
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of a diagnostic structural motif reveals a new reaction intermediate and condensation pathway in kraft lignin formation†
Christopher S.
Lancefield
 a,
Hans L. J.
Wienk
b,
Rolf
Boelens
b,
Bert M.
Weckhuysen
a and
Pieter C. A.
Bruijnincx
*ac
a,
Hans L. J.
Wienk
b,
Rolf
Boelens
b,
Bert M.
Weckhuysen
a and
Pieter C. A.
Bruijnincx
*ac
aInorganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands. E-mail: p.c.a.bruijnincx@uu.nl
bNMR Spectroscopy, Bijvoet Center for Biomolecular Research, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands
cOrganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584 CG Utrecht, The Netherlands
First published on 11th July 2018
Abstract
Kraft lignin, the main by-product of the pulping industry, is an abundant, yet highly underutilized renewable aromatic polymer. During kraft pulping, the lignin undergoes extensive structural modification, with many labile native bonds being replaced by new, more recalcitrant ones. Currently little is known about the nature of those bonds and linkages in kraft lignin, information that is essential for its efficient valorization to renewable fuels, materials or chemicals. Here, we provide detailed new insights into the structure of softwood kraft lignin, identifying and quantifying the major native as well as kraft pulping-derived units as a function of molecular weight. De novo synthetic kraft lignins, generated from (isotope labelled) dimeric and advanced polymeric models, provided key mechanistic understanding of kraft lignin formation, revealing different process dependent reaction pathways to be operating. The discovery of a novel kraft-derived lactone condensation product proved diagnostic for the identification of a previously unknown homovanillin based condensation pathway. The lactone marker is found in various different soft- and hardwood kraft lignins, suggesting the general pertinence of this new condensation mechanism for kraft pulping. These novel structural and mechanistic insights will aid the development of future biomass and lignin valorization technologies.
1 Introduction
Lignin is the most abundant renewable source of aromatic carbon on earth, offering great potential as a raw material for the sustainable production of aromatic chemicals and materials.1–4 Lignin is primarily obtained from lignocellulosic biomass during the preparation of cellulose pulps (pulping) used, for example, to make paper. Currently, the dominant pulping technology is the kraft process, which accounts for 98% of all pulp production (130 Mtpa) resulting in 60 Mtpa of extracted kraft lignin.5 Historically, most kraft lignin has been burnt on-site at pulp mills to provide process energy and for process chemicals recovery;6 however, efficiency improvements and newly introduced technologies such as LignoBoost, LignoForce and SLRP7–9 now allow for the isolation of large volumes of kraft lignin from commercial pulp mills ready for further valorization. At the moment, there are no commercial processes that transform kraft lignin into aromatic chemicals or fuels,1 even though many efforts are directed at kraft lignin depolymerization.10–15 A major impediment to valorization is the highly complex and relatively unknown chemical structure, which has clearly hampered the development of such processes.For a long time, the same could be said of native lignins, but recent efforts, especially using 2D HSQC NMR, have greatly advanced the structural understanding of this material,16–22 in turn allowing for the development of highly selective depolymerization processes.1,23–30 While the native lignin structure is now regarded as complex, but largely understood, our understanding of technical, isolated lignins such as kraft has not followed suit.
One of the first qualitative structural schemes for kraft lignin was proposed by Marton in 1971.31 This structure was proposed based on data accumulated from chemical and spectroscopic analyses of isolated kraft lignins and extensive model compound studies, with a stated understanding that this structure was not to be considered a reliable structural formula for kraft lignin, but was simply consistent with the knowledge at the time and should be refined as new data became available. In the following years significant efforts have been dedicated to achieving this refinement and improving our general understanding of this material. For example, model compound studies have been used to investigate the reactivity of most of the major native lignin linkages (β-O-4, β-5, β–β)33–36 and intermediary structures (epoxides, thiiranes, coniferyl alcohol)37–41 thought to form during kraft pulping and to elucidate some of the key chemical mechanisms underpinning their reactivity both in terms of linkage cleavage and the subsequent condensation chemistry that is the origin of kraft lignin's chemical recalcitrance. In general these studies have reaffirmed the theory that sulfide mediated cleavage of the most abundant β-O-4 linkage in lignin is responsible for the great success of wood delignification by kraft pulping, but also that this is a very complex chemical process.
In addition, many different analytical methods have been employed to study isolated kraft lignins. For example acetylation with aminolysis,42 permanganate oxidation,43 mannich reaction,44 aqueous titration,45 FTIR,45 and oximation with UV-spectroscopy.46 More recently NMR methods have come to the fore in kraft lignin analysis, with 1D 13C,47–5131P NMR,51–53 DOSY54,55 and 2D HSQC/HMQC,32,56–62 methods being used to study the kraft lignin structure and functional group abundance. These analyses have shown that the kraft lignin structure is indeed very different to in planta lignin, with significantly reduced levels of native lignin linkages and increased levels of phenolic groups, consistent with results from other analytical methods and modes of reactivity proposed based on model compound studies.
Recently, an updated kraft lignin structure has being proposed by Crestini and Argyropoulos et al. following extensive analysis of kraft lignins using a combination of lignin fractionation, quantitative 13C and HSQC NMR. In contrast to Marton's proposed structure which suggested the operation of carbanion mediated condensation processes, this work invoked quite extensive radical coupling chemistry (C–C and C–O bond formation) to explain the features of kraft lignin (Fig. 1).32 Despite these efforts and many other recent valuable contributions,59,60,63 still only a relatively small fraction of the kraft lignin structure can be assigned to actual chemical structures or linkages. In fact, although quantification is notoriously difficult and methods used for (semi-)quantification vary somewhat in the literature, we estimate that less than 20% of the kraft lignin structure can currently be assigned to actual chemical structures or linkages (Tables S1–S5†).
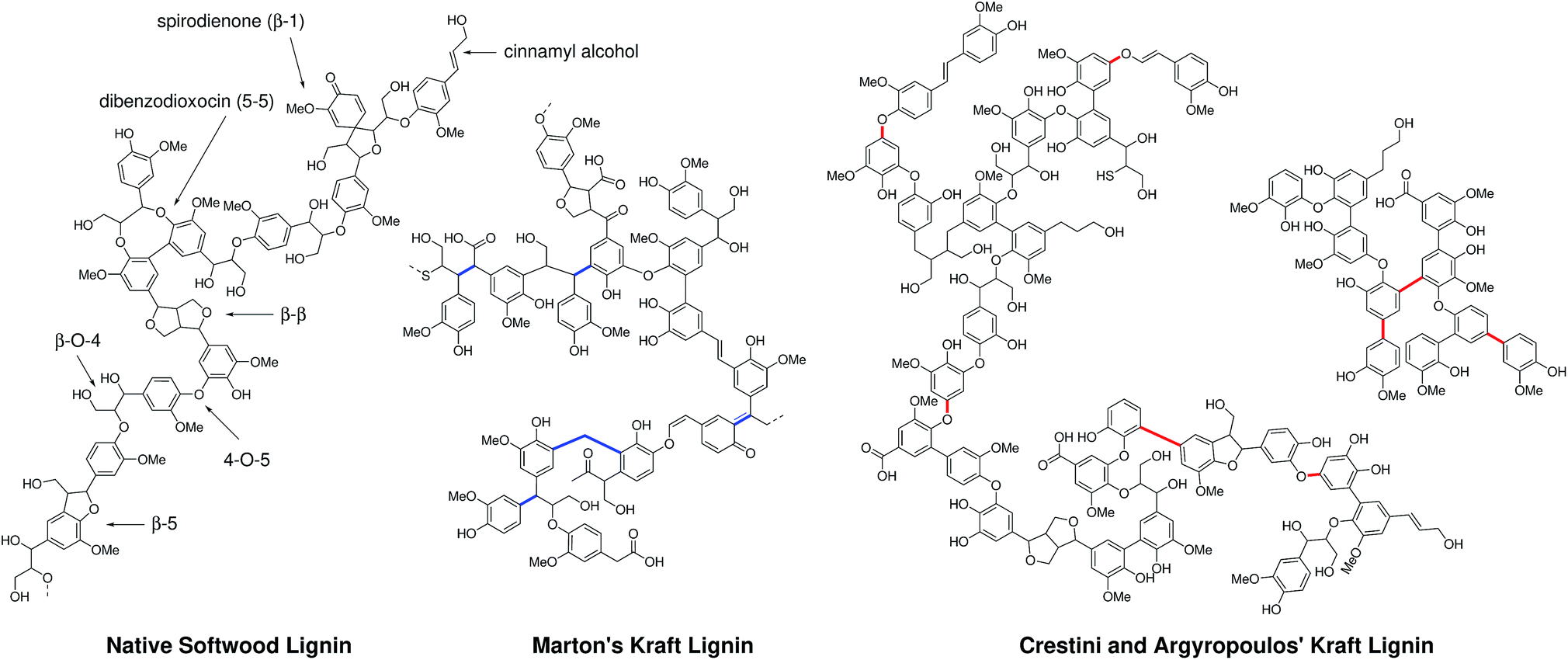 | ||
| Fig. 1 A representation of the structure of a section of native lignin and previously proposed constitutional structural schemes for softwood kraft lignins from Marton31 and Crestini and Argyropoulos et al.32 Non-natural bonding motifs are highlighted in blue (resulting from carbanion-type condensation) and red (radical-type condensation). | ||
This prompted us to examine the kraft lignin structure in more detail following a multipronged approach involving state of the art 2D NMR analysis of crude and fractionated Indulin AT, the prototypical kraft lignin, combined with de novo synthetic kraft synthesis from high fidelity models, including polymeric and isotopically labelled model compounds. Together these analyses provided unprecedented insight into the kraft lignin structure and experimentally validated some of the pathways involved in its formation. This included the first identification of a homovanillin pathway as a significant mode of reactivity for β-O-4 linkages during kraft pulping that is operational next to the established condensation route involving coniferyl alcohol. Based on the identification of a novel lactone species as key condensation product and the analysis of a range of industrial kraft lignins, we show that our findings appear to be widely applicable.
