
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly luminescent phosphine oxide-containing bipolar alkynylgold(III) complexes for solution-processable organic light-emitting devices with small efficiency roll-offs†
Chin-Ho
Lee
,
Man-Chung
Tang
,
Wai-Lung
Cheung
,
Shiu-Lun
Lai
,
Mei-Yee
Chan
and
Vivian Wing-Wah
Yam
 *
*
Institute of Molecular Functional Materials (Areas of Excellence Scheme, University Grants Committee) and Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China. E-mail: wwyam@hku.hk; Fax: +86 852 2857 1586; Tel: +86 852 2859 2153
First published on 28th June 2018
Abstract
We report the synthesis of alkynylgold(III) complexes with an electron-transporting phosphine oxide moiety in the tridentate ligand and hole-transporting triarylamine moieties as auxiliary ligands to generate a new class of phosphine oxide-containing bipolar gold(III) complexes for the first time. Such gold(III) complexes feature high photoluminescence quantum yields of over 70% in 1,3-bis(N-carbazolyl)benzene thin films with relatively short excited-state lifetimes of less than 3.9 μs at a 20 wt% dopant concentration. Highly efficient solution-processable organic light-emitting devices have been prepared with superior current efficiencies of up to 51.6 cd A−1 and external quantum efficiencies of up to 15.3%. Notably, triplet–triplet annihilation has been significantly reduced, as exemplified by a very small efficiency roll-off of ∼1% at a practical brightness of 500 cd m−2.
Introduction
Phosphorescent materials are superior in performance to fluorescent materials because of their capability to harvest all excitons for achieving an internal quantum efficiency of up to its theoretical value of 100%. One of the major challenges for commercialization of full-color all-triplet phosphorescent organic light-emitting devices (OLEDs) is the severe efficiency roll-offs at high brightness, i.e. 1000 cd m−2 and 5000 cd m−2, or even at a practical brightness of around 500–600 cd m−2 for full-color display, limiting their practical applicability in displays and solid-state lighting systems. Such severe efficiency roll-off mostly arises from the imbalance of carrier transporting characteristics in hole- and electron-transporting materials, resulting in a narrow recombination zone for excitons.1 Together with the relatively longer lifetimes of triplet excitons, high density of triplet excitons will be accumulated within the emissive layer,1 which significantly increases the chance of triplet–triplet annihilation (TTA). This in turn will inevitably dissipate energy via a non-radiative pathway, reducing device efficiencies at high current densities.1 Various approaches have been employed to alleviate these limitations, including the use of a graded or mixed emissive layer through doping hole- or electron-transporting materials into host materials to improve the carrier-transporting properties of the emissive layer as well as to broaden the exciton recombination zone,2 and the integration of hole-transporting moieties (such as triphenylamine and carbazole groups) and/or electron-transporting moieties (such as triazine and phosphine oxide groups) into single-molecule hosts to give ambipolar host materials in order to maximize charge balance and a reduced TTA.3Among various carrier-transporting moieties, phosphine oxide has received immense attention owing to its excellent electron-transporting properties.4 Phosphine oxide derivatives are well known to be electron-deficient owing to the presence of a strong electron-withdrawing P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety.4a Unlike the general approach of introducing electronegative atoms into the aromatic ring for enhancing electron-transporting properties,5 the incorporation of phosphine oxide not only can maintain the high triplet energy of host materials, as the tetrahedral phosphorus center in phosphine oxide can break the conjugation to keep the triplet energy of the molecule the same, but also improve the electron-transporting properties of the molecule.4b This distinct geometry of the phosphine oxide moiety also gives rise to the amorphous nature, which is a prerequisite for forming a uniform thin film.6 In addition, triarylphosphine oxide moieties are well known to have high chemical and oxidative stabilities.7 All these are prerequisite and crucial properties of solution-processable materials suitable for OLED applications.
O moiety.4a Unlike the general approach of introducing electronegative atoms into the aromatic ring for enhancing electron-transporting properties,5 the incorporation of phosphine oxide not only can maintain the high triplet energy of host materials, as the tetrahedral phosphorus center in phosphine oxide can break the conjugation to keep the triplet energy of the molecule the same, but also improve the electron-transporting properties of the molecule.4b This distinct geometry of the phosphine oxide moiety also gives rise to the amorphous nature, which is a prerequisite for forming a uniform thin film.6 In addition, triarylphosphine oxide moieties are well known to have high chemical and oxidative stabilities.7 All these are prerequisite and crucial properties of solution-processable materials suitable for OLED applications.
