
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective fluorination of homoallylic alcohols enabled by the tuning of non-covalent interactions†
Jaime A. S.
Coelho‡
a,
Akira
Matsumoto§
a,
Manuel
Orlandi¶
b,
Margaret J.
Hilton||
b,
Matthew S.
Sigman
 *b and
F. Dean
Toste
*a
*b and
F. Dean
Toste
*a
aDepartment of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: fdtoste@berkeley.edu
bDepartment of Chemistry, University of Utah, 315 South 1400 East, Salt Lake City, USA. E-mail: sigman@chem.utah.edu
First published on 3rd August 2018
Abstract
The study of the enantioselective fluorination of homoallylic alcohols via chiral anion phase transfer (CAPT) catalysis using an in situ generated directing group is described. Multivariate correlation analysis, including designer π-interaction derived parameters, revealed key structural features affecting the selectivity at the transition state (TS). Interpretation of the parameters found in the model equation highlights the key differences as well as similarities for the reaction of homoallylic and allylic substrates. A similar T-shaped π-interaction was found to occur between the substrate and the catalyst. The tuning of this crucial interaction by identification of the best combination of phosphoric acid catalyst and boronic acid directing group allowed for the development of a methodology to access γ-fluoroalkenols in typically high enantioselectivities (up to 96% ee).
Introduction
Multivariate linear regression analysis has emerged as a tool in asymmetric catalysis to quantitatively correlate the structure of a chiral catalyst or substrates to selectivity, resulting in predictive equations that can improve a reaction's stereoselectivity.1–15 Moreover, this analysis has been combined with transition state calculations towards mechanistic elucidation and the description of the subtle interplay between reaction components.15,16 We recently applied this approach to uncover the non-covalent interactions (NCIs) between an achiral directing group and a chiral catalyst in the enantioselective electrophilic fluorination of allylic alcohols 1 (Scheme 1A).17–19 This transformation was accomplished using chiral anion phase transfer (CAPT) catalysis19–25 with chiral BINOL-derived phosphate anions (PA) as catalysts in combination with a mixed boronic ester as a transient directing group19,21–24 generated in situ by condensation of the corresponding boronic acid (BA) and the allylic alcohol substrate (Scheme 1A).24 The nature of the BA was found to profoundly influence the stereoselectivity of the reaction. In particular, 3,5-disubstituted phenylboronic acids, such as 3,5-(OMe)2-PhB(OH)2, gave the opposite product enantiomer when compared to other substitution patterns.18,19 Specifically, a T-shaped π-interaction between the aryl ring of the BA and the BINOL backbone of the PA was proposed to be critical for molecular recognition and enantiodivergence of the reaction as dictated by the identity of the BA (Scheme 1A, see proposed TS structure).18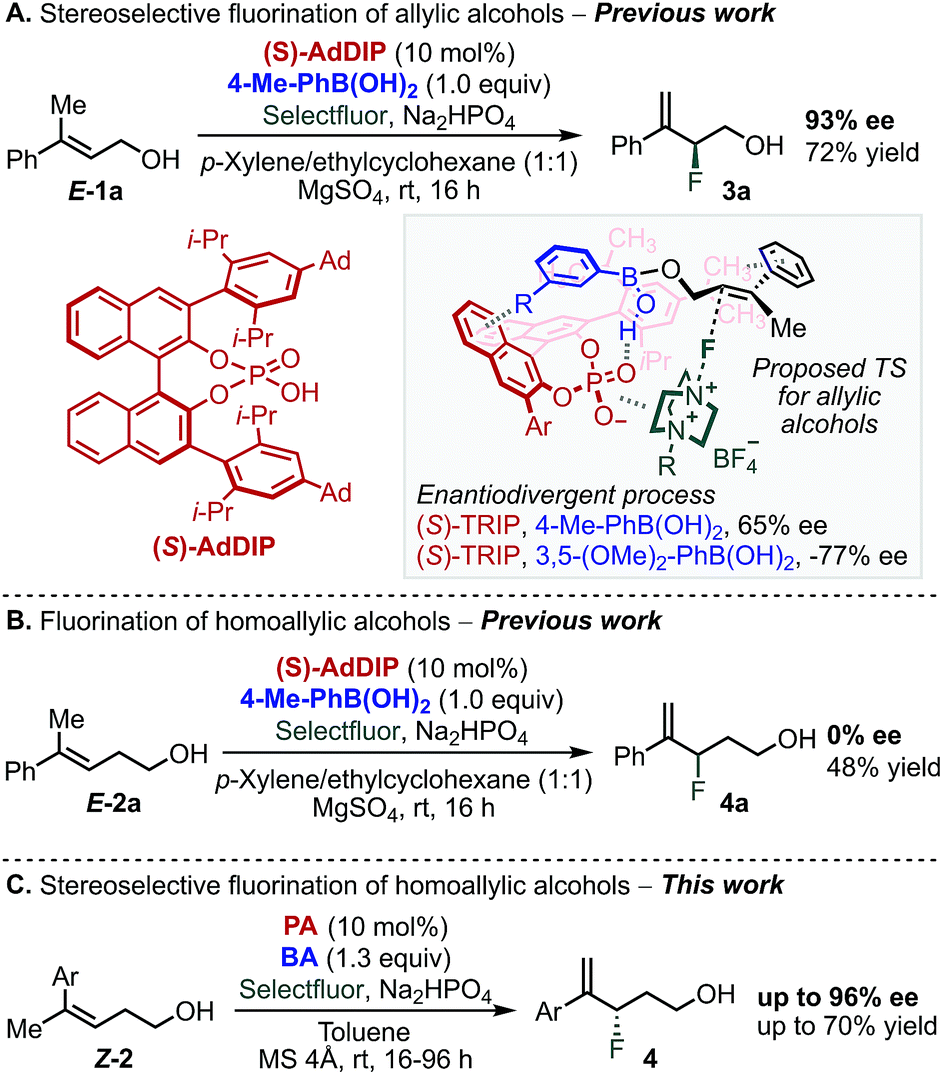 | ||
| Scheme 1 Fluorination of allylic and homoallylic alcohols via CAPT catalysis. | ||
Extension of this transformation to homoallylic alcohol E-2a under the previously established optimized reaction conditions for the allylic substrate (BA: 4-Me-PhB(OH)2 and PA: AdDIP) resulted in the formation of racemic product 4a in 48% yield (Scheme 1B).24 Intrigued by the observed lack of selectivity, and driven by the importance of developing new methods to form carbon-fluorine bonds enantioselectively,26–33 we decided to further study the fluorination of homoallylic alcohols to access valuable γ-fluoroalcohols.34 On the basis of the mechanistic information gained during the investigation of allylic fluorination reaction,18 we anticipated that similar NCIs might be leveraged in the enantiodiscriminating step of the homoallylic fluorination reaction to allow the homologous substrate to undergo fluorination enantioselectively. Moreover, as this catalytic system combines two modular reaction partners, namely the PA and BA, we envisioned that a multivariate regression analysis comparison between the two reactions would inform a strategy to identify the optimal PA/BA combination for the stereoselective fluorination of homoallylic alcohols through fine tuning of weak attractive interactions. Herein, we present the successful application of this strategy to the identification of new combinations of PA and BA directing groups to ultimately promote a highly enantioselective fluorination of homoallylic alcohols (Scheme 1C).