2 Results and discussion
2.1 NMR characterization of kraft lignin and its fractions
The current model of kraft lignin formation proposes that the most important reaction is cleavage of β-aryl ethers, which make up 45–84% of native lignins,1via formation of quinone methides and subsequent sulfide mediated bond cleavage (Scheme 1).34,64 The formed thiirane intermediate 1 is thought to generate coniferyl alcohol (2), via extrusion of S0, which then undergoes poorly understood condensation reactions.37 The fate of the remaining 16–55% of native lignin, which is composed of a range of different linkages, during kraft pulping has been mostly established in model systems35,36,64 but not necessarily always verified or assigned in an actual kraft lignin. Thus, to increase our understanding of the kraft lignin structure it is necessary to assign both remaining native and new kraft-derived linkages resulting from reactions of the major and minor structural elements of lignins.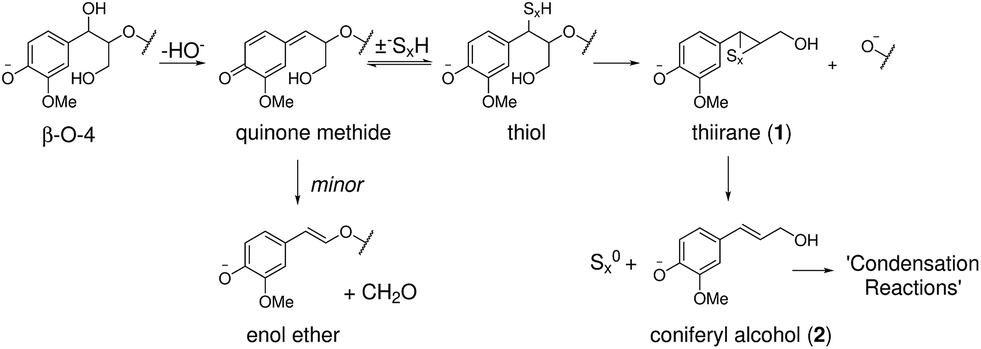 | ||
| Scheme 1 Previously proposed pathway for β-aryl ether cleavage during the kraft process.64 | ||
NMR has proven to be a particularly powerful tool for lignin analysis and for elucidating new structures, with 2D HSQC NMR experiments providing particularly excellent sensitivity and resolution.16,65 We therefore carried out a detailed assignment of the HSQC spectrum of Indulin AT lignin (Fig. 2), allowing for the assignment of 19 different native or kraft-derived lignin units, including, to the best of our knowledge, the first definitive assignments, in an actual kraft lignin sample, of β-5-derived stilbenes (SB5, Fig. S2†), aryl β-hydroxy propanoic acids (HA, Fig. S3†) and reduced β-O-4 structures (Ar, Fig. S4†), all assigned here based on comparison to synthesised model compounds, and diarylmethanes (DAM, Fig. S7†) and arylglycerols (AG, Fig. S9†), which were assigned based on previously reported data for formaldehyde condensed66 and native lignins67,68 respectively. The presence of these newly identified groups is chemically very revealing, for example, DAM's are formed from phenol–formaldehyde reactions,64 which were once thought to be important condensation reactions in kraft pulping47,64,69–72 and HA's are formed through lignin-carbohydrate condensation reactions and the structure identified here is likely to be only one of a number of related compounds resulting from these reactions.73 The presence of Ar's finally implies that formal reduction of quinone methide type intermediates is possible under the pulping conditions.74 Whilst such new assignments in kraft lignin are mechanistically revealing, the AG, HA, DAM and Ar structures are all present in only very small quantities (vide infra) suggesting none represent particularly major reaction pathways. Additionally, we did not find any evidence for a number of previously assigned kraft lignin structural units, including cinnamaldehyde32 and mandelic acid32,60,62,75 groups. Based on our analysis of the reported chemical shifts for these units compared to model compound data collected here and previously,76 these groups appear to have been misassigned previously, highlighting the challenges involved in characterizing this material (Table S1–S5†). Another important observation is that the HSQC cross peak at δH/δC = 4.75 ppm/81.3 ppm is mostly associated with epiresinol structures, rather than dibenzodioxocins77 or lignin-carbohydrate complexes,32,62,78 as previously reported.
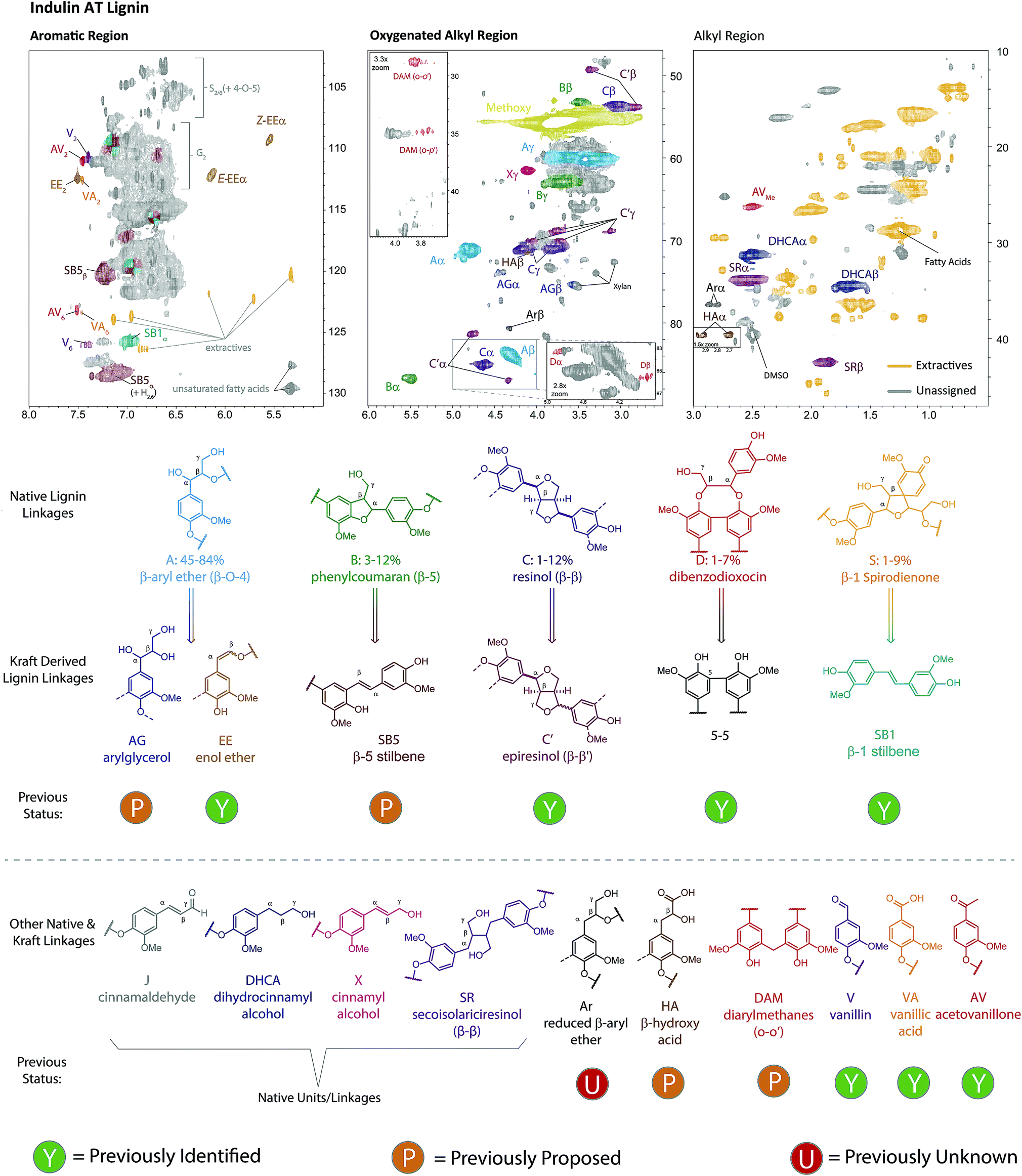 | ||
| Fig. 2 Assignment of the 2D HSQC NMR spectrum of unfractionated Indulin AT kraft lignin. The structures are used to show the native linkages and their kraft-derived products. Structures and contours are color coded to aid interpretation. See Fig. S1–S10† for more detailed assignments of structural units and Fig. S11† for HMBC analysis supporting the assignment of S2/6 units. | ||
To gain further chemical and structural insight a semi-quantitative analysis of HSQC signal intensities was undertaken. To achieve this a defined lignin cross peak in the HSQC spectrum is used as an internal reference, against which a relative, hence semi-quantitative, assessment of structural unit abundance can be made based on volume integration of other characteristic cross peaks. Various different approaches have been used previously to achieve this, including using the whole aromatic region79 or methoxy group60,62 as the reference signals. Here we chose to use the aromatic G2 (and S2/6) peaks as the reference80 as these positions should be relatively unreactive during pulping owing to the high pH of the process. Indeed, quantitative 13C NMR analysis of (fractionated) Indulin AT suggests this to be a reasonable if not perfect assumption, with this integral accounting for approximately 0.80 carbons per C9 unit (Fig. S11–S12† and associated discussion), implying that HSQC linkage quantification results discussed present a small overestimation (ca. 20%). Additional analysis of the C–O and OMe regions relative to the total aromatic region also suggested that neither radical coupling to form diaryl ethers or demethylation reactions are major pathways, at least in the formation of Indulin AT kraft lignin.