In 2014, Yam and co-workers first reported a series of bipolar gold(III) complexes by incorporating electron-transporting benzimidazole moieties and hole-transporting triphenylamine in the alkynyl ligand.8 To allow them to function independently, the electronic communication between these two moieties should be reduced by methyl substitutions on the biphenyl linkage to introduce twisting between the moieties. Alternatively, manipulating the charge transporting properties of the metal complexes via separating the charge transporting groups on the alkynyl ligand and the cyclometalating ligand can overcome the limitation of the conventional method; however, the occurrence of charge transfer emission arising from donor and acceptor moieties being in close proximity may complicate colour tuning. In this work, bipolar gold(III) complexes bearing the phosphine oxide moiety on the cyclometalating ligand and hole-transporting triarylamine moieties on the arylethynyl ligands have been synthesized (Fig. 1), representing the first example to attach simultaneously electron-transporting and hole-transporting groups onto the gold(III) complexes but using the gold metal center to modulate the electronic coupling. These combine both the advantages of excellent electron-transporting properties of the phosphine oxide and the simple molecular design of bipolar alkynylgold(III) complexes. In particular, all of them are thermally stable with decomposition temperatures of >370 °C, as revealed in thermogravimetric analysis (TGA) traces (Fig. S1†). Notably, this class of gold(III) compounds demonstrates high photoluminescence quantum yields (PLQYs) of >70% in solid-state 1,3-bis(N-carbazolyl)benzene (MCP) thin films, almost double that of the structurally related analogue without phosphine oxide.9 Highly efficient solution-processable OLEDs with superior current efficiencies (CE) of up to 51.6 cd A−1 and power efficiencies (PE) of up to 35.9 lm W−1 are recorded, which are also double those of the previously reported bipolar gold(III) complexes.8 Remarkable external quantum efficiencies (EQEs) of up to 15.3% as well as very small efficiency roll-offs of ∼1% have also been recorded at a practical brightness of 500 cd m−2.
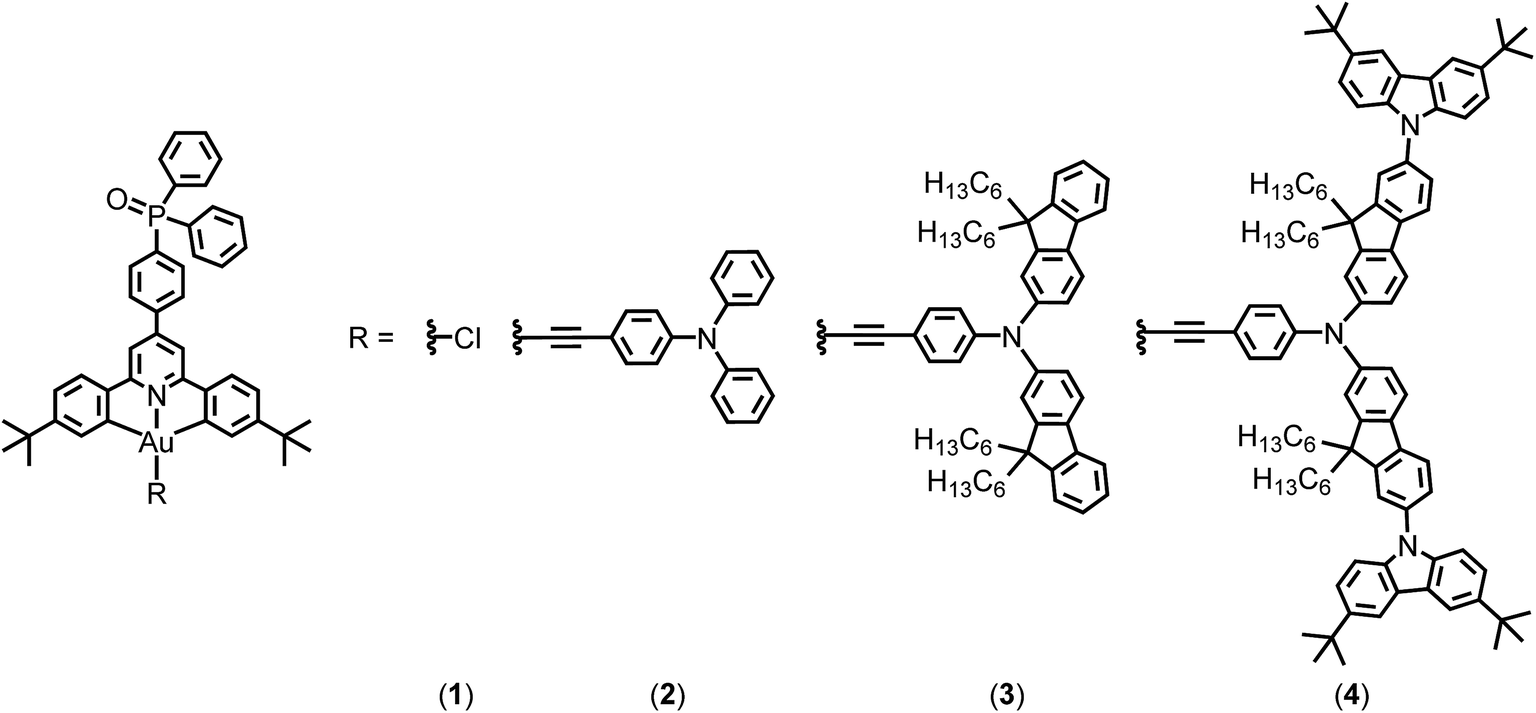 | ||
| Fig. 1 Chemical structures of complexes 1–4. | ||
Synthesis and characterization
The phosphine oxide-containing cyclometalating ligand (tBuC^N(C6H4P(O)Ph2)^CtBu) was synthesized by palladium catalyzed C–P bond formation between tBuC^N(C6H4Br)^CtBu and diphenylphosphine oxide. The resulting cyclometalating ligand was then mercurated and transmetalated to gold(III) metal center to afford cyclometalated chlorogold(III) precursors. Incorporation of alkynes to the cyclometalated chlorogold(III) precursors by means of stirring in the presence of CuI and triethylamine in dichloromethane solution gave rise to a new class of phosphine oxide-containing alkynylgold(III) complexes upon purification. These compounds have been well characterized by 1H, 13C{1H} and 31P{1H} nuclear magnetic resonance (NMR) spectroscopy, electrospray ionization (ESI) or matrix-assisted laser desorption ionization (MALDI) mass spectrometry, elemental analysis as well as infra-red (IR) spectroscopy. The 31P signals of the complexes are in the range of 23.61–26.82 ppm, and the presence of IR stretches in the range of 1118–1121 cm−1 confirms the successful incorporation of the phosphine oxide moiety into the alkynylgold(III) compounds.Photophysical studies
