
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceShape-memory and self-healing functions of DNA-based carboxymethyl cellulose hydrogels driven by chemical or light triggers†
Chen
Wang
 a,
Michael
Fadeev
a,
Junji
Zhang
b,
Margarita
Vázquez-González
a,
Gilad
Davidson-Rozenfeld
a,
He
Tian
b and
Itamar
Willner
*a
a,
Michael
Fadeev
a,
Junji
Zhang
b,
Margarita
Vázquez-González
a,
Gilad
Davidson-Rozenfeld
a,
He
Tian
b and
Itamar
Willner
*a
aInstitute of Chemistry, Center for Nanoscience and Nanotechnology, The Hebrew University of Jerusalem, Jerusalem 91904, Israel. E-mail: itamar.willner@mail.huji.ac.il
bKey Laboratory for Advanced Materials, School of Chemistry and Molecular Engineering, East China University of Science and Technology, Shanghai, China
First published on 23rd July 2018
Abstract
Photoresponsive nucleic acid-based carboxymethyl cellulose (CMC) hydrogels are synthesized, and their application as shape-memory and self-healing functional matrices are discussed. One system involves the preparation of a carboxymethyl cellulose hydrogel crosslinked by self-complementary nucleic acid duplexes and by photoresponsive trans-azobenzene/β-cyclodextrin (β-CD) supramolecular complexes. Photoisomerization of the trans-azobenzene to the cis-azobenzene results in a hydrogel exhibiting lower stiffness due to the separation of the azobenzene/β-CD bridging units. The hydrogel is switched between high and low stiffness states by the cyclic and reversible light-induced isomerization of the azobenzene units between the trans and cis states. The light-controlled stiffness properties of the hydrogel are used to develop a shape-memory hydrogel, where the duplex bridging units act as permanent memory in the quasi-liquid shapeless state of the hydrogel. A second system in the study is a carboxymethyl cellulose hydrogel crosslinked by the K+-stabilized G-quadruplex bridging units and by trans-azobenzene/β-CD complexes. The resulting hydrogel includes dual-trigger functionalities, where the trans-azobenzene/β-CD complexes can be reversibly formed and dissociated through the trans and cis photoisomerization of the azobenzene units, and the K+-stabilized G-quadruplexes can be reversibly dissociated and reformed in the presence of 18-crown-6-ether/K+-ions. The signal-responsive crosslinked hydrogel reveals controlled stiffness properties, where the hydrogel crosslinked by the trans-azobenzene/β-CD and K+-ion-stabilized G-quadruplex reveals high stiffness and the hydrogel crosslinked only by the K+-ion-stabilized G-quadruplexes or only by the trans-azobenzene/β-CD complexes reveals low stiffness properties. The controlled stiffness properties of the hydrogel are used to develop shape-memory hydrogels, where the trans-azobenzene/β-CD complexes or the K+-ion-stabilized G-quadruplexes act as permanent memories in the shapeless and quasi-liquid states of the hydrogels. In addition, the hydrogel that includes two types of stimuli-responsive crosslinking units is used as a self-healing matrix, where each of the triggers guides the self-healing processes.
Introduction
Hydrogels attract growing interest as functional materials for sensing, biomedical applications and tissue engineering.1–3 One broad class of hydrogels consists of hydrophilic polymers crosslinked by supramolecular complexes.4–6 Different crosslinking motifs were reported, and these include receptor/ligand complexes (receptor = cyclodextrin, cucurbituril),7–11 donor–acceptor complexes,12 ionic interactions,13,14 bimolecular recognition complexes6,15 and metal–organic complexes.16–18 One class of hydrogels is the stimuli-responsive hydrogel matrices that undergo gel-to-liquid19 or gel-to-solid20 phase transitions in response to external triggers. Such systems are particularly interesting, as these phase transitions might occur upon the interaction with disease biomarkers21 or under specific cellular conditions (e.g., pH or redox conditions).22 As a result, these stimuli-responsive hydrogels might act as functional sensing matrices,23 targeted drug-delivery systems24 or tissue repair.25 Different external triggers, such as pH,22 heat,26 light,27–30 chemical agents18 or magnetic fields,31,32 were used to induce phase transitions of hydrogels or to control their stiffness. For example, photoresponsive polyacrylamide hydrogels were prepared by mixing polyacrylamide polymer chains modified with trans-azobenzene units and with β-cyclodextrin receptor units.29,30 The supramolecular binding of the trans-azobenzene units to the receptor units resulted in the crosslinking of the polymer chains and the formation of the hydrogel. Photoisomerization of the trans-azobenzene to the cis-azobenzene state that lacks binding affinity to the β-cyclodextrin receptor, led to the separation of the supramolecular bridging units and to the hydrogel-to-liquid transition. In addition, polyacrylamide chains modified with terpyridine ligands were crosslinked by metal ions, e.g. Ru2+, via the formation of Ru2+–terpyridine complexes, to yield redox-responsive polyacrylamide hydrogels.18 A further class of supramolecular hydrogels and, particularly, stimuli-responsive supramolecular hydrogels includes nucleic acid (DNA)-bridged hydrogels.33–35 The crosslinking of the polymer chains (e.g., polyacrylamide chains) by duplex nucleic acids provides a versatile means to generate hydrogels. Furthermore, the signal-triggered reversible reconfiguration of nucleic acid structures provides a rich arsenal of motifs to induce hydrogel-to-solution phase transitions or to control the stiffness of the hydrogel matrices. For example, polyacrylamide hydrogels crosslinked by stimuli-responsive K+-ions-stabilized G-quadruplex units were separated in the presence of 18-crown-6-ether (CE),36 and hydrogels crosslinked by i-motif units at acidic pH values (pH = 5.5) were separated at neutral pH as a resulted of the dissociation of the supramolecular i-motif bridges.37,38 Of particular interest are the hybrid DNA hydrogels bridged by two different bridging motifs, such as duplex/G-quadruplex, duplex/triplex DNA or duplex/i-motif crosslinkers.39 The signal-triggered reversible and switchable control over the stiffness of these hydrogels was demonstrated.Different applications of stimuli-responsive DNA-based hydrogels and, particularly, stimuli-responsive hybrid DNA crosslinked hydrogels were reported, including controlled drug release,24 switchable catalysis and electrocatalysis,40 controlled ion-transport through nanopores41 and stress-induced shape transitions of hydrogel composites.39 In addition, other applications of stimuli-responsive hybrid DNA-based hydrogels include the use of these soft materials as shape-memory matrices and self-healing materials. Shape-memory polymers represent a class of “smart” materials that are processed into a permanent shape, which can be transferred, by an external trigger, into a temporary metastable shape that includes a code (“memory”) to recover the original shape in the presence of a counter-trigger.42–44 By using duplex DNA and stimuli-responsive DNA structures that cooperatively crosslink and stabilize shaped hydrogels, a variety of shape-memory hydrogels were reported.34,45,46 In the presence of the two crosslinking motifs, a stiff shaped hydrogel was generated. The dissociation of the stimuli-responsive bridging units by an appropriate trigger resulted in low-stiffness, quasi-liquid, shapeless hydrogels, which included the residual duplex bridges that retain the entanglement of the polymer chains in the quasi-liquid state as an internal “memory” to recover the shape upon applying the counter trigger that restores the cooperative crosslinking of the stiff hydrogel.