A further complication of using HSQC experiments is that differences in, for example, 1JCH couplings or T1 and T2 relaxation times all affect quantification. For single molecules, two-dimensional extrapolated time-zero 1H–13C HSQC (HSQC0) experiments can correct for all these factors,81–83 however the difference in T2 relaxations resulting from having components of different molecular weights84 in the same sample is not corrected for. Consequently, due to the high polydispersity of kraft lignin, quantification is still biased towards lower molecular weight components. For this reason, Indulin AT kraft lignin was first fractionated using a solvent fractionation method consisting of sequential extraction of the crude lignin with increasingly polar EtOAc/MeOH solvent mixtures, giving 7 distinct molecular weight fractions with considerably lower polydispersities than the starting lignin (Fig. 3). These were then analyzed using both a standard HSQC pulse sequence and a HSQC0 experiment for comparison. Interestingly, both methods gave reasonably similar results for all fractions and units, with 69% of measurement pairs within ±20% of each other and only 6% being more than 30% different (Fig. 4a). Comparison of the abundances determined for each linkage type using the two methods (Fig. S14–S25†), shows that units such as β-O-4, β-5, β-1 stilbenes (SB1) and cinnamyl alcohols (X) give very similar results, whilst units such as secoisolariciresinols (SR) and dihydrocinnamyl alcohols (DHCA) appear to be systematically overestimated by the standard HSQC experiment, however the absolute errors are still quite small and the trends observed in the abundances are the same. Overall this suggests, in our case at least, that the errors associated with using a standard HSQC pulse sequence and short relaxation delay (1 s) is probably quite small, especially considering the practical challenges associated with collecting HSQC0 data for technical lignins.85
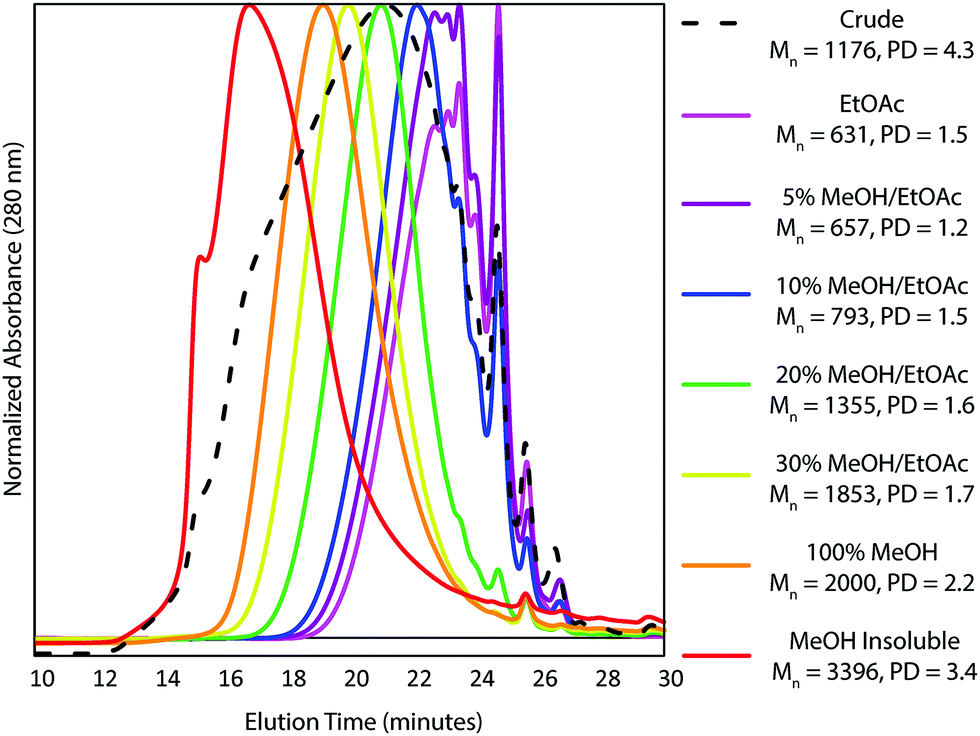 | ||
| Fig. 3 Normalized GPC traces, Mn values and polydispersities (PD) of the crude (dotted) and fractionated (solid colored) Indulin AT kraft lignin samples. | ||
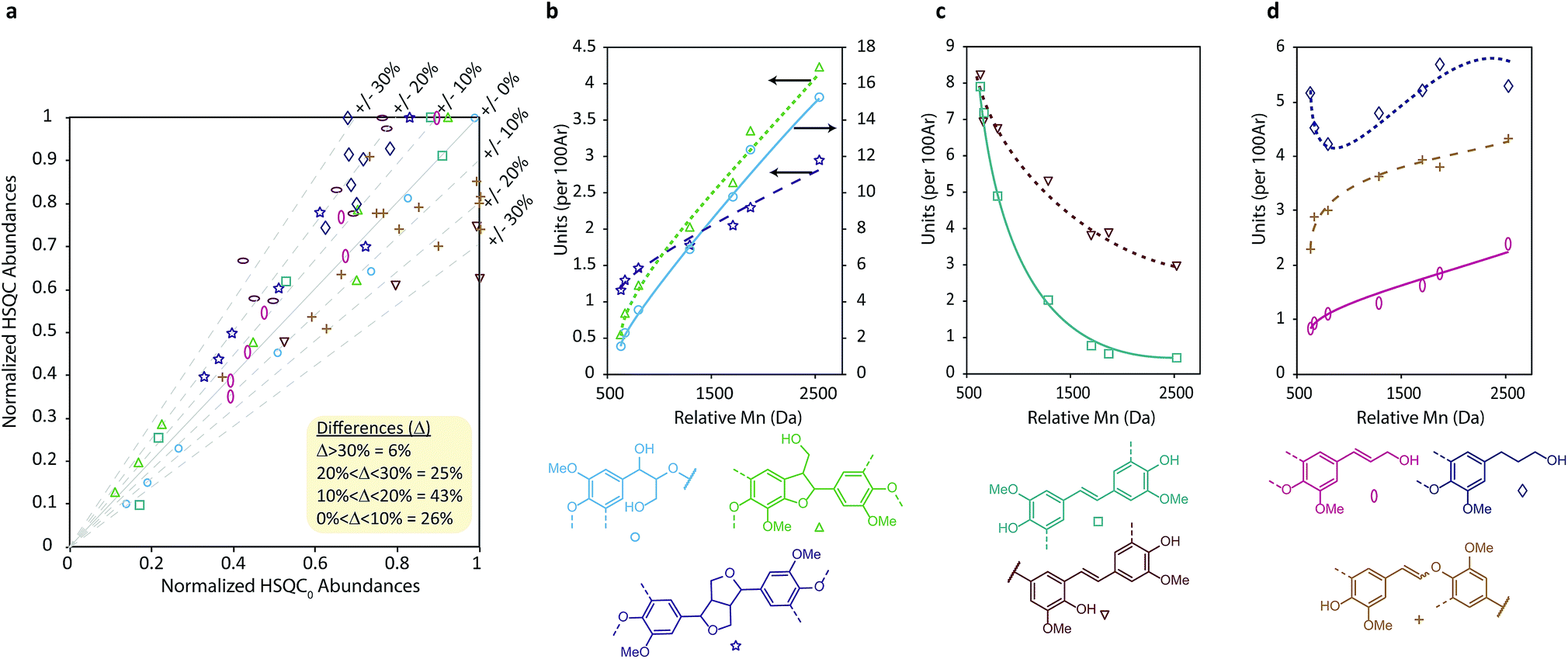 | ||
| Fig. 4 (a) Comparison of normalized lignin unit values across the fractions obtained from HSQC and HSQC0 experiments demonstrating the correlation between the two techniques. Points are color coded to match individual units and each point represents a matched pair of HSQC and HSQC0 measurements. Data was normalised relative to the largest abundance value for each linkage. (b) Abundances of native linkages, (c) kraft-derived stilbenes and (d) lignin end units in the different molecular weight lignin samples obtained from solvent fractionation of Indulin AT Kraft lignin. Lines have been added only to highlight trends and are not regression lines. | ||
In line with recent reports, this HSQC analysis revealed that lignin unit abundance depends heavily on the molecular weight of the kraft lignin fraction and that the structure is significantly modified in all fractions compared to native lignin (Fig. 4b–d).32,60,62 Native linkages were found to be most abundant in higher molecular weight fractions (Fig. 4b) whilst kraft-derived units, specifically stilbenes, were found to be most abundant in the lower molecular weight fractions (Fig. 4c), indicating that depolymerization outweights repolymerization during kraft pulping. Additionally, β-5 stilbenes represent a source of new C5-substituted (condensed) phenols in kraft lignin. Their change in abundance with molecular weight detected by HSQC can therefore help rationalize similar trends observed in 31P NMR analysis for condensed phenolic groups (Fig. S28, Table S12†), without requiring the extensive aromatic repolymerization reactions previously suggested.32 We also found that kraft lignin end groups displayed distinct trends, although less pronounced than for other units, with both cinnamyl alcohol and enol ether groups being most abundant in higher molecular weight fractions (Fig. 4d). This likely reflects the fact that phenolic cinnamyl alcohols are unstable during the kraft process,38 meaning those observed in kraft lignin are likely to be etherified and therefore associated with the lower phenol content, higher molecular weight fractions. In contrast, once formed, enol ethers are thought to be relatively stable,86,87 slowing the depolymerization process and resulting in the observed molecular weight dependence. Additionally, the large number of lignin fractions isolated and analyzed here allowed us to show that the change in kraft lignin structure is continuous across the molecular weight range (see ESI Section 6.3 for more detailed discussions†). This observation differs from previous reports using fewer fractions to suggest that the lower molecular weight kraft lignin fractions are all homogeneous in structure.32
Furthermore, we can start to interpret the observed molecular weight dependences with respect to the chemistry of the kraft process. For example, (phenolic) β-O-4 units are known to be cleaved during the process and so should naturally be depleted in the lower molecular weight (i.e. most cleaved) fractions which is indeed what is observed. β-5 units on the other hand are not cleaved during the process but, if phenolic, do form stilbenes (SB5). Thus, β-5 units should also be depleted in the lowest molecular weight fraction (highest in phenolic groups) but their derived stilbenes should be most abundant, which is also what is found. Similarly, if β–β units are phenolic then they can undergo base catalysed epimerisation to generate an equilibrium mixture of diastereomers, resulting in fewer resinols but more epiresinols in the lower molecular weight fractions. Contrary to initial expectations, in the very lowest molecular weight fractions the abundances of epiresinols actually falls again (Fig. S18†). This, however, is consistent with observations from other degradative analysis methods which have found that resinols are poorly released from softwood lignins88,89 indicating that most (but not