The UV-visible spectra of complexes 1–4 in dichloromethane solution display highly intense absorption bands at ca. 270–350 nm, which could be attributed to the intraligand (IL) π → π* transitions of phosphine oxide, triphenylamine and carbazole moieties, whereas the less intense absorption bands at ca. 380–450 nm are attributed to the metal-perturbed IL π → π* transitions of the cyclometalating ligands, with mixing of some intraligand charge transfer (ILCT) character from the phenyl ring to the pyridine ring (Fig. S2†). Additional Gaussian-shaped absorption bands at ca. 350–400 nm for complexes 3 and 4 are also observed, which could be assigned to IL π → π* transitions of the fluorene/triarylamine hybrids. With the exception of complex 1, all the complexes show absorption tails beyond 450 nm, which can be attributed to the ligand-to-ligand charge transfer (LLCT) π[alkynyl ligand] → π*[tBuC^N(C6H4POPh2)^CtBu] transition. With the extension of the conjugation length of the alkynyl ligands from the triphenylamine moiety to the heterocyclic fused fluorene/triphenylamine hybrid, such absorption tail is found to be red-shifted from complex 2 to 3 and 4. Similar assignments have been made on a related cyclometalated alkynylgold(III) system.4,10 Emission studies of complexes 2–4 have also been carried out in degassed toluene solution (Fig. S3†). Upon excitation with λ ≥ 380 nm, all these complexes exhibit Gaussian-shaped emission bands with peak maxima at ca. 566–580 nm. With the introduction of peripheral carbazole moieties in the alkynyl ligand of complex 4, a slight blue shift of emission wavelength is observed when compared with complex 3. Such a hypsochromic shift is probably due to the electron-withdrawing nature of the nitrogen atoms of the carbazole moieties that could stabilize the highest occupied molecular orbital (HOMO). In addition, the PLQYs of this series of complexes in toluene solution are in the range of 22–48%, very close to those of the tetradentate gold(III) C^N^C^C complexes that were recently reported by Yam and co-workers, representing one of the highest values in the reported literature.11 To further investigate the nature of the excited state of this class of complexes, solvent-dependent emission studies have also been conducted on complex 2 (Fig. S4†). The emission peak maximum is found to exhibit a red shift of ∼3120 cm−1 in response to an increase in solvent polarity from p-xylene (0 D) to dichloromethane (1.6 D). This, together with the fitting of the Lippert–Mataga plot (Fig. S5†), confirms the charge transfer nature of the excited state. Moreover, a significant solvent effect on the PLQYs of complex 2 has been noted. In general, the PLQYs determined in halogenated solvents are significantly lower than that in