Results and discussion
We began our study by evaluating a variety of BAs in combination with PAs 5–7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35 in the fluorination of 2b under the previously optimized conditions (Scheme 1B).19 Among other substitution patterns, 4-alkyl and 3,5-substituted BAs, including methyl and methoxy groups, were selected because they provided the highest enantioselectivities in the fluorination of allylic substrates. Moreover, as previously suggested by TS analysis, including a conformational study, the allylic substrates' aryl group participates in interactions with the para substituent on the PA (proposed TS in Scheme 1A), resulting in control over the alkene's facial selectivity.18 Thus, we hypothesized that the configuration of the alkene might influence the enantioselectivity. Therefore, we included both E- and Z-homoallylic alcohols (E-2 and Z-2) in our study. We envisioned that qualitative and quantitative analysis of this broad data set would allow clear comparison to the allylic system, enhancing insight into the catalyst's mode of action with the homologous substrate in order to ultimately improve the selectivity.
35 in the fluorination of 2b under the previously optimized conditions (Scheme 1B).19 Among other substitution patterns, 4-alkyl and 3,5-substituted BAs, including methyl and methoxy groups, were selected because they provided the highest enantioselectivities in the fluorination of allylic substrates. Moreover, as previously suggested by TS analysis, including a conformational study, the allylic substrates' aryl group participates in interactions with the para substituent on the PA (proposed TS in Scheme 1A), resulting in control over the alkene's facial selectivity.18 Thus, we hypothesized that the configuration of the alkene might influence the enantioselectivity. Therefore, we included both E- and Z-homoallylic alcohols (E-2 and Z-2) in our study. We envisioned that qualitative and quantitative analysis of this broad data set would allow clear comparison to the allylic system, enhancing insight into the catalyst's mode of action with the homologous substrate in order to ultimately improve the selectivity.
Data set design
Evaluation of many of these BA/PA combinations resulted in the enantioselectivities shown in Fig. 1A. Several trends emerged from the graphical analysis of these data (Fig. 1B). Overall, the selectivity observed with homoallylic substrate E-2 (solid lines) with each catalyst was much lower than with allylic alcohol E-1 (dotted lines). For example, PA (R)-5 (green) provided 4b in generally poor enantioselectivity (−28 to 16% ee) when the allylic substrates reacted in up to 90% ee. This difference is likely a result of the greater degrees of conformational freedom of the homoallylic