all) resinols are incorporated into the lignin through chemically resistant linkages and so most are released as trimers or higher order oligomers limiting their abundance in the lowest molecular weight fractions. Based on their greater molecular weight dependence, β-1 stilbenes (SB1) appear to be much more easily released from the lignin polymer during the kraft process than other units, accumulating in the lower molecular weight fractions. Again, similar observations have been made during thioacidolysis of softwood lignins where β-1 derived products appear to be overrepresented in the dimeric fraction based on the apparent abundance of β-1 units in the parent lignin.89 As an explanation, it has been proposed that because β-1 units can only be etherified at one phenolic end during lignification, the probability of releasing a dimeric unit during depolymerisation processes is higher than for other units which require two suitably cleavable uncondensed linkages to be present.89
The extensive NMR assignment of the kraft lignin undertaken in this study allowed for significantly more of its structure to be assigned than in previous studies.32,59,60,62 By combining the lignin fraction yields with the HSQC NMR results (Tables S6 and S7†), molecular weight (MW) adjusted abundances of each lignin unit could be calculated (Table 1).62 This showed that native type lignin units account for 27% and kraft-derived units for 15% of all aromatic groups in Indulin AT lignin, together accounting for approximately double previous assignments (see Tables S1–S5†), even considering the small overestimation anticipated from using this HSQC based method. Interestingly, the same NMR analysis of the as is (i.e. unfractionated) Indulin AT lignin yielded the considerably different figures of 23% and 18% for native and kraft-derived units, respectively, with a particularly large difference seen in the quantification of native β-O-4, β-5 and β–β (underestimated) and kraft stilbenes SB1 and SB5 (overestimated, Table 1). Given that the abundance of these units also showed the strongest molecular weight dependence and that HSQC0 experiments gave relatively similar quantification results (Fig. 4a), it suggests that the dependence of T2 relaxation times, and hence HSQC volume integrals, on molecular weight is indeed a significant factor that needs to be addressed when using HSQC to quantify lignin substructures, especially in technical lignins. This was further supported by analysis of the HSQC0-derived signal attenuation factors which showed increasing signal attenuation as the MW increases (Table S11, Fig. S27†).
| Unita | Spruce CEL | Indulin AT kraft lignin | ||
|---|---|---|---|---|
| Crude | MW adjustedd | MW adjusted + HSQC0d | ||
| a See Fig. 2 for structures. b Determined in acetylated lignin. c Symmetrical units C, C′ and SB1 counted as 2 aromatic units each. d AG, V, AV, HA, Ar units were not included in the fractionation analysis due to their low abundance making quantification challenging. | ||||
| A | 38.5 | 7.0 | 9.8 | 10.3 |
| B | 13.0 | 1.6 | 2.8 | 2.6 |
| C | 3.0 | 1.6 | 2.1 | 1.8 |
| D | 2.4 | 0.2 | — | — |
| X | 5.8 | 1.8 | 1.7 | 1.6 |
| SR | 1.0 | 2.1 | 2.0 | 1.8 |
| DHCA | 5.2 | 4.7 | 4.6 | 4.1 |
| J | 3.7 | 0.0 | 0.0 | 0.0 |
| S | 2.9 | 0.0 | 0.0 | 0.0 |
| C′ | 0.0 | 0.5 | 0.6 | 0.6 |
| Z-EE | 0.0 | 1.1 | 1.0 | 1.0 |
| E-EE | 0.0 | 2.8 | 2.5 | 3.1 |
| SB1 | 0.0 | 2.7 | 1.5 | 1.9 |
| SB5 | 0.0 | 6.5 | 3.8 | — |
| AG | 1.4 | 2.2b | 3.8b | — |
| V | 1.4 | 0.8 | — | — |
| AV | 0.0 | 0.7 | — | — |
| VA | 2.0 | 0.4 | — | — |
| HA | 0.0 | 0.8 | — | — |
| Ar | 0.0 | 0.5 | — | — |
| Totalc | 84.3 | 45.3 | 38.6 | 35.2 |
The NMR analysis above showed that a considerable portion of kraft lignin can be structurally assigned, with the trends in linkage and end group abundance seen for the fractions providing detailed chemical insight into the kraft delignification process. It also showed, however, that more than 50% of the lignin structure is still left unaccounted for. By comparing linkage abundances in kraft lignin and native lignin it becomes clear that a significant portion of the unaccounted structure must result from reactions of the most abundant β-O-4 linkage, which makes up 39% of the aromatic units in native lignin (Spruce CEL) but only 17% in kraft lignin, inclusive of its enol ether and arylglycerol derivatives (Table 1).
2.2 Synthetic kraft studies demonstrate two mechanistically distinct cleavage and condensation regimes
To study the fate of β-O-4 linkages in more detail, we turned to the de novo synthesis of synthetic kraft lignin. As the major kraft reaction pathway for the β-O-4 linkage requires a phenolic group (i.e. most likely involves end groups), phenolic dimeric and polymeric model compounds provide ideal entry points to study kraft chemistry as they should generate the same reactive intermediates as real lignin (Scheme 2). The reaction of phenolic dimeric β-O-4 model (3) under kraft pulping conditions was studied first. This compound has previously been used to study kraft chemistry,34,39,40,86,90 but modern 2D NMR methods were not yet available or applied at the time to analyze the complex product mixtures obtained, making this reaction worthy of revisiting.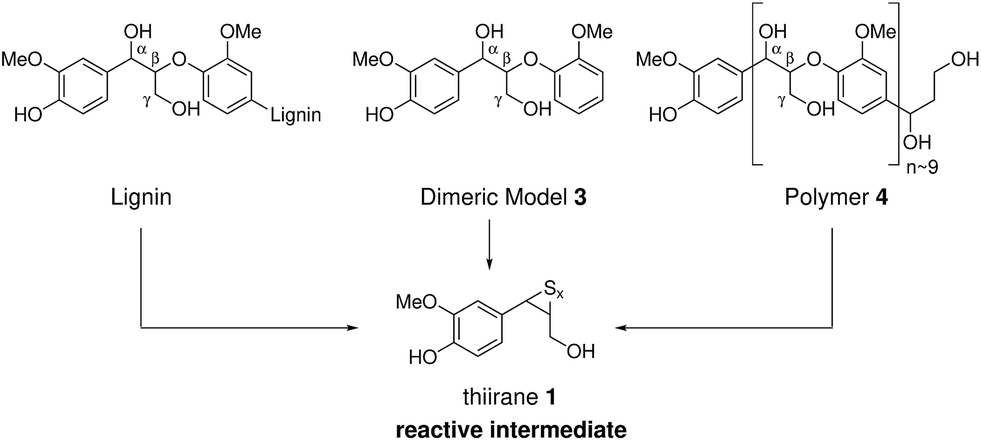 | ||
| Scheme 2 Phenolic β-O-4 models used in this study. | ||
Using conditions that closely mimic the kraft process, namely the use of relatively low NaOH concentrations (0.12 M) in the white liquor and a temperature of 170 °C, 3 was subjected to kraft pulping conditions under both dilute and concentrated substrate concentrations. These conditions were chosen to model the early and late stages of the pulping process when different concentrations of lignin will be present in the pulping liquor. In both cases complex reaction mixtures were obtained (Fig. 5a and c) with relative GPC Mn values indicating formation of oligomeric but not extensively polymerized species. Interestingly, the HSQC spectra of the two reaction mixtures are rather different, demonstrating a substrate concentration dependence of the dominant kraft chemistry pathways.