non-halogenated solvents. However, the PLQY does not decrease with the increasing ease of reduction of the aryl halide solvent (i.e. chlorobenzene (−2.78 V vs. the saturated calomel electrode (SCE)) and 1,2,4-trichlorobenzene (−2.00 V vs. SCE))12 nor with the strength of the C–X bond. The excited state reduction potential of 2 (E(2+/0*)), based on E0–0 energy and electrochemical data,13 was found to be at ca. −1.68 V vs. SCE, which excludes the possibility of the photo-induced electron transfer from the excited species of 2 to aryl halide as the reduction potential of the aryl halide has to be at least less negative than −1.68 V. On the other hand, the drop in the PLQYs correlates with the red-shifting of the emission in various solvents. This, together with the good agreement of the fit to the plot of ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) knrversus Eem, where knr is the non-radiative decay rate constant and Eem is the emission energy, based on the energy gap law (Fig. S6†), suggests that the cause of the PLQY drop could be ascribed to an increase of the non-radiative decay rate upon shifting the emission to lower energy, in accordance with the energy gap law.14 Photophysical properties of the solid-state thin films have also been investigated for complexes 2–4. When doped into MCP–tris(4-carbazoyl-9-ylphenyl)amine (TCTA) (w/w = 3:1) mixed host, their emission bands are found to be red-shifted with increasing dopant concentration (Fig. 2). These emission bands are believed to arise from excimeric emission due to the π–π stacking of the cyclometalating tBuC^N(C6H4P(O)Ph2)^CtBu ligand (Fig. S7†). This unique property of d8 square-planar metal complexes allows us to further control the emission energies in the solid state, beyond the choice of the cyclometalating ligands, as recently communicated by Yam and co-workers.15 Notably, all these complexes are highly emissive in the solid-state thin films with high PLQYs of 46–74%. On the contrary, the PLQY is significantly reduced to ∼30% for the structurally related analogue without the phosphine oxide moiety.9 Such PLQY enhancement can be explained by the presence of the 3-D triphenylphosphine oxide moiety that helps to space out the cyclometalating ligands and thus reduce the chromophoric quenching. The relatively short emission lifetimes in the range of 0.4–9.9 μs in the solid-state thin films can also contribute to such high PLQYs of this class of gold(III) complexes, where the radiative decay is much faster than non-radiative decay pathways such as TTA, which is known to be one of the major dissipative processes of triplet excitons and is one of the major cause of the severe efficiency roll-offs in OLEDs.16 All the photophysical data are summarized in Table S1 in the ESI.†