The self-healing phenomenon of hydrogels represents a related process, where the crosslinking of soft hydrogels stabilized by cooperative hybrid bridges may lead to healed, intact hydrogels.47 In fact, self-healing matrices that use supramolecular ligand–receptor interactions,48,49 Diels–Alder adducts,50 light29,51 or H-bonds52,53 as functional bridges to “heal” hydrogels were reported, and the applications of these processes for tissue engineering and tissue recovery were addressed.25 The self-healing of DNA-based hydrogels and, particularly, the use of DNA-based hydrogels that reveal shape-memory and self-healing functionalities are unprecedented. Furthermore, to date all DNA-based shape-memory hydrogels include two types of DNA as crosslinkers of the hydrogels, where one motif acts as the permanent memory and the other acts as the stimuli-responsive crosslinker that controls the stiffness of the hydrogel. The availability of different non-DNA stimuli-responsive crosslinkers allows, however, combining these triggers with stimuli-responsive DNA bridges. This allows the design of hydrogels exhibiting two different codes for the recovery of shapes or self-healed matrices. Moreover, the incorporation of non-DNA stimuli-responsive triggers as functional units that control the shape-memory and stiffness of the hydrogels introduces new time-domains for the transitions between the hydrogel states.
Results and discussion
In the present paper we report on the preparation of hybrid carboxymethyl cellulose (CMC) hydrogels consisting of nucleic acid bridges and trans-azobenzene/β-cyclodextrin supramolecular crosslinking units as functional matrices for designing shape-memory hydrogels and self-healing materials. Specifically, we introduce hybrid CMC hydrogels crosslinked by duplex nucleic acids and supramolecular trans-azobenzene/β-cyclodextrin photo-responsive units. In addition, we present CMC hydrogels crosslinked by two stimuli-responsive crosslinkers composed of K+-ion-stabilized G-quadruplexes/crown ether and trans/cis-azobenzene/β-cyclodextrin units, respectively.The CMC polymers PA and PB (Fig. 1A), were used to prepare the CMC hydrogel that is crosslinked by the light-active trans-azobenzene/β-cyclodextrin units and the self-complementary nucleic acid-functionalized tethers (1)/(1) associated with the two polymer chains. The polymer PA was synthesized by the coupling of (N-aminoethyl p-carboxamide)-trans-azobenzene (Scheme S1†) via carbodiimide/N-hydroxysulfosuccinimide. In the next step, the aminohexyl-functionalized nucleic acid (1) was tethered to the carboxylic residues of CMC using carbodiimide/N-hydroxysulfosuccinimide coupling. The average loading of the azobenzene units on the CMC scaffold (Mw ≈ 250 kDa, degree of substitution = 0.9) was evaluated to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 by 1H-NMR spectroscopy (Fig. S1 and S2†), and the loading of the nucleic acid tethers (1) on the CMC scaffold was determined spectroscopically to be 1:110, Fig. S3.† The polymer PB was prepared by the primary coupling of aminoethyl-β-cyclodextrin (β-CD) to the CMC scaffold, followed by the coupling of the aminohexyl-modified nucleic acid (1) to the scaffold (for Experimental details see ESI†). The loading of β-CD units and the nucleic acid tethers (1) on the CMC backbone corresponded to 1:10 and 1:110, respectively (see ESI, Fig. S3–S5†). For further spectroscopic characterization of the polymers PA and PB, see ESI Fig. S6 and S7.† The molecular weight of the polymers was derived from the Diffusion Ordered Spectroscopy (DOSY) spectra (Fig. S8 and Table S2†). Fig. S9† shows the Fourier-transform infrared spectroscopy (FTIR) of the polymers. Mixing the polymers PA and PB yielded a hydrogel that exhibits switchable light-responsive stiffness properties, Fig. 1B. The hydrogel crosslinked by the trans-azobenzene/β-CD and the (1)/(1) self-complementary strands reveals a high stiffness (G′ ≈ 62 Pa; G′′ ≈ 13 Pa), whereas the photoisomerization (λ = 365 nm) of the trans-azobenzene to cis-azobenzene that lacks affinity toward β-CD results in the separation of the supramolecular azobenzene/β-CD bridges, and leads to the low stiffness hydrogel crosslinked by the (1)/(1) bridges (G′ ≈ 37 Pa; G′′ ≈ 7 Pa). The stiffness properties of the hydrogel are reversibly cycled between higher and lower values by the cyclic photoisomerization of the azobenzene units between the trans-state (generated under visible light, λ > 420 nm, irradiation) and the cis-state (λ = 365 nm illumination), Fig. 1C. For further support that the photo-induced trans ↔ cis transitions proceed in the hydrogel, see ESI (Fig. S10).† Fig. S10A† depicts the absorption spectra of the trans-azobenzene/β-CD and (1)/(1)-crosslinked hydrogel (curve (a)), and of the (1)/(1)-crosslinked, free β-CD and cis-azobenzene-modified hydrogel (curve (b)). The spectra features are consistent with the light-induced formation of the trans-azobenzene and cis-azobenzene units. The photo-induced transitions between the trans-azobenzene and cis-azobenzene units are switchable and reversible, Fig. S10B.† The association constants of trans-azobenzene to β-CD (Ka = 1100 M−1) and of cis-azobenzene to β-CD (Ka = 4.1 M−1) were reported previously.29 This suggests that the photo-generated cis-azobenzene units indeed demonstrate low affinity towards the β-CD receptor units, leading to the hydrogel of low stiffness. The differences in the stiffness properties of the two hydrogel states are further supported by SEM images, Fig. 1D. The stiffer hydrogel crosslinked by the supramolecular trans-azobenzene/β-CD and the (1)/(1) duplexes demonstrates a high density of small pores, whereas the hydrogel of lower stiffness (quasi-liquid) only crosslinked by the (1)/(1) duplexes reveals substantially larger pores.
