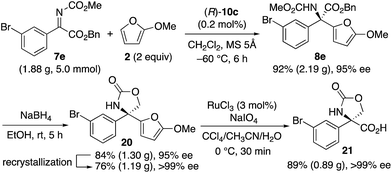 |
| | Scheme 4 Transformation to optically active α-aryl-substituted serine 21. | |
Conclusions
In summary, we have developed chiral BINOL-derived C2- and C1-symmetric bis(phosphoric acid) catalysts. The conjugated double hydrogen bond network was key to increasing the Brønsted acidity and preventing dimerization/deactivation of the catalysts. In particular, we developed a highly enantioselective aza-Friedel–Crafts reaction of 2-methoxyfuran with α-ketimino esters for the first time. By taking advantage of the highly functionalized products, some transformations to versatile N- and O-heterocycles and α-aryl-substituted serine with a chiral quaternary carbon center could be achieved. The further application of these catalysts in other asymmetric catalyses is underway.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support was partially provided by JSPS KAKENHI Grant Numbers JP17H03054, JP15H05755, and JP15H05810 in Precisely Designed Catalysts with Customized Scaffolding. We thank Takuya Mochizuki (Nagoya University) for technical support.
Notes and references
- For reviews on asymmetric hydrogen bond network.
(a) P. M. Pihko, Angew. Chem., Int. Ed., 2004, 43, 2062 CrossRef PubMed;
(b) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef PubMed.
- For reviews on chiral Brønsted acids.
(a) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef PubMed;
(b) M. Terada, Synthesis, 2010, 2010, 1929 CrossRef;
(c) D. Kampen, C. M. Reisinger and B. List, Top. Curr. Chem., 2010, 291, 395 CrossRef PubMed;
(d) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef PubMed;
(e) T. Akiyama and K. Mori, Chem. Rev., 2015, 115, 9277 CrossRef PubMed;
(f) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2017, 117, 10608 CrossRef PubMed;
(g) J. Merad, C. Lalli, G. Bernadat, J. Maury and G. Masson, Chem.–Eur. J., 2018, 24, 3925 CrossRef PubMed.
-
(a) H. Ishibashi, K. Ishihara and H. Yamamoto, Chem. Rec., 2002, 2, 177 CrossRef PubMed;
(b) H. Yamamoto and K. Futatsugi, Angew. Chem., Int. Ed., 2005, 44, 1924 CrossRef PubMed.
-
(a) Y. Huang, A. K. Unni, A. N. Thadani and V. H. Rawal, Nature, 2003, 424, 146 CrossRef PubMed;
(b) N. T. McDougal and S. E. Schaus, J. Am. Chem. Soc., 2003, 125, 12094 CrossRef PubMed;
(c) A. N. Thadani, A. R. Stankovic and V. H. Rawal, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5846 CrossRef PubMed;
(d) A. K. Unni, N. Takenaka, H. Yamamoto and V. H. Rawal, J. Am. Chem. Soc., 2005, 127, 1336 CrossRef PubMed.
- N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2005, 127, 1080 CrossRef PubMed.
- A. Hasegawa, Y. Naganawa, M. Fushimi, K. Ishihara and H. Yamamoto, Org. Lett., 2006, 8, 3175 CrossRef PubMed.
-
(a) T. Hashimoto and K. Maruoka, J. Am. Chem. Soc., 2007, 129, 10054 CrossRef PubMed;
(b) T. Hashimoto, H. Kimura, H. Nakatsu and K. Maruoka, J. Org. Chem., 2011, 76, 6030 CrossRef PubMed.
-
(a) X.-H. Chen, W.-Q. Zhang and L.-Z. Gong, J. Am. Chem. Soc., 2008, 130, 5652 CrossRef PubMed;
(b) J. Yu, L. He, X.-H. Chen, J. Song, W.-J. Chen and L.-Z. Gong, Org. Lett., 2009, 11, 4946 CrossRef PubMed;
(c) J. Yu, W.-J. Chen and L.-Z. Gong, Org. Lett., 2010, 12, 4050 CrossRef PubMed;
(d) C. Guo, J. Song and L.-Z. Gong, Org. Lett., 2013, 15, 2676 CrossRef PubMed;
(e) L. He, X.-H. Chen, D.-N. Wang, S.-W. Luo, W.-Q. Zhang, J. Yu, L. Ren and L.-Z. Gong, J. Am. Chem. Soc., 2011, 133, 13504 CrossRef PubMed.
- M. Hatano, T. Maki, K. Moriyama, M. Arinobe and K. Ishihara, J. Am. Chem. Soc., 2008, 130, 16858 CrossRef PubMed.
-
(a) N. Momiyama, T. Konno, Y. Furiya, T. Iwamoto and M. Terada, J. Am. Chem. Soc., 2011, 133, 19294 CrossRef PubMed;
(b) N. Momiyama, T. Narumi and M. Terada, Chem. Commun., 2015, 51, 16976 RSC;
(c) N. Momiyama, K. Funayama, H. Noda, M. Yamanaka, N. Akasaka, S. Ishida, T. Iwamoto and M. Terada, ACS Catal., 2016, 6, 949 CrossRef.