knrversus Eem, where knr is the non-radiative decay rate constant and Eem is the emission energy, based on the energy gap law (Fig. S6†), suggests that the cause of the PLQY drop could be ascribed to an increase of the non-radiative decay rate upon shifting the emission to lower energy, in accordance with the energy gap law.14 Photophysical properties of the solid-state thin films have also been investigated for complexes 2–4. When doped into MCP–tris(4-carbazoyl-9-ylphenyl)amine (TCTA) (w/w = 3:1) mixed host, their emission bands are found to be red-shifted with increasing dopant concentration (Fig. 2). These emission bands are believed to arise from excimeric emission due to the π–π stacking of the cyclometalating tBuC^N(C6H4P(O)Ph2)^CtBu ligand (Fig. S7†). This unique property of d8 square-planar metal complexes allows us to further control the emission energies in the solid state, beyond the choice of the cyclometalating ligands, as recently communicated by Yam and co-workers.15 Notably, all these complexes are highly emissive in the solid-state thin films with high PLQYs of 46–74%. On the contrary, the PLQY is significantly reduced to ∼30% for the structurally related analogue without the phosphine oxide moiety.9 Such PLQY enhancement can be explained by the presence of the 3-D triphenylphosphine oxide moiety that helps to space out the cyclometalating ligands and thus reduce the chromophoric quenching. The relatively short emission lifetimes in the range of 0.4–9.9 μs in the solid-state thin films can also contribute to such high PLQYs of this class of gold(III) complexes, where the radiative decay is much faster than non-radiative decay pathways such as TTA, which is known to be one of the major dissipative processes of triplet excitons and is one of the major cause of the severe efficiency roll-offs in OLEDs.16 All the photophysical data are summarized in Table S1 in the ESI.†
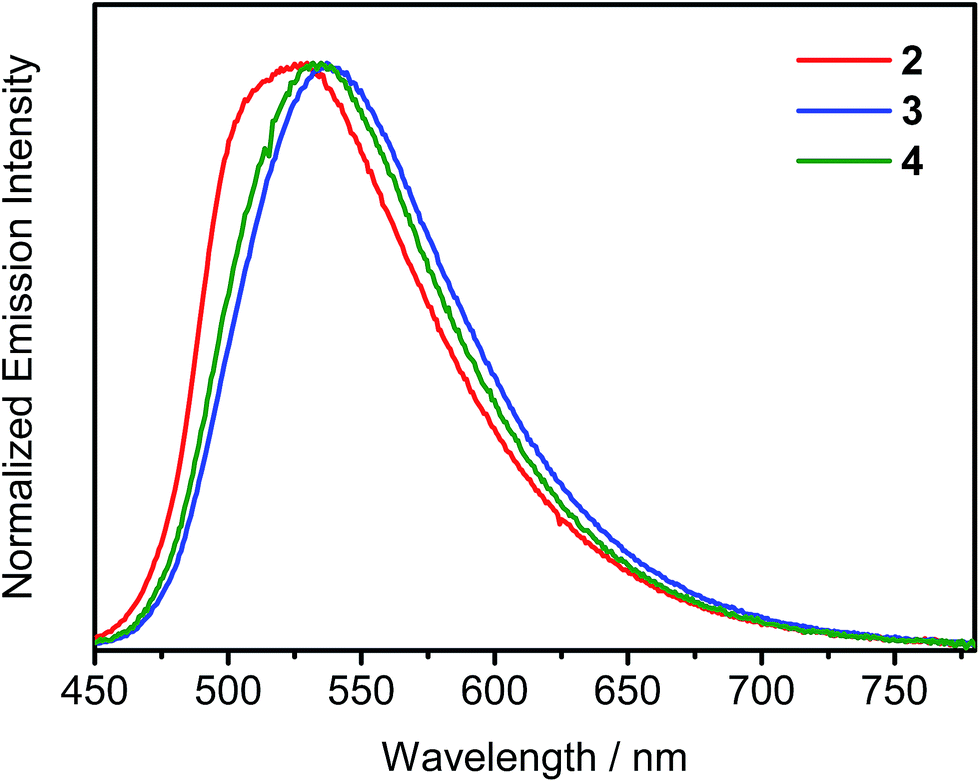 | ||
| Fig. 2 Emission spectra of thin MCP–TCTA mixed host films doped with 5 wt% complexes 2–4 at 298 K. | ||
Electrochemical studies
To estimate the energy levels of their HOMO and lowest unoccupied molecular orbitals (LUMO), cyclic voltammetric studies have been carried out for complexes 2–4 and their cyclic voltammograms and electrochemical data have been shown and summarized in Fig. S8 and Table S2, respectively, in the ESI.† All the complexes show one quasi-reversible reduction couple at −1.35 V vs. SCE and one irreversible reduction wave at −1.81 V vs. SCE. All these reduction couples are found to be independent of the nature of the alkynyl ligands and hence they are attributed to the reduction of the cyclometalating ligands. In the oxidative scan, all the complexes show a first quasi-reversible oxidation couple at +0.75 V to +0.92 V vs. SCE. This oxidation couple can be attributed to the alkynyl ligand-centered oxidation. The attachment of the fluorene moieties at the peripheral of –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–C6H4–N(C6H4)2– in complexes 3 and 4 further extends the conjugation length, resulting in the destabilization of HOMO levels, yielding less positive potentials for the first oxidation of 3 and 4 (both occur at +0.75 V vs. SCE) than that of 2 (+0.92 V vs. SCE). In addition, oxidation couples at higher potentials (+1.15 V and +1.25 V vs. SCE) are observed for complex 4 bearing carbazole moieties at the periphery. These multiple quasi-reversible oxidation couples can be attributed to carbazole-centered oxidation.