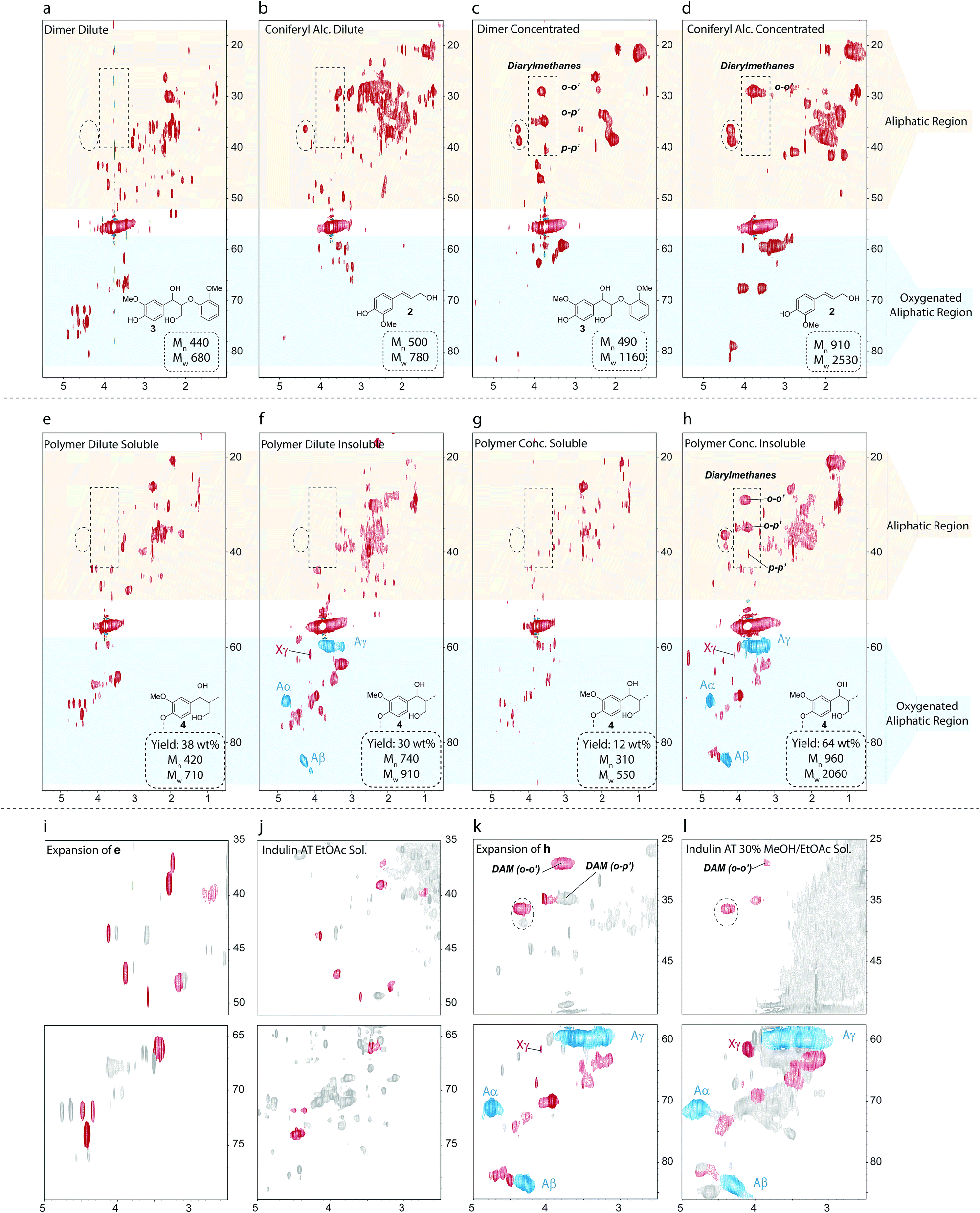 | ||
| Fig. 5 1H–13C HSQC spectra of the reaction products obtained from kraft treatment of (a–d) guaiacylglycerol-β-guaiacyl ether (3) and (e–h) a β-O-4 phenolic model polymer (4). (i–l) Comparisons of selected regions of the HSQC NMR spectra from the reactions of the model compounds and of kraft lignin fractions. The structure in each panel shows the starting materials used to generate each synthetic kraft lignin. All Mn and Mw values are rounded to the nearest 10. Starting polymer Mn = 1490, Mw = 2780. The red colored contours in (i–l) indicate similarities between spectra. Blue and pink contours represent β-O-4 linkages and cinnamyl alcohol groups, respectively. The cross peak at δH/δC = 4.4/36.3 ppm (highlighted by the dashed circle) appears to be particularly diagnostic for a coniferyl alcohol pathway. | ||
Importantly, comparison of the HSQC spectra of the reaction mixtures with Indulin kraft lignin showed several structural similarities in the oxygenated alkyl and aliphatic regions (Fig. S32†), highlighting that both the choice of model and the reaction conditions selected really are relevant to the kraft process. Current theory suggests most β-O-4 degradation proceeds through a coniferyl alcohol type intermediate and therefore the reaction products from the β-O-4 models should logically resemble those obtained from the kraft treatment of 2. Indeed, this turns out to be at least partly true, but only under concentrated conditions, with several similar cross peaks being observed in the aliphatic region in both cases (cf.Fig. 5c and d). Under dilute conditions, however, the synthetic kraft products obtained from 3 and 2 were quite different (cf.Fig. 5a and b), suggesting that 2 is not a major intermediate at the early stages of pulping.
Model 3 thus provided valuable insight, but has its limitations in mimicking the chemistry of polymeric native lignin during kraft pulping as it offers an unrealistically high proportion of phenols and releases guaiacol upon cleavage, which has an additional reactive position on the aromatic ring. The polymeric β-O-4 model (4) better captures the complexity of lignin5,91 and was subjected to kraft pulping conditions for the first time. Two distinct fractions, consisting of acidic water insoluble and soluble products, could be obtained upon work-up. For both the dilute and concentrated cases, the soluble products (Fig. 5e and g) were very similar to those obtained from 3, sharing the same structural similarities with kraft lignin (Fig. 5i and j). The HSQC spectra of the water insoluble products were, however, quite different (Fig. 5f and h) but shared many additional similarities with kraft lignin not seen using 3 (Fig. 5k and l), reflecting the greater fidelity and utility of such models in terms of overall correspondence with the structural features of real lignin. As in actual kraft lignin, small amounts of β-O-4 units were still found in the insoluble products, indicating the formation of new linkages that block further depolymerization during the reaction. As found for 3, only a high substrate concentration of polymer 4 yielded a product mixture similar to the coniferyl alcohol reaction (c.f.Fig. 5d and h). Another notable feature is that o-p′-diarylmethanes are detected in the polymer-derived products, indicating that free guaiacol type species are generated, probably via retro-aldol reactions, as previously suggested.64 This helps to explain the trace amounts of these units that are observed in kraft lignin (Fig. 2). Surprisingly, small amounts of cinnamyl alcohols could also be observed in these polymer reactions. As coniferyl alcohol (2), a free phenolic cinnamyl alcohol, is highly reactive under the applied conditions, we believe the observed cinnamyl alcohol signatures have originated from etherified units, which would give stable etherified cinnamyl alcohols. Indeed, treatment of an etherified β-O-4 model under the same conditions yielded small amounts of etherified (stable) cinnamyl alcohol, as well as arylglycerols (Fig. S33†), supporting this theory.
The advanced model-derived synthetic kraft lignins thus allowed two distinct kraft chemistry regimes to be identified. Under concentrated (late stage) conditions, coniferyl alcohol (2) appears to be an important intermediate, in line with current understanding of kraft pulping; however, under dilute conditions (early stage) an alternative pathway for cleavage and/or condensation operates that does not extensively involve 2. Given that signatures of both the dilute and concentrated regimes are found in the actual Indulin kraft spectra, mechanistic insight into this second, unknown regime and the pathways operating under these conditions need to be elucidated to further improve our understanding of kraft lignin structure.
2.3 Diagnostic new structural motif revealed a novel reaction intermediate
Previous investigations have shown the value in analyzing the low molecular weight compounds contained in kraft black liquors as a proxy to study kraft pulping chemistry, but such analyses are complicated by the highly complex nature of the mixtures obtained and the difficulty in determining the origin of each compound.92 For example, determining which compounds form from lignin or carbohydrate degradation, or identifying from which lignin linkage a compound originates, may not be trivial. In our model systems many of these ambiguities are removed, suggesting that new insights into this complex process could be obtained via this approach.Thus, purification of a synthetic kraft sample obtained from 3 yielded a chromatographically pure fraction containing two diastereomeric compounds. Detailed NMR analysis allowed assignment of these compounds as novel lactones 5a and 5b with the key NMR correlations used for their assignment shown in Scheme 3a. Based on quantitative 1H NMR analysis, the yield of these previously unidentified kraft components was only 3%, but they proved highly diagnostic for the identification of an alternative kraft formation pathway.
 | ||
| Scheme 3 (a) Formation of lactones 5a and 5b during kraft pulping of dimeric model 3. (b) Retrosynthetic analysis of 5. (c) A derivative of 5 previously identified in a kraft black liquor.92 | ||
Based on a retrosynthetic analysis of 5 (Scheme 3b) homovanillin (6) and formaldehyde (7) are proposed as its logical precursors, which themselves can form from the classical thiirane intermediate 1 by a retro-aldol and hydrolysis reaction. In the forward sense, the reaction is envisaged to proceed by condensation of formaldehyde with 6 followed by dehydration and 1,4-addition of a second homovanillin molecule. A formal intramolecular disproportionation reaction and lactonization upon acidic workup would give 5a/b.
As the formation of 5 requires a multistep, three-component reaction between highly reactive precursors, 6 and 7 must then be present in appreciable quantities during the reaction and this pathway could therefore be responsible for much of the chemistry observed under dilute reaction conditions. Indeed, de novo kraft lignin synthesis starting from 7 and acetyl homovanillin (8), used as a precursor which generates 6 during the reaction, not only resulted in the formation of lactones 5a and 5b, but also produced a synthetic kraft material remarkably similar to those obtained from the β-O-4 models, based on their HSQC NMR signatures (Fig. S34†).