:10 by 1H-NMR spectroscopy (Fig. S1 and S2†), and the loading of the nucleic acid tethers (1) on the CMC scaffold was determined spectroscopically to be 1:110, Fig. S3.† The polymer PB was prepared by the primary coupling of aminoethyl-β-cyclodextrin (β-CD) to the CMC scaffold, followed by the coupling of the aminohexyl-modified nucleic acid (1) to the scaffold (for Experimental details see ESI†). The loading of β-CD units and the nucleic acid tethers (1) on the CMC backbone corresponded to 1:10 and 1:110, respectively (see ESI, Fig. S3–S5†). For further spectroscopic characterization of the polymers PA and PB, see ESI Fig. S6 and S7.† The molecular weight of the polymers was derived from the Diffusion Ordered Spectroscopy (DOSY) spectra (Fig. S8 and Table S2†). Fig. S9† shows the Fourier-transform infrared spectroscopy (FTIR) of the polymers. Mixing the polymers PA and PB yielded a hydrogel that exhibits switchable light-responsive stiffness properties, Fig. 1B. The hydrogel crosslinked by the trans-azobenzene/β-CD and the (1)/(1) self-complementary strands reveals a high stiffness (G′ ≈ 62 Pa; G′′ ≈ 13 Pa), whereas the photoisomerization (λ = 365 nm) of the trans-azobenzene to cis-azobenzene that lacks affinity toward β-CD results in the separation of the supramolecular azobenzene/β-CD bridges, and leads to the low stiffness hydrogel crosslinked by the (1)/(1) bridges (G′ ≈ 37 Pa; G′′ ≈ 7 Pa). The stiffness properties of the hydrogel are reversibly cycled between higher and lower values by the cyclic photoisomerization of the azobenzene units between the trans-state (generated under visible light, λ > 420 nm, irradiation) and the cis-state (λ = 365 nm illumination), Fig. 1C. For further support that the photo-induced trans ↔ cis transitions proceed in the hydrogel, see ESI (Fig. S10).† Fig. S10A† depicts the absorption spectra of the trans-azobenzene/β-CD and (1)/(1)-crosslinked hydrogel (curve (a)), and of the (1)/(1)-crosslinked, free β-CD and cis-azobenzene-modified hydrogel (curve (b)). The spectra features are consistent with the light-induced formation of the trans-azobenzene and cis-azobenzene units. The photo-induced transitions between the trans-azobenzene and cis-azobenzene units are switchable and reversible, Fig. S10B.† The association constants of trans-azobenzene to β-CD (Ka = 1100 M−1) and of cis-azobenzene to β-CD (Ka = 4.1 M−1) were reported previously.29 This suggests that the photo-generated cis-azobenzene units indeed demonstrate low affinity towards the β-CD receptor units, leading to the hydrogel of low stiffness. The differences in the stiffness properties of the two hydrogel states are further supported by SEM images, Fig. 1D. The stiffer hydrogel crosslinked by the supramolecular trans-azobenzene/β-CD and the (1)/(1) duplexes demonstrates a high density of small pores, whereas the hydrogel of lower stiffness (quasi-liquid) only crosslinked by the (1)/(1) duplexes reveals substantially larger pores.
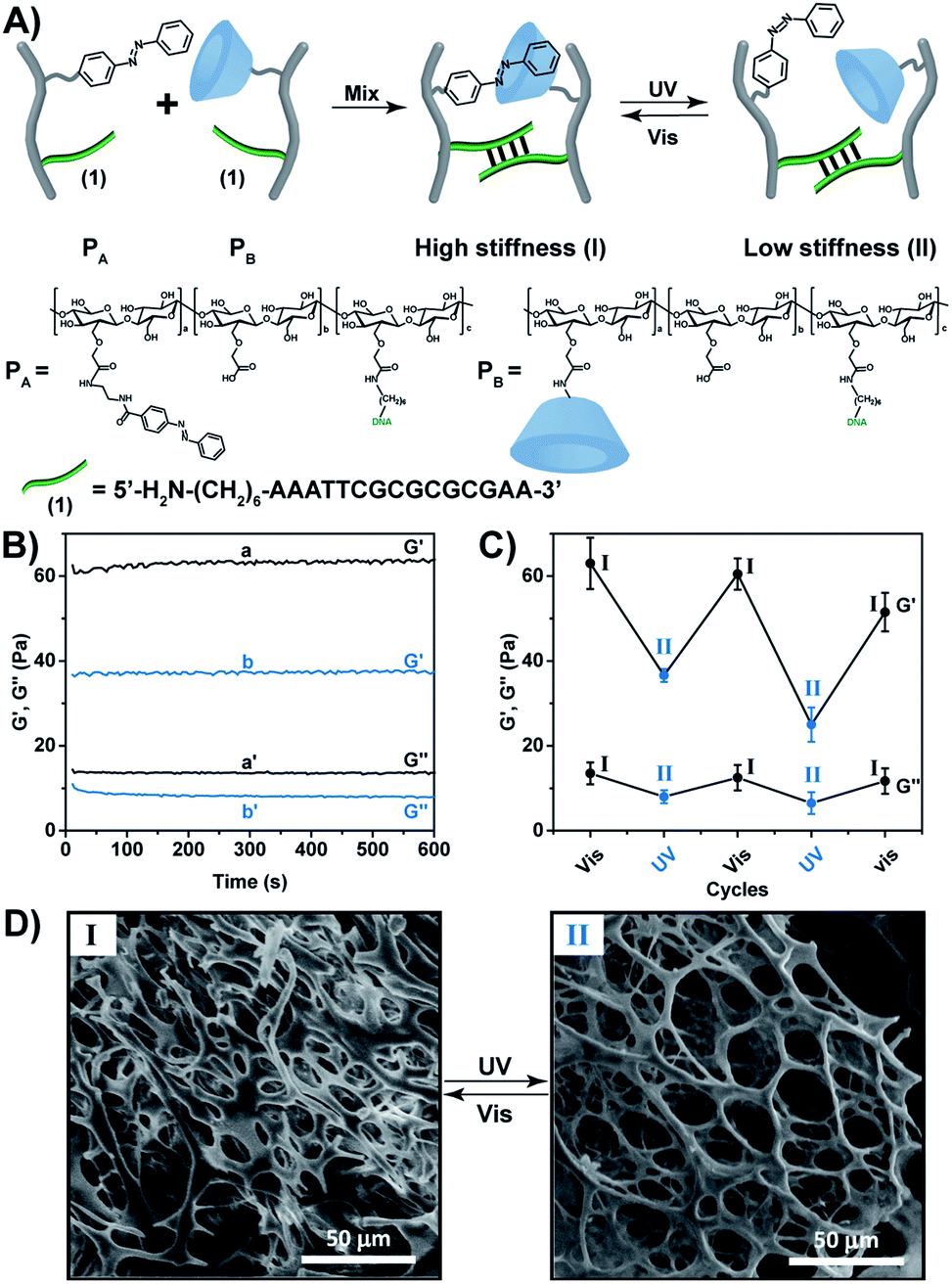 | ||
| Fig. 1 (A) Schematic composition of a photo-responsive CMC hydrogel crosslinked by self-complementary duplexes (1)/(1) and trans-azobenzene/β-CD complexes. (B) Rheometry analysis corresponding to: (a and a′) the G′ and G′′ of the CMC hydrogel crosslinked by the (1)/(1) and trans-azobenzene/β-CD complexes; (b and b′) the G′ and G′′ of the (1)/(1)-crosslinked hydrogel in the presence of the cis-azobenzene tethers. (C) The reversible and switchable changes of the G′ and G′′ upon the photoisomerization of the (1)/(1)-crosslinked hydrogel in the presence of the trans-azobenzene/β-CD complex (state I) and in the presence of the cis-azobenzene and β-CD units (state II). (D) SEM images of: (I) the stiff hydrogel in state I; (II) the hydrogel of low stiffness in state II. | ||
The light-induced control over the stiffness properties of the hydrogel was then applied to develop a shape-memory hydrogel, Fig. 2. In this system, the stiff hydrogel crosslinked by the trans-azobenzene/β-CD and the (1)/(1) duplex crosslinkers was prepared in the mold and extruded from it as the triangle shape, Fig. 2A. Subjecting the hydrogel to UV irradiation (λ = 365 nm) resulted in the dissociation of the supramolecular crosslinking units due to the isomerization of the trans-azobenzene to cis-azobenzene that lacks affinity toward β-CD. The separation of the trans-azobenzene/β-CD bridges leads to a shapeless, quasi-liquid hydrogel. The resulting hydrogel includes, however, the (1)/(1) duplex units that provide an internal memory code for the reshaping of the hydrogel. The entangled polymer chain network