C–C6H4–N(C6H4)2– in complexes 3 and 4 further extends the conjugation length, resulting in the destabilization of HOMO levels, yielding less positive potentials for the first oxidation of 3 and 4 (both occur at +0.75 V vs. SCE) than that of 2 (+0.92 V vs. SCE). In addition, oxidation couples at higher potentials (+1.15 V and +1.25 V vs. SCE) are observed for complex 4 bearing carbazole moieties at the periphery. These multiple quasi-reversible oxidation couples can be attributed to carbazole-centered oxidation.
Electroluminescence studies
Taking advantage of their excellent solubility in a variety of common organic solvents and high PLQYs, solution-processable OLEDs based on these bipolar gold(III) complexes 2–4 as phosphorescent dopants have been made. In good agreement with the emission studies, the electroluminescence (EL) spectra of the devices are identical to the photoluminescence measured in solid-state thin films (Fig. S9†). A plot of EQEs of the OLEDs doped with 20 wt% of complexes 2–4 at different luminances is shown in Fig. 3. In addition, superior device performance has been realized with extraordinarily high CEs and EQEs. Notably, the optimized device doped with complex 2 (i.e. 20 wt%) gives a maximum CE of 51.6 cd A−1 and a PE of 35.9 lm W−1. These values correspond to a maximum EQE of 15.3% at a current density of 0.45 mA cm−2. Surprisingly, the introduction of fluorene and carbazole moieties to generate higher generation complexes (i.e.3 and 4, respectively) does not cause a severe degradation of the device performance. High CEs of up to 33.6 cd A−1 (corresponding to an EQE of 10.6%) and 32.0 cd A−1 (corresponding to an EQE of 9.8%) can be achieved for the devices based on 20 wt% of 3 and 4. In addition, the device based on 3 exhibits a higher PE (i.e. 39.0 lm W−1), double that of the device based on 2 (i.e. 19.1 lm W−1). Such a high PE can be ascribed to the HOMO destabilization in complex 3 that can greatly improve the hole injection from the hole-transporting layer into the emissive layer (particularly directly into the guest molecules), as shown in the electrochemical studies. More importantly, a small efficiency roll-off has been observed for the optimized device based on 2. Notably, the EQE has only dropped by ∼1% and ∼8% at a practical brightness of 500 cd m−2 and 1000 cd m−2, respectively. This is not the case for most other iridium(III) complexes.17 Such small efficiency roll-offs can be attributed to the presence of phosphine oxide moieties that balance charge carrier transport, as well as the relatively short excited-state lifetime of <3.9 μs in the 20 wt% MCP doped solid-state thin film. Further introduction of hole-transporting fluorene moieties in 3 and carbazole moieties in 4 could also demonstrate such bipolar properties, as exemplified in the 20 wt% devices. Table S3† summarizes the key parameters for devices based on 2–4. Nevertheless, such high CEs and EQEs are close to the highest values for both vacuum-deposited and solution-processable OLEDs based on gold(III) complexes.18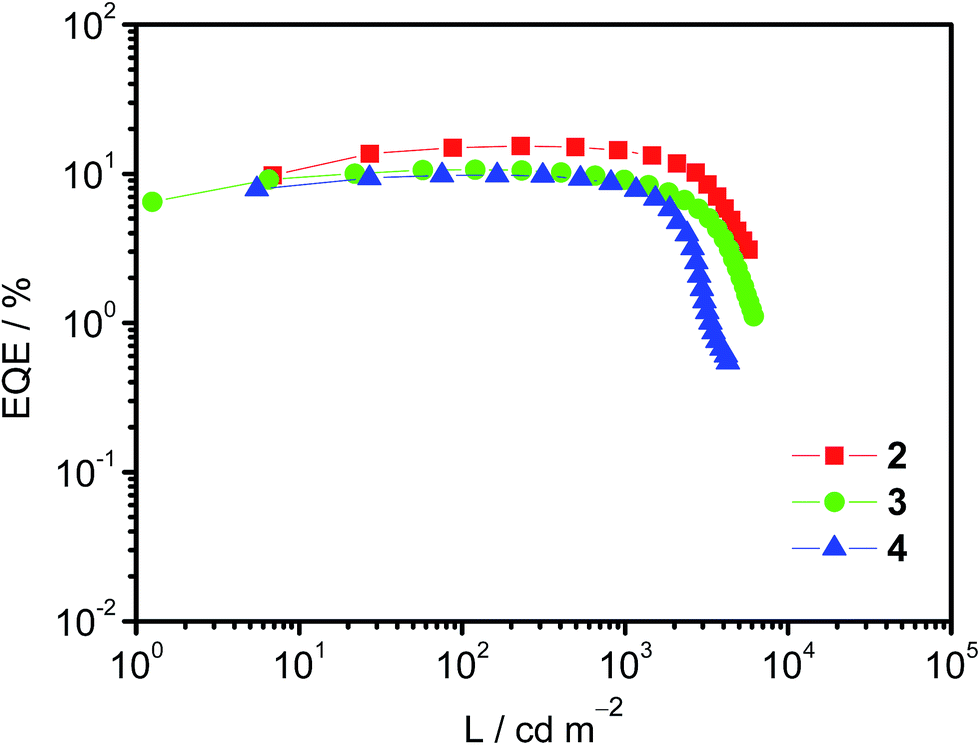 | ||
| Fig. 3 A plot of EQE of solution-processable devices at different luminances doped with 20 wt% complexes 2–4. | ||