substrate relative to its allylic counterpart, which results in a more flexible conformational space at the TS and thus accessing an ensemble of competitive transition states leading to the poorer observed selectivity. A second difference between the allylic and homoallylic cases is the stereodivergence trend observed with (R)-6 (blue) and (R)-7 (red). While these two catalysts presented similar behaviour when used with substrate E-1 (both provided an inversion of selectivity with 3,5-substituted BAs), with E-2 they reacted with the opposite π-face of the prochiral alkene. In other words, (R)-7 provided product 4b with opposite optical rotation when switching from 2- or 4-substituted BAs to 3,5-substituted BAs, while the use of catalyst (R)-6 yields product 4a with the opposite configuration (see outliers, Fig. 1B). These observations are consistent with the requirement for a sterically more demanding phosphoric acid ((R)-7vs.(R)-6) capable of geometrically constraining the homoallylic mixed boronic ester into a similarly active conformation that leads to the same facial selectivity as observed with E-1 and (R)-7. Hence, trend analysis in Fig. 1B revealed that fluorination of the homoallylic substrate E-2 required greater catalyst imposed conformational constraints to achieve acceptable enantioselectivities compared to allylic alcohol substrate E-1.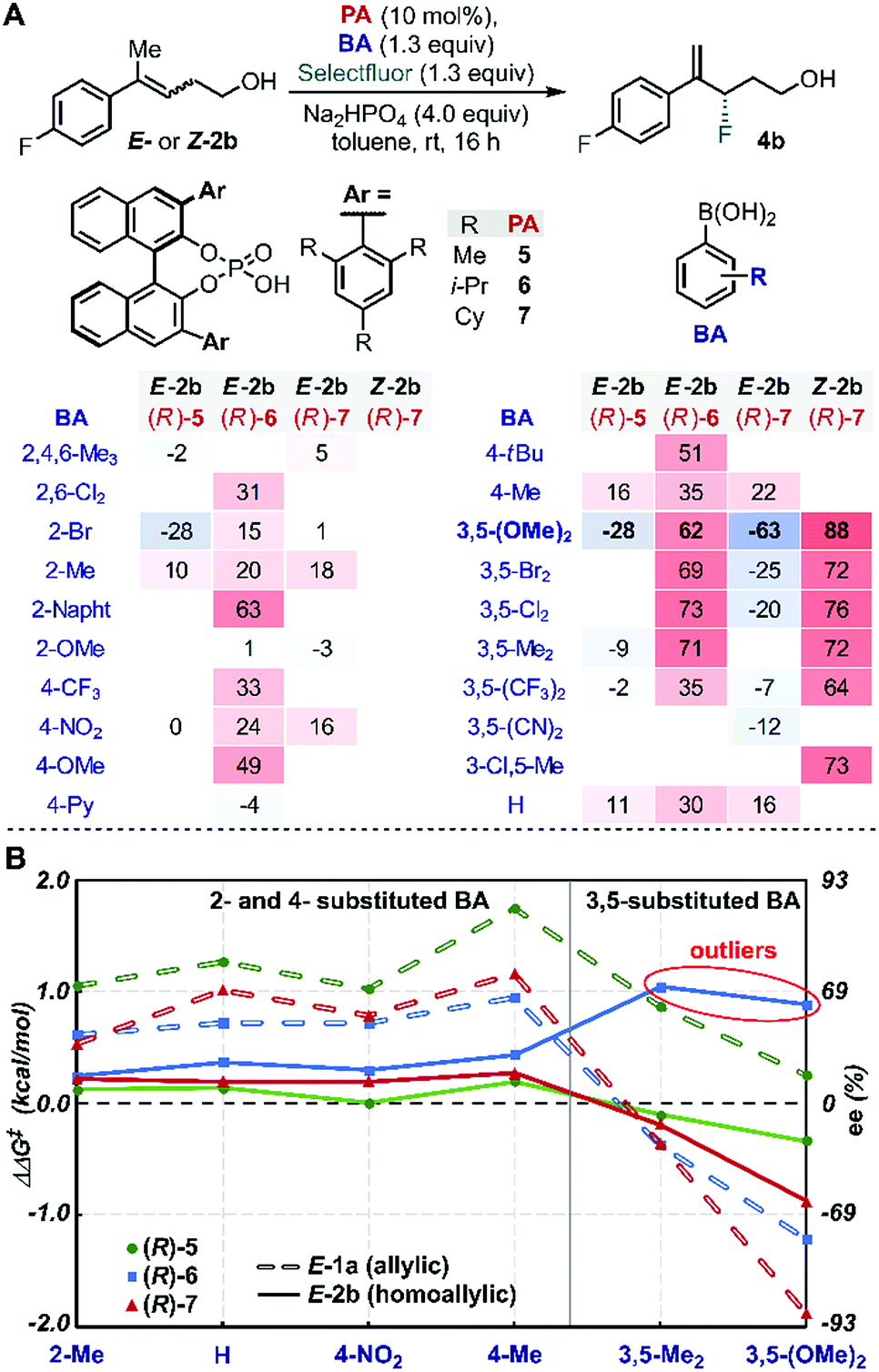 | ||
| Fig. 1 (A) Enantioselectivity data for the fluorination of homoallylic alcohol 2b obtained by variation of PA, BA and alkene configuration. All reactions were conducted on 0.05 mmol scale with respect to homoallylic alcohol 2b. (B) PA structure-selectivity trends as a function of BA structure for the fluorination of E-1 (allylic) and E-2 (homoallylic). | ||
Inversion of stereoselectivity was also observed when the alkene (2) configuration was changed. Specifically, 88% and −63% ee were measured employing Z-2b and E-2b as substrates, respectively, when (R)-7 PA was used in combination with 3,5-(OMe)2-PhB(OH)2 (Fig. 1A, bold). Interestingly, the level of selectivity achieved with substrate Z-2b was higher for all BAs tested. The formation of opposite product enantiomers from the Z- and E-alkene implies that fluorination occurs on the same π-face of the substrates. This result suggests a potential interaction between the homoallylic substrates' aryl group and the para substituent on the PA. Furthermore, the high dependence of the ee on the BA (Fig. 1) further highlights the importance of the subtle NCIs present in the TS, as previously observed within the allylic system. Therefore, in order to uncover an optimal PA/BA combination for high enantioselectivity with homoallylic substrates, we sought to gain greater insights into these influential putative NCIs.