- N. Momiyama, H. Tabuse, H. Noda, M. Yamanaka, T. Fujinami, K. Yamanishi, A. Izumiseki, K. Funayama, F. Egawa, S. Okada, H. Adachi and M. Terada, J. Am. Chem. Soc., 2016, 138, 11353 CrossRef PubMed.
- Terada reported a pioneering work on the enantioselective aza-FC reaction of 2-methoxyfuran with aldimines and ketimines by chiral phosphoric acid catalysts. See:
(a) D. Uraguchi, K. Sorimachi and M. Terada, J. Am. Chem. Soc., 2004, 126, 11804 CrossRef PubMed;
(b) A. Kondoh, Y. Ota, T. Komuro, F. Egawa, K. Kanomata and M. Terada, Chem. Sci., 2016, 7, 1057 RSC.
- Shao recently reported catalytic enantioselective aza-FC-type reactions with alkynyl aldimines by using chiral phosphoric acid catalysts. Y. Wang, L. Jiang, L. Li, J. Dai, D. Xiong and Z. Shao, Angew. Chem., Int. Ed., 2016, 55, 15142 CrossRef PubMed.
- Substrates 1 would have an E-geometry. See the ESI† for details. Also see our previous report: M. Hatano, K. Yamashita, M. Mizuno, O. Ito and K. Ishihara, Angew. Chem., Int. Ed., 2015, 54, 2707 CrossRef PubMed.
- The preparation of (R)-5a is described in detail in the ESI.†.
- To consider the acidity of the catalysts, we performed preliminary theoretical calculations in a working model by using molecular electrostatic potential (MEP). As a result, even among the same simple units, the conjugated double hydrogen bond network of bis(phosphoric acid)s showed higher acidity than the corresponding normal (phosphoric acid)s. See the ESI† for details.
- Other optimizations, such as for the solvent, reaction temperature, concentration, drying agents, etc., are shown in the ESI.†.
- A linear relationship was observed for less Brønsted-basic (R)-4b, which might act as a monomeric catalyst. See the ESI† for details.
-
(a) N. Li, X.-H. Chen, S.-M. Zhou, S.-W. Luo, J. Song, L. Ren and L.-Z. Gong, Angew. Chem., Int. Ed., 2010, 49, 6378 CrossRef PubMed;
(b) M. R. Monaco, G. Pupo and B. List, Synlett, 2016, 27, 1027 CrossRef.
- Indeed, ESI-MS analysis of (R)-5b showed an almost exclusive peak at m/z = 957.2763, which might be a monomer species as [M − H]−. In contrast, achiral phosphoric acids and conventional chiral phosphoric acids showed some peaks, which might be monomer as well as dimer, trimer, tetramer, and so on. See the ESI† for details.
- The preparation of (R)-10c is described in detail in the ESI.†.
- D. G. Manly and E. D. Amstutz, J. Org. Chem., 1956, 21, 516 CrossRef.
- T. Hashimoto, K. Yamamoto and K. Maruoka, Chem. Lett., 2011, 40, 326 CrossRef.
- The structure of 19a was confirmed by X-ray analysis. A transformation of the Diels–Alder adduct to 1,4-naphthoquinol-derived 1,3-oxazinan-2-one 19a involved a completely stereoselective H+-promoted 1,2-migration. See the ESI† for details.
-
(a) J. E. Saxton, Nat. Prod. Rep., 1993, 10, 349 RSC;
(b) S. E. O'Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532 RSC.
-
(a) N. L. Dirlam, B. S. Moore and F. J. Urban, J. Org. Chem., 1987, 52, 3587 CrossRef;
(b) A. Küpfer, R. Patwardhan, S. Ward, S. Schenker, R. Preisig and R. A. Branch, J. Pharmacol. Exp. Ther., 1984, 230, 28 Search PubMed;
(c) A. Olma, Polish J. Chem., 1996, 70, 1442 Search PubMed;
(d) J. A. Marco, M. Carda, J. Murga, F. González and E. Falomir, Tetrahedron Lett., 1997, 38, 1841 CrossRef. Also see a review.
(e) C. Cativiela and M. D. Díaz-de-Villegas, Tetrahedron: Asymmetry, 1998, 9, 3517 CrossRef.
-
(a) F. Köhler, H.-J. Gais and G. Raabe, Org. Lett., 2007, 9, 1231 CrossRef PubMed;
(b) H. Liu, J. Xu and D.-M. Du, Org. Lett., 2007, 9, 4725 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization data, additional control experiments, copies of 1H NMR and 13C NMR spectra of all new compounds. CCDC 1520624, 1520625, 1834631 and 1834632. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02290a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Haruka
Okamoto
a,
Taro
Kawakami
a,
Kohei
Toh
a,
Hidefumi
Nakatsuji
a,
Akira
Sakakura
a,
Haruka
Okamoto
a,
Taro
Kawakami
a,
Kohei
Toh
a,
Hidefumi
Nakatsuji
a,
Akira
Sakakura