provides the code to retain the spatial positions of the azobenzene and β-CD functionalities and the configuration of these polymer network to recover the original shape. That is, upon the reverse photoisomerization of the cis-state to the trans-state (λ > 420 nm), the network-dictated formation of the trans-azobenzene/β-CD complexes recovers the triangle-shaped stiff hydrogel. Fig. 2B shows the cyclic shape-memory properties of the hydrogel. Irradiation of the stiff triangle-shaped hydrogel in state I (λ = 365 nm) leads to the separation of the azobenzene/β-CD bridges and to the formation of the quasi-liquid, shapeless hydrogel in state II. Further visible-light illumination of the quasi-liquid hydrogel (λ > 420 nm) restores the triangle shape of the stiff hydrogel crosslinked by the trans-azobenzene/β-CD and (1)/(1) tethers (state I). Using the Image-Pro Plus software, we estimate that the transition between the shaped (stiff) state and the quasi-liquid state involves a volume increase of ca. 25%.
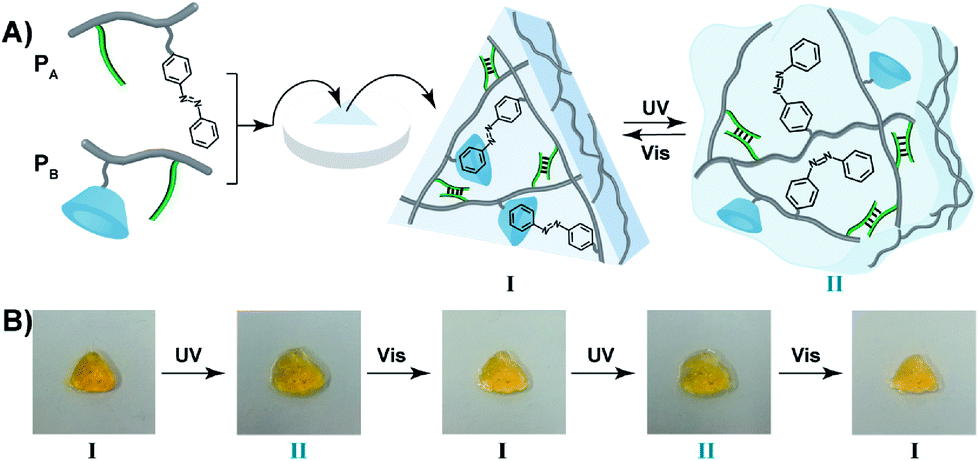 | ||
| Fig. 2 (A) Schematic preparation of a triangle-shaped CMC hydrogel crosslinked by the (1)/(1) duplexes and trans-azobenzene/β-CD complexes, and light-induced reversible transitions between the stiff shaped hydrogel I and the shapeless quasi-liquid hydrogel II that is only crosslinked by the (1)/(1) bridges. (B) Macroscopic images of the reversible light-induced transitions of the hydrogel between the triangle-shaped hydrogel I and the quasi-liquid shapeless hydrogel II, respectively. | ||
In the next step, we designed a hybrid hydrogel that includes two stimuli-triggered crosslinking units, where each of these bridges may act as a shape-memory code. Two CMC polymer chain PC and PD were synthesized, Fig. 3A. The CMC polymer chain PC was modified with the photoisomerizable trans-azobenzene units and the guanosine-rich tethers (2) that represent the sequences of G-quadruplex subunits. The CMC polymer scaffold PD includes β-cyclodextrin receptor units and guanosine-rich tethers (2). The average loading of the trans-azobenzene units and the guanosine-rich tethers (2) on the polymer chains PC was evaluated to be 1:10 and 1:87 by 1H-NMR and absorption spectroscopy, respectively (see Fig. S1, S11 and S13†). Similarly, the average loading of the β-cyclodextrin receptors and of the guanosine-rich tethers (2) on the polymer chains PD was evaluated to be 1:10 and 1:87 by 1H-NMR and absorption spectroscopy, respectively (Fig. S3, S12 and S13†). For further characterization of the polymers PC and PD, see ESI (Fig. S9, S14, S15 and Table S2).† The mixture of the polymers PC and PD yields the stimuli-responsive hydrogel in the presence of K+-ions. The hydrogel is crosslinked by the supramolecular trans-azobenzene/β-CD bridges and by the K+-ion-stabilized G-quadruplex units (that are composed by the two half G-rich subunits (2)). Rheometry experiments show that the storage modulus G′ and loss modulus G′′ of the hydrogel in state I correspond to 83 Pa and 12 Pa, respectively (Fig. 3B). Photoisomerization of the trans-azobenzene to cis-azobenzene (λ = 365 nm) separates the supramolecular azobenzene/β-CD bridges, leading to the hydrogel of low stiffness in state II that is crosslinked by the K+-ion-stabilized G-quadruplex units (G′ ≈ 51 Pa, G′′ ≈ 7 Pa). Further illumination of the hydrogel in state II with visible light (λ > 420 nm) restores the hydrogel in state I (G′ ≈ 82 Pa; G′′ ≈ 11 Pa). Subjecting the stiff hydrogel in state I to 18-crown-6-ether dissociates the G-quadruplex bridges by the elimination of K+-ions that stabilize the G-quadruplex. This results in the hydrogel of low stiffness in state III, that is only crosslinked by the trans-azobenzene/β-CD supramolecular bridges (G′ ≈ 56 Pa, G′′ ≈ 8 Pa). By the reversible treatment of the hydrogel in state III with K+-ions, the stiff hydrogel in state I is regenerated. The hydrogel can be cycled between high stiffness and low stiffness states by subjecting the stiff hydrogel in state I to reversible photoisomerization steps between the trans-azobenzene/cis-azobenzene states (λ > 420 nm/λ = 365 nm) or G-quadruplex/random strands (in the presence of K+-ions/crown ether), Fig. 3C. Additional SEM images of the hydrogels in their different states support the stiffness properties of the hydrogels, Fig. 3D. The hydrogel in state I shows highly crosslinked structures of small pores (I), whereas the triggered transition of the hydrogel in state I to the hydrogel in state II or III generates hydrogels with lower crosslinking and large pores (images II and III, respectively).