Stereochemical model
With this goal in mind, a multidimensional correlation analysis strategy was employed in order to gain a quantitative understanding of the relationship between the structural variation of the substrates, BAs and PA catalysts on the enantioselectivity. Furthermore, the obtained mathematical equation, connecting molecular properties of the reaction components to enantioselectivity, could be leveraged for virtual evaluation of a number of BAs, a diverse variety of which are readily accessible compared to PAs, aiming for improved reaction enantioselectivity.As NCIs are hypothesized to drive the enantioselective catalysis, we aimed to apply our recently reported computed interaction energies (Eπ) and distances (Dπ) as parameters in multidimensional correlation analysis15 as they are descriptive of π-stacking and CH–π interactions at the TS level.12,18,36 This method provides a direct analysis tool that enables data driven hypotheses of weak interactions likely involved in the stereochemistry determining step.12,37 Additionally, this approach allows for a union of the knowledge gained from multidimensional, experimentally-derived statistical analysis and TS modeling. Since π-stacking and/or CH–π interactions are likely at play between the BA and PA in this reaction manifold, we computed TEπ and TDπ parameters, according to the protocol previously described, for several BAs by using benzene as a probe to represent both types of interactions.18 Additional descriptors for the BAs and PAs were also computed, including Sterimol values (B1, B5 and L) to describe steric bulk, and NBO charges and IR intensities/frequencies (i,ν) to account for electronic effects. Additionally, δ was introduced to distinguish the alkene configuration of Z-2b and E-2b, which is necessary to specifically describe the stereoselectivity inversion associated with the alkene stereochemistry (δ is a binary parameter with value +1 for Z-2b and −1 for E-2b, Fig. 2A). The dataset used included 44 data points derived from combinations of PA 5–7 with the BAs listed in Fig. 2A.38
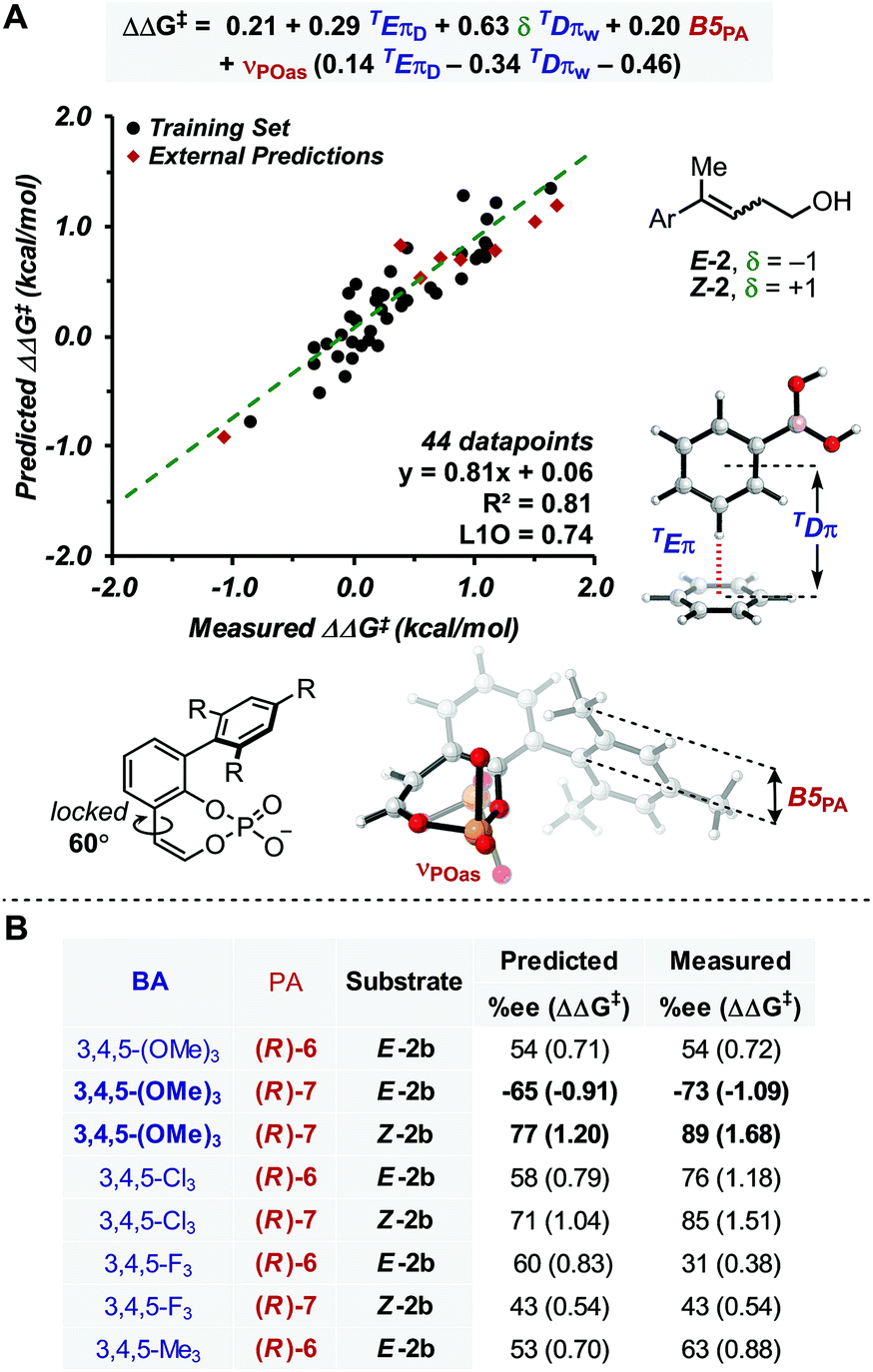 | ||
| Fig. 2 (A) Model showing correlation between measured ΔΔG‡ and molecular properties of reaction components, including π-parameters (B) external predictions of 3,4,5-substituted BAs. | ||
Preliminary single-parameter correlations with data obtained from each catalyst separately suggested that a T-shaped π-stacking interaction plays an important role with homoallylic alcohol substrates.38 Indeed, TDπw was the only parameter that provided qualitative trends with the measured selectivities, expressed as ΔΔG‡ (kcal mol−1). When data from all catalysts were subjected to multivariate analysis, the model in Fig. 2A was obtained (R2 = 0.81, intercept = 0.06). Leave-one-out cross validation test showed that the model is statistically sound (L1O = 0.74). The parameters that appear in the equation are B5PA (maximum width of the catalyst arene), νPOas (phosphate asymmetric stretching frequency), TDπw, δ, and TEπD. In comparison to the multidimensional correlation analysis obtained for the allylic case, with the exception of δ and TEπD parameters, these are the same parameters, suggesting that a similar molecular recognition mode of the substrate by the catalyst (i.e., the previously hypothesized T-shaped interaction between the BA aryl group and the naphthyl backbone of PA) is likely functioning. Moreover, νPOas may account for the ability of the phosphate moiety to engage the mixed boronic ester OH group in an H-bond and Selectfluor in an electrostatic interaction. The cross terms δTDπw, νPOasTDπw and νPOasTEπD are consistent with synergistic effects derived from the concomitant interaction of the three reaction components (PA, BA and alkene) in the TS.