Conclusion
In conclusion, a novel class of cyclometalated alkynylgold(III) complexes displaying high PLQYs of up to 70% in solid-state thin films has been designed and synthesized. Notably, this is the first successful demonstration of direct incorporation of an electron-transporting phosphine oxide moiety into luminescent alkynylgold(III) complexes. The 3-D property of phosphine oxide moieties not only provides steric hindrance to reduce chromophoric quenching, doubling the PLQY than those of the structurally related analogues, but also improves the charge carrier balance. Superior device performance with maximum CEs of up to 51.6 cd A−1 and EQEs of up to 15.3% can be realized for solution-processable OLEDs based on these alkynylgold(III) complexes. Together with the relatively short emission lifetimes in thin films, the degree of TTA has been significantly reduced as exemplified by the small efficiency roll-offs of ∼1% and ∼8% at a practical brightness of 500 and 1000 cd m−2, respectively. These excellent device performances are by virtue of the distinct electron-transporting and geometric properties of the phosphine oxide moiety. These findings have provided insights into the design of new types of phosphorescent emitters that are capable of suppressing efficiency roll-offs to enhance the OLED performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. W.-W. Y. acknowledges UGC funding administered by The University of Hong Kong (HKU) for supporting the Matrix-Assisted Laser Desorption Ionization Time-of-Flight Mass Spectrometry and Electrospray Ionization Quadrupole Time-of-Flight Mass Spectrometry Facilities under the support for Interdisciplinary Research in Chemical Science, and the support from URC Strategically Oriented Research Theme on Functional Materials for Molecular Electronics of HKU. The work described in this paper was fully supported by a grant from the University Grants Committee Areas of Excellence Scheme of the Hong Kong Special Administrative Region, China (Project No. AoE/P-03/08). C.-H. L. and W.-L. C. acknowledge the receipt of postgraduate studentships from HKU, and C.-H. L. also acknowledges the receipt of a Hung Hing Ying Scholarship from HKU.References
- C. Murawski, K. Leo and M. C. Gather, Adv. Mater., 2013, 25, 6801 CrossRef PubMed.
- (a) A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556 CrossRef; (b) N. Chopra, J. Lee, Y. Zheng, S.-H. Eom, J. Xue and F. So, ACS Appl. Mater. Interfaces, 2009, 1, 1169 CrossRef PubMed; (c) D. Zhang, L. Duan, Y. Li, H. Li, Z. Bin, D. Zhang, J. Qiao, G. Dong, L. Wang and Y. Qiu, Adv. Funct. Mater., 2014, 24, 3551 CrossRef.
- (a) Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943 RSC; (b) F. May, M. Al-Helwi, B. Baumeier, W. Kowalsky, E. Fuchs, C. Lennartz and D. Andrienko, J. Am. Chem. Soc., 2012, 134, 13818 CrossRef PubMed; (c) D. Wagner, S. T. Hoffmann, U. Heinemeyer, I. Münster, A. Köhler and P. Strohriegl, Chem. Mater., 2013, 25, 3758 CrossRef; (d) H. Sasabe, H. Nakanishi, Y. Watanabe, S. Yano, M. Hirasawa, Y. J. Pu and J. Kido, Adv. Funct. Mater., 2013, 23, 5550 CrossRef; (e) K. S. Yook and J. Y. Lee, Adv. Mater., 2014, 26, 4218 CrossRef PubMed.