Given that industrial kraft process conditions can vary quite substantially94 we were interested to see whether we could identify markers for this chemistry in a range of different industrial kraft lignins. Indeed, structures 5a and 5b could be identified, in small, but significant amounts (1–2 units per 100 Ar) in all 7 different kraft lignins examined by us (Fig. 6 and S36†). This includes both Indulin AT and BioChoice lignins (Fig. 6b and d) which are produced from softwoods but under very different pulping conditions,60 and lignins produced from hardwoods such as eucalyptus (Fig. 6c). Interestingly, compound 9 (Scheme 2c), a derivative of 5, had previously been isolated and identified from a kraft black liquor, however its origin at the time was unclear and was postulated to possibly arise from carbohydrate-lignin condensation.92 We can now say that 5 (and its derivative 9) constitute a novel, non-native and solely kraft process-derived lignin linkage, which serves as an important diagnostic marker for the operation of a homovanillin pathway in most, if not all, kraft pulping processes.
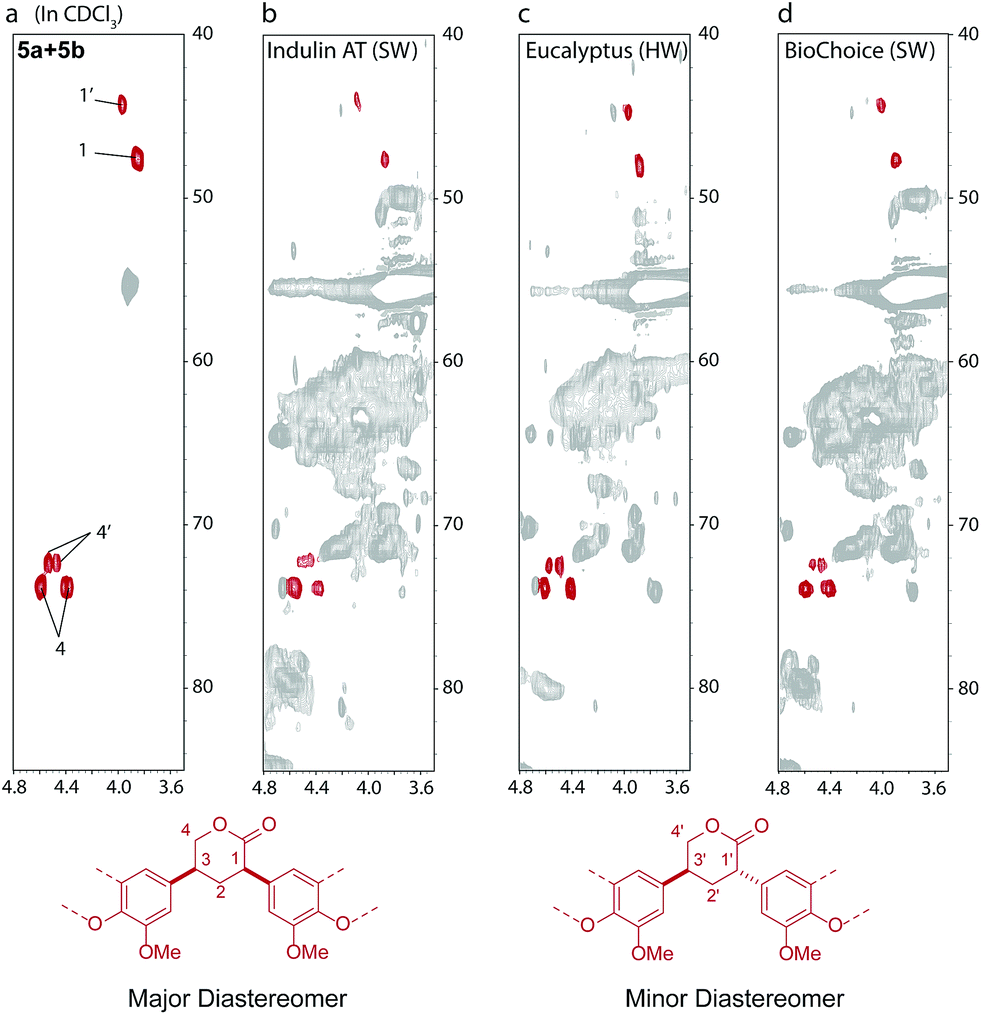 | ||
| Fig. 6 1H–13C HSQC spectra (in CDCl3) showing the identification of 5a and 5b type lactones in a range of different acetylated kraft lignins: (a) a mixture of 5a and 5b; (b) acetylated Indulin AT (mainly softwood origin, SW); (c) acetylated eucalyptus (hardwood origin, HW) and (d) acetylated BioChoice (SW) kraft lignins. | ||
2.4 13C labelled models show the fate of the β-O-4 linkage
As 2, 6 and 7 are all highly reactive species, multiple condensation pathways are available and expected to occur, in addition to the one leading to 5. The products of these condensation reactions would contribute to the current ‘structure gap’ in the description of kraft lignin. Given the anticipated complexity of condensation, β- and γ-13C labelled versions of dimeric model compound 3 were employed to track the fate of β-O-4 linkages by HSQC and HMBC NMR under dilute and concentrated kraft pulping conditions.The spectra illustrate the complexity and non-specificity of the condensation processes operating under the applied kraft pulping conditions (Fig. 7). For example, the HSQCs show that under dilute reaction conditions the β- and γ-label can be found in approximately 60+ and 100+ unique new protonated carbon chemical environments, respectively, based on manual peak analysis. For the β-label most cross peaks were associated with oxygenated aliphatic species (Fig. 7a), whilst the γ-label was found mainly in aliphatic species (Fig. 7b). In both cases, only small amounts of new aromatic/olefinic resonances (excluding enol ethers) were observed (Fig. S38†), suggesting that the formation of new mono or polyaromatic moieties containing these carbons is not a major reaction pathway. HMBC analysis additionally showed many correlations for the β-label in the carboxylic acid/ester region with a smaller number of carbonyl and aromatic/olefinic correlations. For the γ-label, most HMBC correlations were observed to carbons in the aliphatic region, with only a small number of correlations to carbonyls, carboxylic acid/esters and aromatic/olefinic carbons. These observations imply disproportionation reactions involving homovanillin, and its derivatives, generating carboxylic acids/esters and alcohols containing the β-label, together with extensive carbanion-mediated condensation reactions involving formaldehyde and homovanillin, which can generate the observed aliphatic and oxygenated alkyl structures.
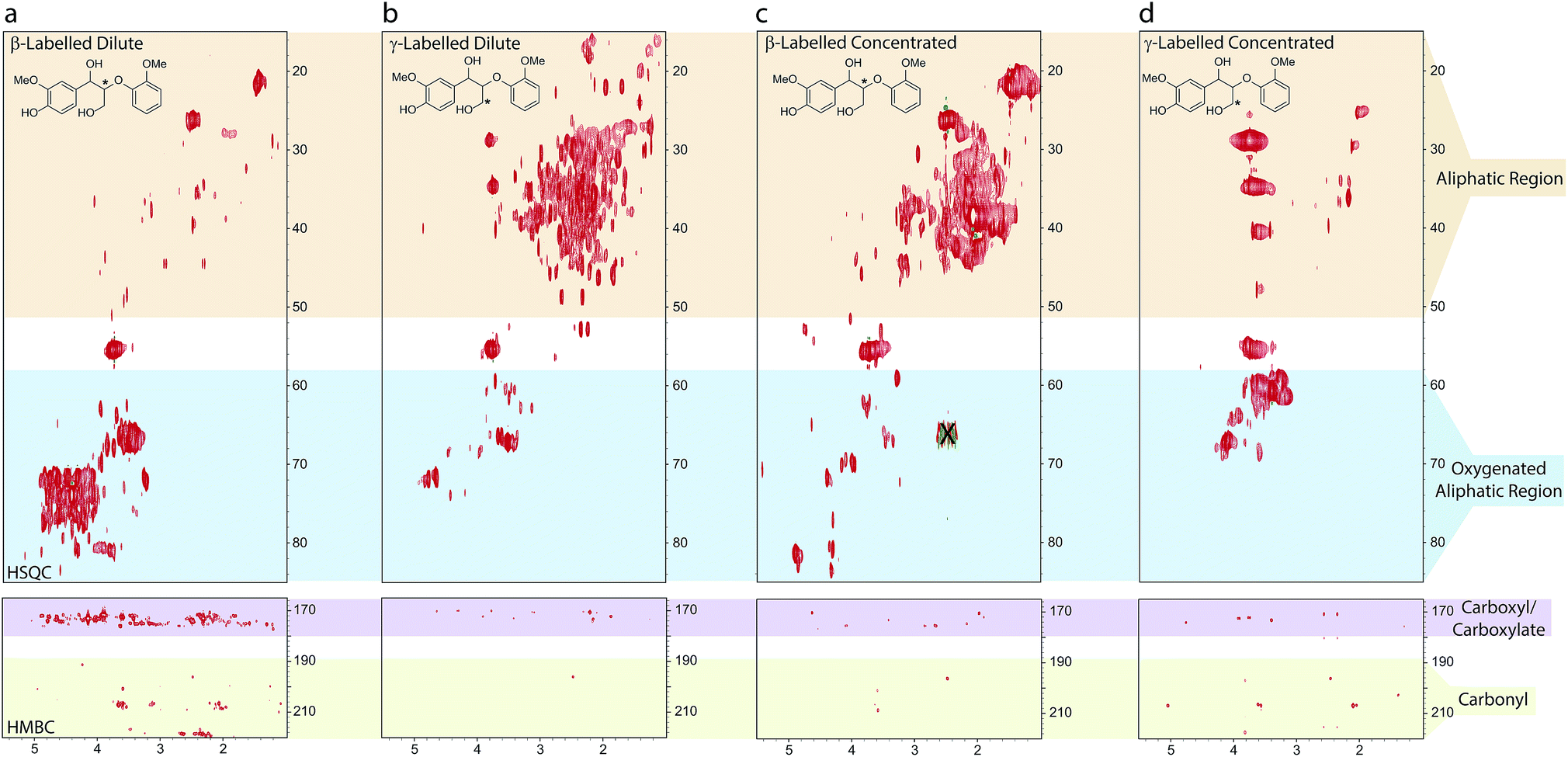 | ||
| Fig. 7 Sections of the 1H–13C HSQC and HMBC spectra of the aliphatic and oxygenated aliphatic (HSQC) and the carboxyl/carbonyl (HMBC) regions of the synthetic kraft lignins obtained from either β- or γ-13C labelled models 3: (a) β-labelled dilute; (b) γ-labelled dilute; (c) β-labelled concentrated; (d) γ-labelled concentrated. See ESI Fig. S37–S39† for spectra plotted at different contour levels, the aromatic regions and full HMBC spectra. X indicates a spectral artifact.93 Reagents and conditions: 0.25 M Na2S, 0.12 M NaOH, H2O, 10 or 100 mg mL−1, 170 °C, 2 h. | ||
Under concentrated reaction conditions relatively fewer new peaks were observed. The β-label was found to give predominantly aliphatic resonances with particularly intense cross peaks being observed at approximately δH/δC = 2.0/39 and 1.5/22 ppm (Fig. 7c), which were also very prominent in the coniferyl alcohol reaction products (c.f.Fig. 5d). For the γ-label (Fig. 7d) intense cross peaks were observed for the expected diarylmethanes, however another very intense signal was observed at δH/δC = 3.4/61 ppm, i.e. in an oxygenated aliphatic environment. In both cases far fewer carboxyl groups were observed in the HMBC spectra compared to the dilute reactions, results again consistent with a switch away from homovanillin as a reaction intermediate. These results highlight the remarkable complexity of the products being generated from the β-O-4 linkage during the kraft process and help to explain why the fate of this linkage has largely remained elusive to date.