In order to evaluate the model's ability to make predictions, a series of 3,4,5-substituted BAs, which were not previously tested in the allylic reaction were evaluated virtually. This substitution pattern was selected because the BAs were expected to have identical TDπw values to their 3,5-substituted partners but different TEπD values, a significant parameter in the model. Remarkably, the enantioselectivities of eight 3,4,5-substituted BAs were reasonably predicted by this equation (Fig. 2), which also provided another validation mode of the model. From this study, only 3,4,5-(OMe)3-PhB(OH)2 was predicted to outperform the other BAs with substrate E-2 and PA (R)-7. This prediction was experimentally accurate, resulting in the best enantioselectivity for the homoallylic fluorination to date (89% ee).
Overall, this model is congruent with the molecular recognition pattern proposed for the allylic system, although the main additional feature in the homoallylic variant is the presence of TEπD. This parameter is defined as the interaction energy of the benzene–BA complex conformer with longer TDπ value (where more than one conformation is accessible);18 thus, the model suggests that interactions between the catalyst and the reaction partners occur at a greater distance. This is likely due to the longer alkyl chain of the substrate, which perturbs the optimal geometrical recognition pattern of the allylic system, where the much more rigid geometric features allow the postulated NCIs to occur in the TS. This hypothesis is consistent with the generally lower ee's obtained with the homoallylic variant. Hence, comparison of the allylic and homoallylic models suggests that the energy of the T-shaped π-interaction is not as crucial for the allylic system as it is for the homoallylic variant, where the increased flexibility requires stronger NCIs for efficient substrate recognition. In other words, in order to avoid competition of other less selective pathways, the enantioselective fluorination of homoallylic alcohols relies on the ability of the catalyst to effectively associate the substrate-BA adduct via a stronger π-interaction than that required for allylic alcohols. Thus, the TEπD parameter is necessary to accurately describe and predict the enantioselectivity of various BA/PA combinations.
Chiral anion phase transfer control
After identifying the optimal PA and BA, we next studied the influence of the solvent by multivariate correlation analysis towards reaction conditions optimization. During the optimization, we noted that the fluorination of homoallylic alcohol E-2b under homogenous conditions (in acetonitrile instead of toluene) afforded exclusively the undesired halocyclization39 product 8b as a mixture of diastereoisomers (dr 1.7:1, Scheme 2). The low dr obtained under these conditions is suggestive of the formation of a stable, benzylic β-fluorocarbenium cation. Thus, this experiment not only highlights the need for CAPT catalysis in order to achieve a stereoselective process, but also that it also imparts chemoselectivity that allows access to the fluorinated homoallylic alcohols 4.
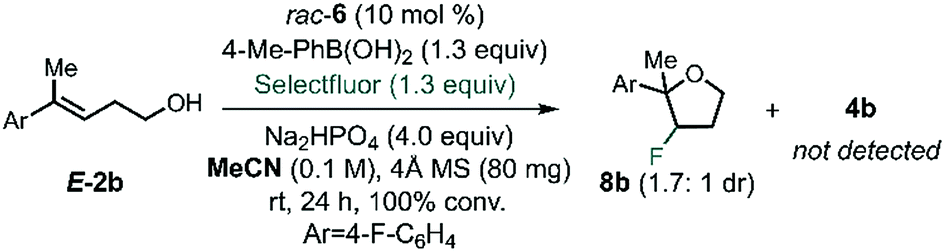 | ||
| Scheme 2 Fluorination homoallylic alcohols under homogeneous conditions. | ||
CAPT catalysis efficiency relies on the minimization of the uncatalysed background reaction due to reagents' solubility. In other words, solvent and reagents are carefully selected so that one of the reactants is insoluble in the reaction media. In this manner, the background reaction outside the chiral environment provided by the catalyst is diminished. To obtain additional insight into the CAPT process as a function of the reaction media, we applied multidimensional correlation analysis of enantioselectivity data from 11 different aromatic solvents and their corresponding properties. A simple model resulted (8 values in training set and 3 in validation set), in which polarity and the partition coefficient in octanol–water (logP) were used as parameters (Fig. 3, R2 = 0.91, intercept = 0.06, L1O = 0.84). This analysis revealed that as the solvent became more polar, the reaction became less selective. Due to the complex NCIs needed for an enantioselective reaction, this result may be expected, as such attractive interactions are favored in a less polar environment.40–42 However, this quantitative evaluation emphasizes the subtle, dynamic nature of the interactions that are responsible for enantioselectivity, as a range of ca. 0.7 kcal mol−1 was observed depending on the solvent used. Based on the presented analysis, we selected toluene as an optimal solvent for this transformation.