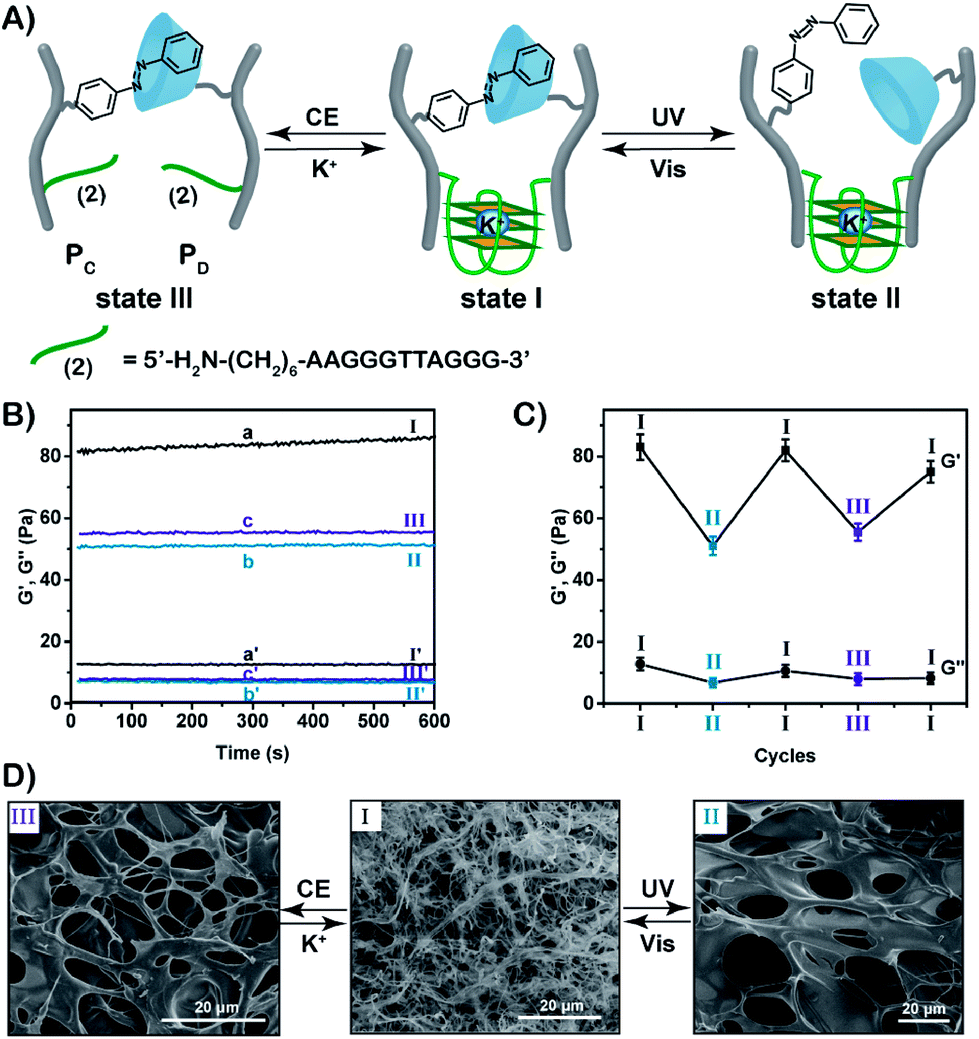 | ||
| Fig. 3 (A) Schematic composition of a dually signal-triggered CMC hydrogel that is crosslinked by K+-ion-stabilized G-quadruplexes and trans-azobenzene/β-CD stimuli-responsive bridges. Scheme presents the reversible light-induced transitions of the stiff hydrogel to the hydrogel of low stiffness that includes the cis-azobenzene. The scheme also presents the reversible and switchable transitions of the stiff hydrogel to a hydrogel of lower stiffness in the presence of 18-crown-6-ether and K+ ions. (B) Rheometry results corresponding to: (a and a′) the G′ and G′′ of the hydrogel crosslinked by the trans-azobenzene/β-CD bridges and the K+-ion-stabilized G-quadruplex bridges, respectively; (b and b′) the G′ and G′′ of the hydrogel crosslinked only by K+-ions-stabilized G-quadruplexes, respectively. (c and c′) the G′ and G′′ of the hydrogel crosslinked only by the trans-azobenzene/β-CD units, respectively. (C) Cyclic and reversible transitions of the hydrogel in state I to the hydrogel in state II and state III. (D) The SEM images corresponding to the CMC hydrogel in state I, II and III. | ||
The bimodal control over the stiffness properties of the G-quadruplex and trans-azobenzene/β-CD-crosslinked hydrogel by means of two alternative trigger, i.e., 18-crown-6-ether or light, were then applied to induce the controlled release of a load from the hydrogel, Fig. 4. Tetramethylrhodamine (TMR)-modified dextran was incorporated in the stiff hydrogel crosslinked by the G-quadruplex and trans-azobenzene/β-CD complex. No release of the TMR-dextran load from the hydrogel was observed after a time-interval of 4 h, Fig. 4, curve (a). The triggered photoisomerization of the trans-azobenzene to cis-azobenzene (λ = 365 nm, irradiation time 10 min) resulted in the hydrogel of lower stiffness, due to the separation of the azobenzene/β-CD crosslinking units. The resulted hydrogel that exhibits lower stiffness allowed the release of the TMR-dextran load, Fig. 4, curve (b). Similarly, the stiff hydrogel was treated with 18-crown-6-ether. Fig. 4, curve (c), shows the fluorescence spectrum of the released TMR-dextran after a time-interval of 4 h. Evidently, the 18-crown-6-ether-induced dissociation of the G-quadruplex units and the accompanying low stiffness of the hydrogel resulted in the release of the load. Note that the release of the load from the G-quadruplex-crosslinked hydrogel is of lower efficiency, as compared to the trans-azobenzene/β-CD crosslinked hydrogel. These results are consistent with the rheometry features of the hydrogel (state II and state III).