3 Conclusions
Kraft lignins are potentially available on megaton scales as co-products from the pulp and paper industry, far exceeding the volumes of any lignin available from other industries and processes. Detailed understanding of their structure is imperative, however, to achieve successful and efficient valorization of this renewable feedstock. We have shown for the prototypical industrial kraft lignin ‘Indulin AT’ that the fate of the minor linkages can be well traced and that approximately 45% of the lignin structure can be assigned to a mixture of native and new kraft linkages based on fractionation and HSQC NMR analysis. Investigations into the fate of the most abundant native β-O-4 linkages showed that the chemistry of this unit is particularly complex. Model compound studies revealed that their fragmentation and repolymerization can follow two main paths; either the previously well recognized coniferyl alcohol pathway or a newly identified homovanillin–formaldehyde one. Identification of a novel lactone product of this pathway in different kraft lignins, including soft- and hardwood ones, suggests that this finding is generally applicable. 13C labelling experiments provided additional insight, demonstrating that the highly reactive intermediates generated from this linkage are involved in complex condensation reactions, resulting in well over 100 new chemical structures. Although these are easily detected by 2D NMR for the labelled system, this is not necessarily the case in actual kraft lignin owing to their low abundance and spectral dispersion, which helps to explain why the fate of the β-O-4 linkage remained largely unknown so far. The fact that defining a single constitutional scheme for the structure of kraft lignin might prove elusive, can thus be considered as another recalcitrant feature of this material. We do anticipate though that the detailed new knowledge gained in terms of structure composition, the fraction-dependent linkage abundance and the pathways of formation can greatly aid future valorization efforts, be it for materials application or by catalytic depolymerization or modification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank UPM, RAIZ - Forest and Paper Research Institute (Portugal), Celbi Pulp and Paper (Portugal), ECN (Netherlands) and Smurfit Kappa (Netherlands) for kindly donating lignin and black liquor samples. This project has been performed within the framework of the CatchBio program. The authors gratefully acknowledge financial support of NWO, the Smart Mix Program of the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture and Science. The NWO Large grant 175.107.301.10 is also gratefully acknowledged.Notes and references
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- T. Renders, S. Van den Bosch, S.-F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2016, 9, 1544–1558 CrossRef PubMed.
- C. S. Lancefield and N. J. Westwood, Green Chem., 2015, 17, 4980–4990 RSC.
- E. K. Vakkilainen, Kraft recovery boilers: Principles and practice, Suomen Soodakattilayhdistys r.y, Helsinki, Finland, 2005, p. 246 Search PubMed.
- M. A. Lake and J. C. Blackburn, Cellul. Chem. Technol., 2014, 48, 799–804 Search PubMed.
- F. Oehman, H. Theliander, P. Tomani and P. Axegard, US Pat., US20100325, 2009.
- L. Kouisni, P. Holt-Hindle, K. Maki and M. Paleologou, Pulp and Paper Canada, 2014, 115, 18–22 Search PubMed.
- C. R. Kumar, N. Anand, A. Kloekhorst, C. Cannilla, G. Bonura, F. Frusteri, K. Barta and H. J. Heeres, Green Chem., 2015, 17, 4921–4930 RSC.
- T. Rinesch, J. Mottweiler, M. Puche, P. Concepción, A. Corma and C. Bolm, ACS Sustainable Chem. Eng., 2017, 5, 9818–9825 CrossRef.
- J. Löfstedt, C. Dahlstrand, A. Orebom, G. Meuzelaar, S. Sawadjoon, M. V. Galkin, P. Agback, M. Wimby, E. Corresa, Y. Mathieu, L. Sauvanaud, S. Eriksson, A. Corma and J. S. M. Samec, ChemSusChem, 2016, 9, 1392–1396 CrossRef PubMed.
- J. Mottweiler, M. Puche, C. Räuber, T. Schmidt, P. Concepción, A. Corma and C. Bolm, ChemSusChem, 2015, 8, 2106–2113 CrossRef PubMed.
- J. Zakzeski, A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5, 1602–1609 CrossRef PubMed.
- X. Ma, R. Ma, W. Hao, M. Chen, F. Yan, K. Cui, Y. Tian and Y. Li, ACS Catal., 2015, 5, 4803–4813 CrossRef.
- J. Ralph and L. L. Landucci, in Lignin and Lignans: Advances in Chemistry, ed. C. Heitner, D. Dimmel and J. A. Schmidt, CRC Press, Taylor & Francis, Boca Raton, FL, USA, 2010, pp. 137–244 Search PubMed.
- Y. Tobimatsu, F. Chen, J. Nakashima, L. L. Escamilla-Treviño, L. Jackson, R. A. Dixon and J. Ralph, Plant Cell, 2013, 25, 2587–2600 CrossRef PubMed.
- F. Yue, F. Lu, S. Ralph and J. Ralph, Biomacromolecules, 2016, 17, 1909–1920 CrossRef PubMed.
- Y. Li, T. Akiyama, T. Yokoyama and Y. Matsumoto, Biomacromolecules, 2016, 17, 1921–1929 CrossRef PubMed.
- J. Carlos Del Río, J. Rencoret, A. Gutiérrez, H. Kim and J. Ralph, Plant Physiol., 2017, 174, 2072–2082 CrossRef PubMed.
- S. D. Karlen, R. A. Smith, H. Kim, D. Padmakshan, A. Bartuce, J. K. Mobley, H. C. A. Free, B. G. Smith, P. J. Harris and J. Ralph, Plant Physiol., 2017, 175, 1058–1067 CrossRef PubMed.
- T.-Q. Yuan, S.-N. Sun, F. Xu and R.-C. Sun, J. Agric. Food Chem., 2011, 59, 10604–10614 CrossRef PubMed.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef PubMed.
- R. Jastrzebski, S. Constant, C. S. Lancefield, N. J. Westwood, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2016, 9, 2074–2079 CrossRef PubMed.
- C. S. Lancefield, O. S. Ojo, F. Tran and N. J. Westwood, Angew. Chem., Int. Ed., 2015, 54, 258–262 CrossRef PubMed.
- P. J. Deuss, C. S. Lancefield, A. Narani, J. G. de Vries, N. J. Westwood and K. Barta, Green Chem., 2017, 19, 2774–2782 RSC.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994 RSC.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Science, 2016, 354, 329–333 CrossRef PubMed.
- M. V. Galkin and J. S. M. Samec, ChemSusChem, 2014, 7, 2154–2158 CrossRef PubMed.
- J. Marton, in Lignins: occurrence, formation, structure and reactions, Wiley-Interscience, Toronto, 1971, ch. 16, pp. 639–689 Search PubMed.
- C. Crestini, H. Lange, M. Sette and D. S. Argyropoulos, Green Chem., 2017, 19, 4104–4121 RSC.
- J. Gierer and I. Noren, Acta Chem. Scand., 1962, 16, 1713–1729 CrossRef.
- J. Gierer, B. Lenz and N. H. Wallin, Acta Chem. Scand., 1964, 18, 1469–1476 CrossRef.