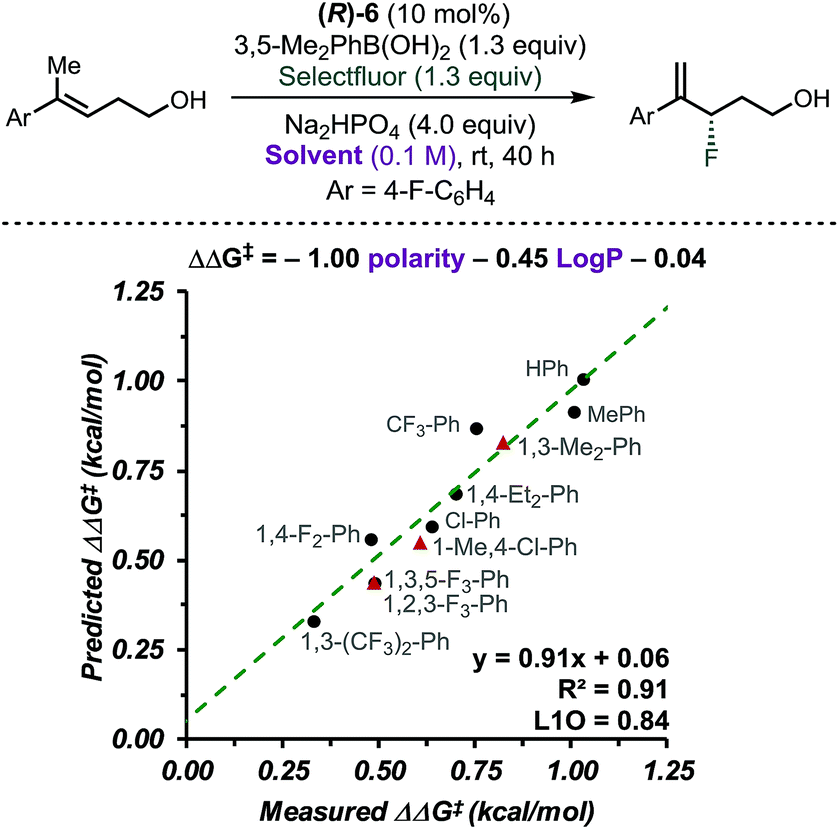 | ||
| Fig. 3 Multidimensional correlation analysis showing the dependence of the reaction enantioselectivity on logP and polarity of the aromatic solvent. The dataset includes 8 data points (black) and 3 external validations (red). | ||
Substrate evaluation
In order to further evaluate the validity of the conclusion presented above, reaction for both E- and Z-homoallylic alcohols was evaluated employing the optimized PA/BA combinations (Scheme 3).43 While moderate yields were generally obtained for the fluorination of Z-homoallylic alcohols (40% avg. yield), the reaction tolerated different aryl substitutions. In particular, Z-alkenols bearing electron-withdrawing or neutral groups were fluorinated in 85–95% ee (9a–9d, 9h and 9f). Electron-rich aryl groups were also compatible with this transformation, as Z-2e, Z-2g and Z-2h reacted in 89, 81% and 94% ee, respectively. Finally, fluorination of Z-2i in 85 and 64% ee demonstrated that groups in ortho-position negatively affect the stereoselectivity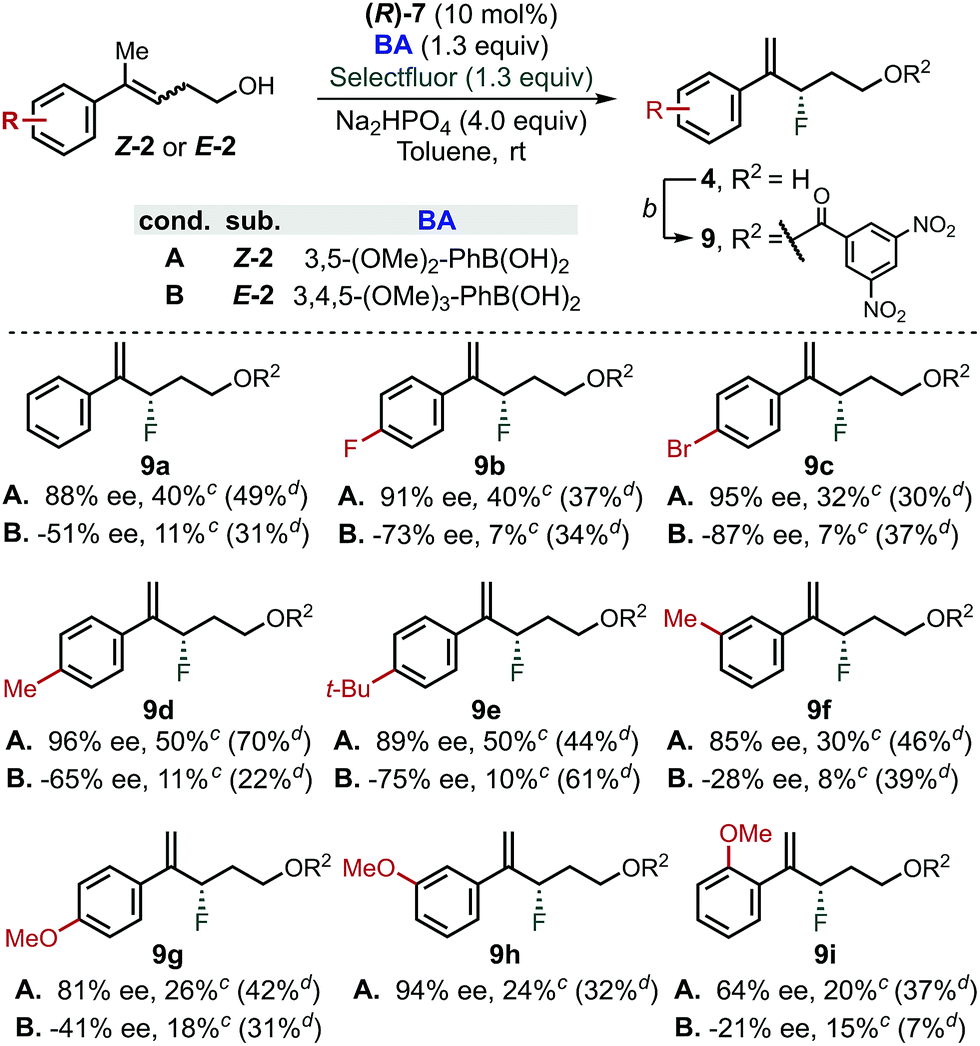 | ||
| Scheme 3 Substrate scope for the fluorination of homoallylic alcohols via CAPT catalysis using an in situ directing group. aGeneral conditions: (R)-7 (10 mol%), BA (1.3 equiv), Na2HPO4 (4.0 equiv), Selectfluor (1.3 equiv), 4 Å MS (80 mg/0.1 mmol of substrate), toluene (0.1 M), rt, 4 days. 3,5-(MeO)2-PhB(OH)2 was used for the reaction with Z-homoallylic alcohol whereas 3,4,5-(MeO)3-PhB(OH)2 was used for the reaction of E-isomers. bEsterification conditions: 3,5-dinitrobenzoyl chloride (1.5 equiv), DMAP (1.5 equiv), Et3N (1.5 equiv), DCM (0.1 M), 20 min. cNMR yield of 4a–h using 1-fluoro-3-nitrobenzene as internal standard. dIsolated yield of 9a–h from the fluorination reaction using PA/BA = (R)-6/4-Me-PhB(OH)2.38 | ||
In contrast, the reaction of E-alkenols was more substrate dependent, although the general trend observed for the Z-isomers was maintained. Specifically, E-2a–2e reacted in moderate to good enantioselectivities (−51 to −87% ee), while ortho- and meta-substituted and electron rich aryl groups led to −28 to −41% ee (9f–9i). The efficiency of the fluorination of E-homoallylic alcohols was lower compared to the Z-analogues, which is a result of the low conversion of the starting material.38 We attribute this divergent behaviour to the use of different BAs (i.e., 3,5-(OMe)2vs. 3,4,5-(OMe)3), as we found that fluorination of Z- and E-substrates using PA/BA = (R)-6/4-Me-PhB(OH)2 gave similar isolated yields (Scheme 3, yields in parenthesis).
Conclusions
The enantioselective fluorination of homoallylic alcohols was studied by multivariate linear regression analysis of a dataset of PAs, BAs and alkene configuration. The transformation relies on CAPT catalysis in combination with a directing group generated in situ in order to access the enantioenriched γ-fluoroalkenol products. The reaction proved to be stereodivergent depending on the PA/BA combinations. Specifically, our analysis highlighted the presence of crucial NCIs that affect