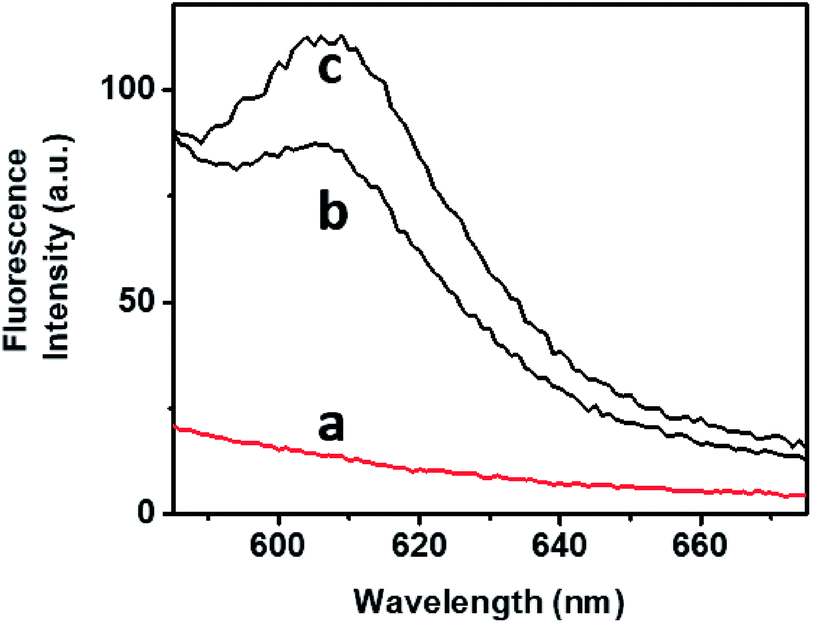 | ||
| Fig. 4 Fluorescence spectra of the released TMR-dextran from the hydrogel (state I) (a) without irradiation or without adding CE, (b) upon UV light irradiation, (c) upon treatment of the hydrogel with CE. | ||
The bifunctional stimuli-responsive hydrogel crosslinked by the trans-azobenzene/β-CD and K+-ion-stabilized G-quadruplex bridging units, was then applied to develop a shape-memory hydrogel, where each of the crosslinking elements is alternately applied as a shape-memory code, Fig. 5. The stiff hydrogel crosslinked by the trans-azobenzene/β-CD and K+-ion-stabilized G-quadruplex was prepared in a triangle-shaped mold. The extruded triangle-shaped hydrogel was subjected to UV light irradiation (λ = 365 nm) that dissociated the trans-azobenzene/β-CD bridging units. The low stiffness of the resulting hydrogel yielded a quasi-liquid shapeless hydrogel. The K+-ion-stabilized G-quadruplex bridging units provide, however, an internal memory for restoring the triangle-shaped hydrogel. The G-quadruplex units provide the instructive memory of chain entanglement in the hydrogel to restore the shaped hydrogel upon the reverse photoisomerization of the cis-azobenzene units associated with the quasi-liquid hydrogel (λ > 420 nm). The light-induced reversible shape-memory functions of the hydrogel are presented in Fig. 5A. Alternatively, the treatment of the triangle-shaped hydrogel in state I with 18-crown-6-ether dissociates the G-quadruplex bridges, leading to the shapeless, quasi-liquid hydrogel in state III. The trans-azobenzene/β-CD bridges of the low stiffness hydrogel provide the memory code for re-shaping the hydrogel. That is, the dictated entanglement of the polymer chains by means of the trans-azobenzene/β-CD provides the instructive information for the dictated re-assembly of the triangle-shaped hydrogel upon the re-addition of K+ ions, Fig. 5A. The cyclic and reversible shape-memory functions of the hydrogel upon the stepwise addition of crown ether (CE) and K+-ions are shown in Fig. 5B. Using the Image-Pro Plus software, we estimate that the light-induced transition of the stiff and shaped trans-azobenzene/β-CD and K+-ion-stabilized hydrogel to the quasi-liquid state that is crosslinked only by the G-quadruplexes involves a volume change of ca. 45%. Similarly, the 18-crown-6-ether-stimulated transition of the shaped hydrogel to the quasi-liquid state crosslinked by the trans-azobenzene/β-CD involves a volume increase of ca. 55%.
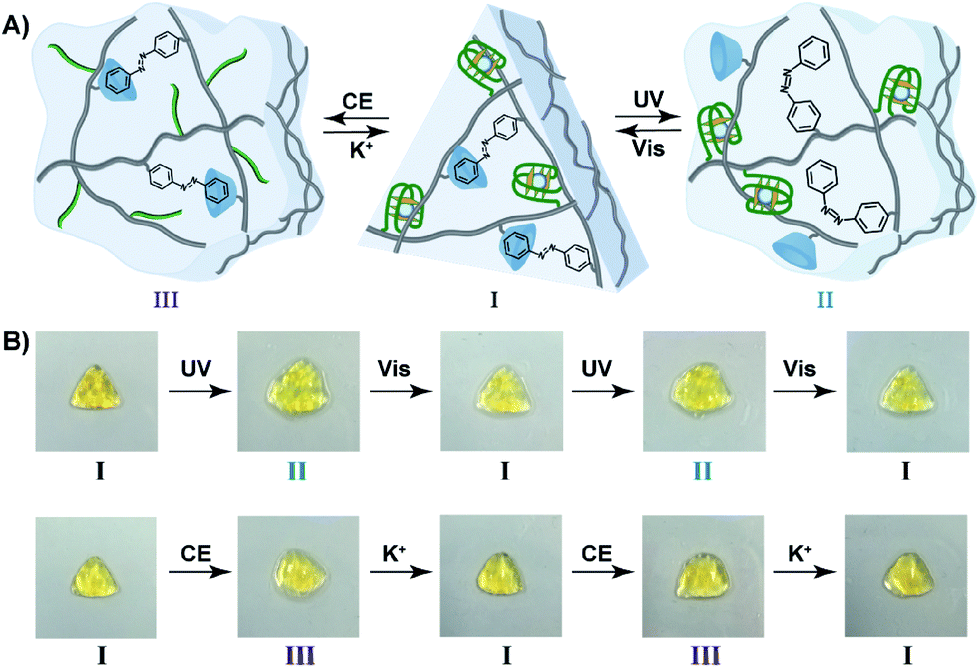 | ||
| Fig. 5 (A) Schematic preparation of the stiff triangle-shaped CMC hydrogel crosslinked by the trans-azobenzene/β-CD and K+-ion-stabilized G-quadruplex bridges. The scheme describes the dually-triggered reversible transitions of the hydrogel between the shaped triangle hydrogel in state I and the quasi-liquid hydrogel in state II, and the reversible transitions between the shaped hydrogel in state I and the quasi-liquid hydrogel in state III, respectively. (B) Macroscopic images corresponding to shape-memory hydrogel transitions, (top) reversible and switchable light-induced transitions between a triangle-shaped hydrogel in state I and the shapeless quasi-liquid hydrogel in state II; (bottom) reversible and switchable transitions between the triangle-shaped hydrogel in state I and the quasi-liquid shapeless hydrogel in state III, using K+ ions and 18-crown-6-ether as triggers. | ||
For further experimental support on the reversible re-configuration of the K+-stabilized G-quadruplex-crosslinked hydrogel and its separation, in the presence of 18-crown-6-ether, into random-coil nucleic acid-modified hydrogel matrices, see Fig. S16† and the accompanying discussion. Fig. S16† shows the method and experimental results to follow the reversible reconfiguration of the G-quadruplex and its separation in the hydrogel matrix in the presence of K+-ions and 18-crown-6-ether, respectively. We make use of the fact that the K+-stabilized G-quadruplex binds hemin, and the resulting hemin/G-quadruplex exhibits horseradish peroxidase DNAzyme activities, where the catalyzed oxidation of Amplex Red by H2O2 generates the fluorescent resorufin product (Fig. S16A†). The hydrogel crosslinked by the K+-stabilized G-quadruplexes and the trans-azobenzene/β-CD units reveals, in the presence of hemin and H2O2, catalytic functions reflected by the formation of resorufin, Fig. S16B,† curve (a). In contrast, treatment of the K+-stabilized G-quadruplex-crosslinked hydrogel with 18-crown-6-ether leads to the separation of the G-quadruplex units, and to the loss of the catalytic activity towards the oxidation of Amplex Red to resorufin by H2O2, Fig. S16B,† curve (b). These results confirm that the G-quadruplexes are presented as crosslinkers in the hydrogel matrix, in the presence of K+-ions. Furthermore, the results imply that in the presence of 18-crown-6-ether, the G-quadruplex crosslinking units are separated. It should be noted that the transition of the shaped hydrogel to the quasi-liquid state, in the presence of 18-crown-6-ether, and the reverse re-assembly of the quasi-liquid state into the shaped state are quite slow, and each of the transitions proceeds for a time-interval of ca. 4 h after the application of the respective trigger. However, the photo-induced shape-memory transitions are faster. The irradiation (λ = 365 nm) of the shaped stiff hydrogel for a time-interval of 10 min, resulted in the quasi-liquid state. Similarly, illumination (λ > 420 nm) of the quasi-liquid state for 10 min recovered the original triangle-shaped hydrogel.