- J. Gierer and L.-A. Smedman, Acta Chem. Scand., 1971, 25, 1461–1467 CrossRef.
- E. Adler, J. Marton and I. Falkehag, Acta Chem. Scand., 1964, 18, 1311–1312 CrossRef.
- J. Gierer, I. Pettersson, L.-A. Smedman and I. Wenneberg, Acta Chem. Scand., 1973, 27, 2083–2094 CrossRef.
- J. Gierer and O. Lindeberg, Acta Chem. Scand., Ser. B, 1978, 32, 577–587 CrossRef.
- R. Kondo and K. V. Sarkanen, J. Wood Chem. Technol., 1984, 4, 301–311 CrossRef.
- F. Berthold, E.-L. Lindfors and G. Gellerstedt, Holzforschung, 1998, 52, 481–489 CrossRef.
- J. Gierer and N. H. Wallin, Acta Chem. Scand., 1965, 19, 1502–1503 CrossRef.
- D. R. Robert, M. Bardet, G. Gellerstedt and E. L. Lindfors, J. Wood Chem. Technol., 1984, 4, 239–263 CrossRef.
- G. Gellerstedt and K. Gustafsson, J. Wood Chem. Technol., 1987, 7, 65–80 CrossRef.
- H. Mikawa, K. Sato and C. Takasaki, Bull. Chem. Soc. Jpn., 1956, 29, 259–265 CrossRef.
- C. G. Boeriu, D. Bravo, R. J. Gosselink and J. E. van Dam, Ind. Crops Prod., 2004, 20, 205–218 CrossRef.
- O. Faix, B. Andersons and G. Zakis, Holzforschung, 1998, 52, 268–274 CrossRef.
- G. Gellerstedt, D. Robert, V. D. Parker, M. Oivanen and L. Eberson, Acta Chem. Scand., Ser. B, 1987, 41, 541–546 CrossRef.
- K. P. Kringstad and R. Mörck, Holzforschung, 1983, 37, 237–244 CrossRef.
- M. Y. Balakshin and E. A. Capanema, RSC Adv., 2015, 5, 87187–87199 RSC.
- Z. Xia, L. G. Akim and D. S. Argyropoulos, J. Agric. Food Chem., 2001, 49, 3573–3578 CrossRef PubMed.
- D. S. Argyropoulos, J. Wood Chem. Technol., 1994, 14, 65–82 CrossRef.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef.
- C. Cui, R. Sun and D. S. Argyropoulos, ACS Sustainable Chem. Eng., 2014, 2, 959–968 CrossRef.
- J. Rönnols, A. Jacobs and F. Aldaeus, Holzforschung, 2017, 71, 563–570 Search PubMed.
- J. R. D. Montgomery, C. S. Lancefield, D. M. Miles-Barrett, K. Ackermann, B. E. Bode, N. J. Westwood and T. Lebl, ACS Omega, 2017, 2, 8466–8474 CrossRef.
- T. Liitiä, S. Maunu and B. Hortling, J. Agric. Food Chem., 2003, 51, 2136–2143 CrossRef PubMed.
- L. Zhang, G. Henriksson and G. Gellerstedt, Org. Biomol. Chem., 2003, 1, 3621–3624 RSC.
- M. Y. Balakshin, E. A. Capanema, C. L. Chen and H. S. Gracz, J. Agric. Food Chem., 2003, 51, 6116–6127 CrossRef PubMed.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- Z. Hu, X. Du, J. Liu, H. M. Chang and H. Jameel, J. Wood Chem. Technol., 2016, 36, 432–446 CrossRef.
- C. Mattsson, S. I. Andersson, T. Belkheiri, L. E. Åmand, L. Olausson, L. Vamling and H. Theliander, Biomass Bioenergy, 2016, 95, 364–377 CrossRef.
- X. Jiang, D. Savithri, X. Du, S. Pawar, H. Jameel, H. M. Chang and X. Zhou, ACS Sustainable Chem. Eng., 2017, 5, 835–842 CrossRef.
- E. A. Capanema, M. Y. Balakshin, C. L. Chen, J. S. Gratzl and H. Gracz, Holzforschung, 2001, 55, 302–308 Search PubMed.
- J. Gierer, Wood Sci. Technol., 1980, 14, 241–266 CrossRef.
- J.-L. Wen, S.-L. Sun, B.-L. Xue and R.-C. Sun, Materials, 2013, 6, 359–391 CrossRef PubMed.
- D. J. Yelle and J. Ralph, Int. J. Adhes. Adhes., 2016, 70, 26–36 CrossRef.
- J. Ralph, T. Akiyama, H. Kim, F. Lu, P. F. Schatz, J. M. Marita, S. A. Ralph, M. S. S. Reddy, F. Chen and R. A. Dixon, J. Biol. Chem., 2006, 281, 8843–8853 CrossRef PubMed.
- D. J. Yelle, D. Wei, J. Ralph and K. E. Hammel, Environ. Microbiol., 2011, 13, 1091–1100 CrossRef PubMed.
- B. Ahvazi, G. Pageau and D. S. Argyropoulos, Adv. Lignocellul. Chem. Ecol. Friendly Pulping Bleaching Technol., 1998, 76, 506–512 Search PubMed.
- V. L. Chiang and M. Funaoka, Holzforschung, 1988, 42, 385–391 CrossRef.
- V. L. Chiang and M. Funaoka, Holzforschung, 1990, 44, 147–156 CrossRef.
- V. L. Chiang and M. Funaoka, Holzforschung, 1990, 44, 309–313 CrossRef.
- J. Gierer and S. Wännström, Holzforschung, 1984, 38, 181–184 CrossRef.
- A. Holmgren, G. Brunow, G. Henriksson, L. Zhang and J. Ralph, Org. Biomol. Chem., 2006, 4, 3456 RSC.
- D. Ibarra, M. I. Chávez, J. Rencoret, J. C. Del Río, A. Gutiérrez, J. Romero, S. Camarero, M. J. Martínez, J. Jiménez-Barbero and A. T. Martínez, J. Agric. Food Chem., 2007, 55, 3477–3490 CrossRef PubMed.
- S. Ralph, L. Landucci and J. Ralph, NMR Database of Lignin and Cell Wall Model Compounds, 2009, www.glbrc.org/databases_and_software/nmrdatabase/ Search PubMed.
- The α position correlation for the dibenzodioxocin structure is found at δH/δC = 4.84 ppm/83.1 ppm.
- Nishimura et al. have recently reported direct evidence for LCC complexes in softwood lignin. The key α position correlation for the α-ether-type LCC is found at δH/δC = 4.50 ppm/80.2 ppm – this cross peak is not observed in Indulin AT kraft lignin. See: H. Nishimura, A. Kamiya, T. Nagata, M. Katahira and T. Watanabe, Sci. Rep., 2018, 8, 6538 CrossRef PubMed.
- F. Tran, C. S. Lancefield, P. C. J. Kamer, T. Lebl and N. J. Westwood, Green Chem., 2015, 17, 244–249 RSC.
- L. Zhang and G. Gellerstedt, Magn. Reson. Chem., 2007, 45, 37–45 CrossRef PubMed.
- K. Hu, W. M. Westler and J. L. Markley, J. Am. Chem. Soc., 2011, 133, 1662–1665 CrossRef PubMed.
- M. Sette, H. Lange and C. Crestini, Comput. Struct. Biotechnol. J., 2013, 6, e201303016 CrossRef PubMed.
- F. Fardus-Reid, J. Warren and A. Le Gresley, Anal. Methods, 2016, 8, 2013–2019 RSC.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Phys. Rev., 1948, 73, 679 CrossRef.
- Obtaining sufficient S/N for the 3rd HSQC0 increment for technical lignins requires very long experiment times. Here we used spectral aliasing, as well as only using 2 increments for back extrapolation, to reduce experiment times. Comparison to 3 increment experiments showed that similar quantification results are obtained for most linkages, however differences of up to ca. 20% are observed in some cases (Table S9†).
- F. Berthold, E.-L. Lindfors and G. Gellerstedt, Holzforschung, 1998, 52, 398–404 CrossRef.
- F. Berthold and G. Gellerstedt, Holzforschung, 1998, 52, 490–498 CrossRef.
- J. Ralph, K. Lundquist, G. Brunow, F. Lu, H. Kim, P. F. Schatz, J. M. Marita, R. D. Hatfield, S. A. Ralph, J. H. Christensen and W. Boerjan, Phytochem. Rev., 2004, 3, 29–60 CrossRef.
- F. Yue, F. Lu, M. Regner, R. Sun and J. Ralph, ChemSusChem, 2017, 10, 830–835 CrossRef PubMed.
- E. Adler, I. Falkehag, J. Marton and H. Halvarson, Acta Chem. Scand., 1964, 18, 1313–1314 CrossRef.
- T. Kishimoto, Y. Uraki and M. Ubukata, Org. Biomol. Chem., 2006, 4, 1343 RSC.
- J. Gierer, O. Lindeberg, K. Daasvatn, J. Krane and H. Glaumann, Acta Chem. Scand., Ser. B, 1980, 34, 161–170 CrossRef.
- These spectral artifacts are only observed for the most intense cross peaks in the HSQC spectra obtained for the 13C labelled reaction mixtures. They are mixed-phase artifacts which occur at regular spacings in the F1 dimension from the real cross peak, but with varying intensities. The exact origin of these is unknown but is thought to relate to the very high signal intensities observed from the 13C labelled compounds.
- M. Macleod, Pap. Timber, 2007, 89, 1–7 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02000k |
| This journal is © The Royal Society of Chemistry 2018 |