selectivity within a stereochemical model that shares similarities with the previously reported fluorination of allylic alcohols. However, dramatic differences were also observed that are due to the higher conformational freedom of homoallylic alcohols with respect to their allylic counterparts. Most notably, the fluorination of homoallylic alcohols requires a stronger T-shaped π-interaction between the substrate and catalysts than the allylic variant to access high enantioselectivity.In addition, the role of the solvent in CAPT catalysis was quantitatively described via multivariate correlations for the first time, emphasizing the sensitive nature of the CAPT process and its influence on the stereodetermining step. The insights gained in these studies were successfully applied to the development of a method to access γ-fluoroalkenol products in typically high ee (up to 96%). Even though moderate yields were obtained, this transformation is not accessible without a CAPT process, as the uncatalysed reaction produced cyclized furan products.
More broadly, the extension of the fluorination reaction scope from allylic to homoallylic alcohols illustrates a strategy to address substrate scope limitations using a data-driven mechanistic approach. Data and insights gained from multivariate analysis of the original transformation informed the investigation of the desired extension in scope, enabling the transformation of a previous nonselective fluorination into a highly enantioselective process. The protocol outline herein can be generalized and applied to the expansion of the scope of catalytic reactions in which NCIs govern the selectivity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. D. T and M. S. S. thank the NIH (1 R01 GM121383) for support of this work. J. A. S. C. thanks Fundação para a Ciência e a Tecnologia (SFRH/BPD/100433/2014) and M. O. thanks the Ermenegildo Zegna Group for postdoctoral fellowships. We acknowledge Dr Andrew J. Neel for helpful discussions and running preliminary experiments, Regev Parnes for preparation of some substrates and Alec H. Christian for assistance with MS.Notes and references
- K. C. Harper and M. S. Sigman, Science, 2011, 333, 1875–1878 CrossRef PubMed.
- K. C. Harper, S. C. Vilardi and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 2482–2485 CrossRef PubMed.
- A. Milo, E. N. Bess and M. S. Sigman, Nature, 2014, 507, 210–214 CrossRef PubMed.
- E. N. Bess, R. J. DeLuca, D. J. Tindall, M. S. Oderinde, J. L. Roizen, J. Du Bois and M. S. Sigman, J. Am. Chem. Soc., 2014, 136, 5783–5789 CrossRef PubMed.
- E. N. Bess, A. J. Bischoff and M. S. Sigman, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14698–14703 CrossRef PubMed.
- A. Milo, A. J. Neel, F. D. Toste and M. S. Sigman, Science, 2015, 347, 737–743 CrossRef PubMed.
- E. N. Bess, D. M. Guptill, H. M. L. Davies and M. S. Sigman, Chem. Sci., 2015, 6, 3057–3062 RSC.
- V. Mougel, C. B. Santiago, P. A. Zhizhko, E. N. Bess, J. Varga, G. Frater, M. S. Sigman and C. Coperet, J. Am. Chem. Soc., 2015, 137, 6699–6704 CrossRef PubMed.