The dually triggered transitions of the trans-azobenzene/β-CD and G-quadruplex-crosslinked hydrogel between high and low stiffness were not only used as a hydrogel exhibiting shape-memory functions, but also as a functional matrix that reveals self-healing properties, Fig. 6. The hydrogel of low stiffness that was crosslinked by the trans-azobenzene/β-CD units was cut into two pieces that were physically inter-connected, Fig. 6A. The inter-connected hydrogel was subjected to K+-ions for a time-interval of 4 h. After this time-interval, the gel was self-healed into a non-separable, united hydrogel crosslinked by the K+-ion-stabilized G-quadruplexes and the trans-azobenzene/β-CD supramolecular complexes. Then the resulting self-healed hydrogel could be cycled between low and high stiffness states. A control experiment of treating the two physically connected hydrogel pieces without K+ ions did not lead to the healing of the hydrogel, and gentle shaking of the inter-connected hydrogel led to the separation of the hydrogel pieces, Fig. S17A.† In analogy, the light-induced self-healing of the hydrogel was demonstrated, Fig. 6B. The K+-ion-stabilized G-quadruplex-bridged hydrogel including the non-bridged cis-azobenzene and β-cyclodextrin units was cut into two pieces. The pieces were physically inter-connected and subjected to visible irradiation (λ > 420 nm) for a time-interval of 15 min. The resulting hydrogel revealed self-healing properties, and the light-induced process led to the formation of a non-separable hydrogel matrix crosslinked by the two crosslinking motifs (the K+-stabilized G-quadruplex and the trans-azobenzene/β-CD complex). Then, the resulting non-separable hydrogel could be cycled between lower and higher stiffness states by reversible photoisomerization of the azobenzene units between the cis and trans isomer states, respectively. In a control experiment, the physically connected two pieces were not irradiated, and gentle shaking of the pieces led to their separation (Fig. S17B†). Furthermore, UV irradiation of the two inter-connected pieces did not yield a healed matrix, implying that possible heating of the matrix is not the source for the healing process. It should be noted that the initial inter-connection of the hydrogel pieces is not mandatory to reach the self-healing effect. Fig. S18† describes the self-healing of two separated pieces of low-stiffness hydrogel crosslinked by the trans-azobenzene/β-CD units upon shaking and addition of K+-ions as the healing trigger.
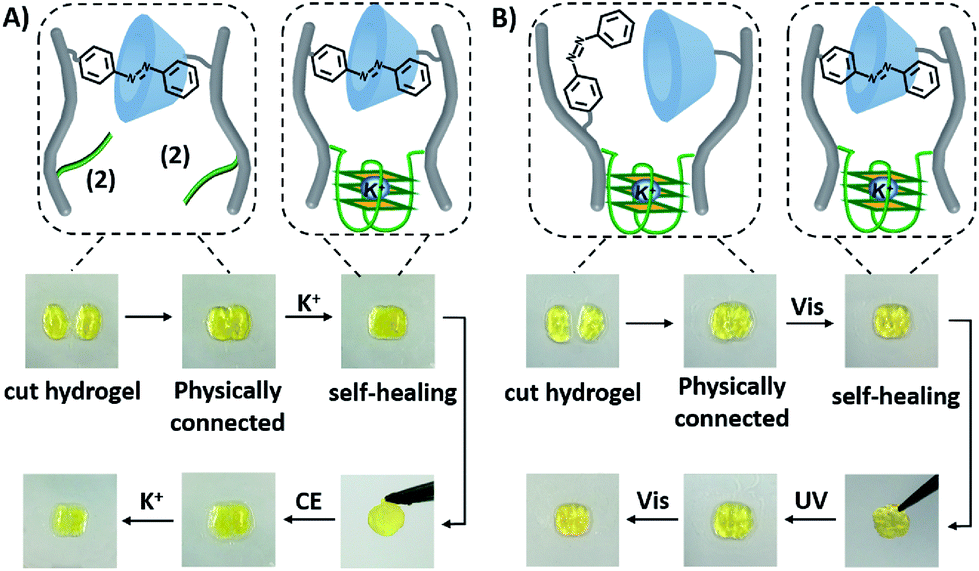 | ||
| Fig. 6 Self-healing functions of the CMC hydrogel crosslinked by the trans-azobenzene/β-CD and the K+-ion-stabilized G-quadruplex units: (A) Self-healing of physically connected CMC hydrogel pieces crosslinked by the trans-azobenzene/β-CD units via the cooperative formation of the healed K+-ion-stabilized G-quadruplexes. Images show the steps of self-healing and the switchable properties of the resulting healed hydrogel. (B) Self-healing of physically connected CMC hydrogel pieces crosslinked by the K+-ions-stabilized G-quadruplexes via the cooperative formation of the healed trans-azobenzene/β-CD units. Images show the steps of the self-healing and the switchable properties of the resulting healed hydrogel. | ||
Conclusions
The present study has introduced hybrid hydrogels that are cooperatively stabilized by photoisomerizable trans-azobenzene/β-cyclodextrin and nucleic acids as crosslinkers. In view of the G′ and G′′ values of the different hydrogels, we conclude that all hydrogel matrices are soft materials with moderate stiffness properties. In contrast to most previous stimuli-responsive DNA-based hydrogels that applied polyacrylamide as the polymeric backbone of the hydrogels, the present study introduced carboxymethyl cellulose (CMC) as the backbone matrix of the hydrogels. The availability of carboxylic acid residues on the CMC matrix provides effective anchoring sites for the conjugation of a variety of functional tethers. One system included the crosslinking of the hydrogel by trans-azobenzene/β-cyclodextrin supramolecular complexes and by duplex nucleic acid bridges. The stiffness of the hydrogel was shifted between high and low stiffness values by the cyclic switchable photoisomerization of the trans-azobenzene units to the cis-azobenzene units that lack binding affinity toward β-CD. These features were used to apply the photo-responsive hydrogel as a shape-memory. The second hydrogel system included a carboxymethyl cellulose backbone crosslinked by two stimuli-responsive units composed of trans-azobenzene/β-cyclodextrin and K+-ion-stabilized G-quadruplex. The two types of crosslinkers were triggered individually to be separated by light (photoisomerization of the trans-azobenzene to cis-azobenzene) or by chemical means (separation of the G-quadruplexes by 18-crown-6-ether). These led to the development of a hydrogel that exhibited switchable and cyclic stiffness properties controlled either by light or chemical agents. The control over the stiffness properties enables the development of shape-memory or self-healing hydrogels that use two different codes to stimulate these processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Israel Science Foundation.Notes and references
- Y. Lu, A. A. Aimetti, R. Langer and Z. Gu, Nat. Rev. Mater., 2016, 2, 16075 CrossRef.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef PubMed.