- (a) S. O. Jeon, K. S. Yook, C. W. Joo and J. Y. Lee, J. Mater. Chem., 2009, 19, 5940 RSC; (b) S. O. Jeon and J. Y. Lee, J. Mater. Chem., 2012, 22, 4233 RSC; (c) G. Zhou, C.-L. Ho, W.-Y. Wong, Q. Wang, D. Ma, L. Wang, Z. Lin, T. B. Marder and A. Beeby, Adv. Funct. Mater., 2008, 18, 499 CrossRef; (d) G. Zhou, Q. Wang, C.-L. Ho, W.-Y. Wong, D. Ma, L. Wang and Z. Lin, Chem.–Asian J., 2008, 3, 1830 CrossRef PubMed; (e) L. Deng, T. Zhang, R. Wang and J. Li, J. Mater. Chem., 2012, 22, 15910 RSC; (f) X.-K. Liu, C.-J. Zheng, M.-F. Lo, J. Xiao, C.-S. Lee, M.-K. Fung and X.-H. Zhang, Chem. Commun., 2014, 50, 2027 RSC.
- (a) Y.-C. Chiu, J.-Y. Hung, Y. Chi, C.-C. Chen, C.-H. Chang, C.-C. Wu, Y.-M. Cheng, Y.-C. Yu, G.-H. Lee and P.-T. Chou, Adv. Mater., 2009, 21, 2221 CrossRef; (b) Y. You and S. Y. Park, Dalton Trans., 2009, 1267 RSC.
- A. L. Von Ruden, L. Cosimbescu, E. Polikarpov, P. K. Koech, J. S. Swensen, L. Wang, J. T. Darsell and A. B. Padmaperuma, Chem. Mater., 2010, 22, 5678 CrossRef.
- P. E. Burrows, A. B. Padmaperuma, L. S. Sapochak, P. Djurovich and M. E. Thompson, Appl. Phys. Lett., 2006, 88, 183503 CrossRef.
- M.-C. Tang, D. P.-K. Tsang, Y.-C. Wong, M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2014, 136, 17861 CrossRef PubMed.
- V. K.-M. Au, D. P.-K. Tsang, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, J. Organomet. Chem., 2015, 792, 109 CrossRef.
- (a) V. W. W. Yam, K. M. C. Wong, L. L. Hung and N. Zhu, Angew. Chem., Int. Ed., 2005, 44, 3107 CrossRef PubMed; (b) V. W. W. Yam, K. M. C. Wong, L. L. Hung and N. Zhu, Angew. Chem., 2005, 117, 3167 CrossRef; (c) K. M.-C. Wong, L.-L. Hung, W. H. Lam, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2007, 129, 4350 CrossRef PubMed; (d) M.-C. Tang, A. K.-W. Chan, M.-Y. Chan and V. W.-W. Yam, Top. Curr. Chem., 2016, 374, 46 CrossRef PubMed; (e) C.-H. Lee, M.-C. Tang, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 10539 CrossRef PubMed.
- (a) B. Y.-W. Wong, H.-L. Wong, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, Angew. Chem., Int. Ed., 2016, 56, 302 CrossRef PubMed; (b) B. Y.-W. Wong, H.-L. Wong, Y.-C. Wong, M.-Y. Chan and V. W.-W. Yam, Angew. Chem., 2017, 129, 308 CrossRef.
- S. O. Farwell, F. A. Beland and R. D. Geer, J. Electroanal. Chem., 1975, 61, 303 CrossRef.
- C. R. Bock, J. A. Connor, A. R. Gutierrez, T. J. Meyer, D. G. Whitten, B. P. Sullivan and J. K. Nagle, J. Am. Chem. Soc., 1979, 101, 4815 CrossRef.
- J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1983, 87, 952 CrossRef.
- (a) M. C. Tang, C. H. Lee, M. Ng, Y. C. Wong, M. Y. Chan and V. W. W. Yam, Angew. Chem., 2018, 130, 5561 CrossRef; (b) M. C. Tang, C. H. Lee, M. Ng, Y. C. Wong, M. Y. Chan and V. W. W. Yam, Angew. Chem., Int. Ed., 2018, 57, 5463 CrossRef PubMed.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622 CrossRef.
- T. Qin, J. Ding, L. Wang, M. Baumgarten, G. Zhou and K. Müllen, J. Am. Chem. Soc., 2009, 131, 14329 CrossRef PubMed.
- (a) W. P. To, D. Zhou, G. S. M. Tong, G. Cheng, C. Yang and C. M. Che, Angew. Chem., 2017, 129, 14224 CrossRef; (b) W.-P. To, D. Zhou, G. S. M. Tong, G. Cheng, C. Yang and C.-M. Che, Angew. Chem., Int. Ed., 2017, 56, 14036 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02265h |
| This journal is © The Royal Society of Chemistry 2018 |