- T. Piou, F. Romanov-Michailidis, M. Romanova-Michaelides, K. E. Jackson, N. Semakul, T. D. Taggart, B. S. Newell, C. D. Rithner, R. S. Paton and T. Rovis, J. Am. Chem. Soc., 2017, 139, 1296–1310 CrossRef PubMed.
- J. M. Crawford, E. A. Stone, A. J. Metrano, S. J. Miller and M. S. Sigman, J. Am. Chem. Soc., 2018, 140, 868–871 CrossRef PubMed.
- Y. Park, Z. L. Niemeyer, J. Q. Yu and M. S. Sigman, Organometallics, 2018, 37, 203–210 CrossRef.
- M. Orlandi, M. J. Hilton, E. Yamamoto, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 12688–12695 CrossRef PubMed.
- J. Y. Guo, Y. Minko, C. B. Santiago and M. S. Sigman, ACS Catal., 2017, 7, 4144–4151 CrossRef.
- B. P. Woods, M. Orlandi, C. Y. Huang, M. S. Sigman and A. G. Doylet, J. Am. Chem. Soc., 2017, 139, 5688–5691 CrossRef PubMed.
- M. S. Sigman, K. C. Harper, E. N. Bess and A. Milo, Acc. Chem. Res., 2016, 49, 1292–1301 CrossRef PubMed.
- C. B. Santiago, J.-Y. Guo and M. S. Sigman, Chem. Sci., 2018, 9, 2398–2412 RSC.
- F. D. Toste, M. S. Sigman and S. J. Miller, Acc. Chem. Res., 2017, 50, 609–615 CrossRef PubMed.
- M. Orlandi, J. A. S. Coelho, M. J. Hilton, F. D. Toste and M. S. Sigman, J. Am. Chem. Soc., 2017, 139, 6803–6806 CrossRef PubMed.
- A. J. Neel, A. Milo, M. S. Sigman and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 3863–3875 CrossRef PubMed.
- V. Rauniyar, A. D. Lackner, G. L. Hamilton and F. D. Toste, Science, 2011, 334, 1681–1684 CrossRef PubMed.
- R. J. Phipps, K. Hiramatsu and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 8376–8379 CrossRef PubMed.
- T. Honjo, R. J. Phipps, V. Rauniyar and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 9684–9688 CrossRef PubMed.
- J. Wu, Y. M. Wang, A. Drljevic, V. Rauniyar, R. J. Phipps and F. D. Toste, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13729–13733 CrossRef PubMed.
- W. W. Zi, Y. M. Wang and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 12864–12867 CrossRef PubMed.
- X. Y. Yang, R. J. Phipps and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 5225–5228 CrossRef PubMed.
- P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073–9174 CrossRef PubMed.
- X. Y. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826–870 CrossRef PubMed.
- V. Bizet and D. Cahard, in Stereoselective Synthesis of Drugs and Natural Products, John Wiley & Sons, Inc., 2013 Search PubMed.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef PubMed.
- Y. J. Zhao, Y. H. Pan, S. B. D. Sim and C. H. Tan, Org. Biomol. Chem., 2012, 10, 479–485 RSC.
- J. A. Ma and D. Cahard, Chem. Rev., 2008, 108, Pr1–Pr43 CrossRef PubMed.
- N. Shibata, T. Ishimaru, S. Nakamura and T. Toru, J. Fluorine Chem., 2007, 128, 469–483 CrossRef.
- C. Bobbio and V. Gouverneur, Org. Biomol. Chem., 2006, 4, 2065–2075 RSC.
- To the best of our knowledge, a general methodology for the preparation of enantioenriched γ-fluoroalcohols has yet to be developed.
- PAs 5–7 were selected for this study because they showed a linear relationship between their optical purity and the product ee in the previously reported fluorination of allylic alcohols, while other catalysts provided a nonlinear effect. Indeed, this discrepancy between 5–7 and other less bulky PAs suggests that mechanistic differences are present between these two subclasses of compounds.
- M. Orlandi, F. D. Toste and M. S. Sigman, Angew. Chem., Int. Ed., 2017, 56, 14080–14084 CrossRef PubMed.
- S. Biswas, K. Kubota, M. Orlandi, M. Turberg, D. H. Miles, M. S. Sigman and F. D. Toste, Angew. Chem., Int. Ed., 2018, 57, 589–593 CrossRef PubMed.
- See ESI† for further details.
- S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938–10953 CrossRef PubMed.
- L. Yang, C. Adam and S. L. Cockroft, J. Am. Chem. Soc., 2015, 137, 10084–10087 CrossRef PubMed.
- B. U. Emenike, S. N. Bey, B. C. Bigelow and S. V. S. Chakravartula, Chem. Sci., 2016, 7, 1401–1407 RSC.
- S. L. Cockroft and C. A. Hunter, Chem. Commun., 2009, 3961–3963 RSC.
- Further derivatization of products 4 into the corresponding 3,5-dinitrobenzoylesters 9 was necessary in order to isolate the product. Hence, the yields and ee's reported herein refer to the corresponding derivatives 9 over two reaction steps.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02223b |
| ‡ Present Address: Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Av. Prof. Gama Pinto, 1649-003, Lisbon, Portugal. |
| § Present Address: Department of Material Chemistry, Graduate School of Engineering, Kyoto University, Kyotodaigaku-Katsura, Nishikyo, Kyoto 615-8510, Japan. |
| ¶ Present Address: Department of Chemistry, Università degli Studi di Milano, Via Camillo Golgi, 19, 20133, Milan, Italy. |
| || Present Address: Department of Chemistry, Yale University, P.O. Box 208107, New Haven, Connecticut 06520-8107, United States. |
| This journal is © The Royal Society of Chemistry 2018 |