- M. P. Lutolf, Nat. Mater., 2009, 8, 451–453 CrossRef PubMed.
- S. Kiyonaka, K. Sada, I. Yoshimura, S. Shinkai, N. Kato and I. Hamachi, Nat. Mater., 2004, 3, 58–64 CrossRef PubMed.
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- X. Guo, J. Wang, L. Li, D. T. Pham, P. Clements, S. F. Lincoln, B. L. May, O. Chen, L. Zheng and R. K. Prud'Homme, Macromol. Rapid Commun., 2010, 31, 300–304 CrossRef PubMed.
- E. a. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251–14260 CrossRef PubMed.
- J. Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef PubMed.
- A. Harada, Y. Takashima and M. Nakahata, Acc. Chem. Res., 2014, 47, 2128–2140 CrossRef PubMed.
- C. Koopmans and H. Ritter, Macromolecules, 2008, 41, 7416–7422 CrossRef.
- S. Datta, N. Dey and S. Bhattacharya, Chem. Commun., 2017, 53, 2371–2374 RSC.
- Q. Wang, J. L. Mynar, M. Yoshida, E. Lee, M. Lee, K. Okuro, K. Kinbara and T. Aida, Nature, 2010, 463, 339–343 CrossRef PubMed.
- J. N. Hunt, K. E. Feldman, N. A. Lynd, J. Deek, L. M. Campos, J. M. Spruell, B. M. Hernandez, E. J. Kramer and C. J. Hawker, Adv. Mater., 2011, 23, 2327–2331 CrossRef PubMed.
- Y. Cao and H. Li, Chem. Commun., 2008, 4144–4146 RSC.
- R. M. Meudtner and S. Hecht, Macromol. Rapid Commun., 2008, 29, 347–351 CrossRef.
- N. Holten-Andersen, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef PubMed.
- M. Fadeev, G. Davidson-Rozenfeld, Y. Biniuri, R. Yakobi, R. Cazelles, M. A. Aleman-Garcia and I. Willner, Polym. Chem., 2018, 9, 2905–2912 RSC.
- D. Yang, M. R. Hartman, T. L. Derrien, S. Hamada, D. An, K. G. Yancey, R. Cheng, M. Ma and D. Luo, Acc. Chem. Res., 2014, 47, 1902–1911 CrossRef PubMed.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef.
- W. H. Chen, W. C. Liao, Y. S. Sohn, M. Fadeev, A. Cecconello, R. Nechushtai and I. Willner, Adv. Funct. Mater., 2018, 28, 1705137 CrossRef.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- H. H. Yang, H. P. Liu, H. Z. Kang and W. H. Tan, J. Am. Chem. Soc., 2008, 130, 6320–6321 CrossRef PubMed.
- J. Ren, Y. Hu, C.-H. Lu, W. Guo, M. A. Aleman-Garcia, F. Ricci and I. Willner, Chem. Sci., 2015, 6, 4190–4195 RSC.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef PubMed.
- K. Kuroiwa, T. Shibata, A. Takada, N. Nemoto and N. Kimizuka, J. Am. Chem. Soc., 2004, 126, 2016–2021 CrossRef PubMed.
- Y. L. Zhao and J. Fraser Stoddart, Langmuir, 2009, 25, 8442–8446 CrossRef PubMed.
- A. M. Rosales, K. M. Mabry, E. M. Nehls and K. S. Anseth, Biomacromolecules, 2015, 16, 798–806 CrossRef PubMed.
- S. Tamesue, Y. Takashima, H. Yamaguchi, S. Shinkai and A. Harada, Angew. Chem., Int. Ed., 2010, 49, 7461–7464 CrossRef PubMed.
- I. Tomatsu, A. Hashidzume and A. Harada, J. Am. Chem. Soc., 2006, 128, 2226–2227 CrossRef PubMed.
- C. Yu, C. F. Wang and S. Chen, Adv. Funct. Mater., 2014, 24, 1235–1242 CrossRef.
- Y. Zhang, B. Yang, X. Zhang, L. Xu, L. Tao, S. Li and Y. Wei, Chem. Commun., 2012, 48, 9305–9307 RSC.
- J. Liu, Soft Matter, 2011, 7, 6757–6767 RSC.
- J. S. Kahn, Y. Hu and I. Willner, Acc. Chem. Res., 2017, 50, 680–690 CrossRef PubMed.
- Y. H. Roh, R. C. H. Ruiz, S. Peng, J. B. Lee and D. Luo, Chem. Soc. Rev., 2011, 40, 5730–5744 RSC.
- C. H. Lu, X. J. Qi, R. Orbach, H. H. Yang, I. Mironi-Harpaz, D. Seliktar and I. Willner, Nano Lett., 2013, 13, 1298–1302 CrossRef PubMed.
- W. Guo, C. H. Lu, X. J. Qi, R. Orbach, M. Fadeev, H. H. Yang and I. Willner, Angew. Chem., Int. Ed., 2014, 53, 10134–10138 CrossRef PubMed.
- T. Liedl, H. Dietz, B. Yurke and F. Simmel, Small, 2007, 3, 1688–1693 CrossRef PubMed.
- C. H. Lu, W. Guo, Y. Hu, X. J. Qi and I. Willner, J. Am. Chem. Soc., 2015, 137, 15723–15731 CrossRef PubMed.
- J. S. Kahn, A. Trifonov, A. Cecconello, W. Guo, C. Fan and I. Willner, Nano Lett., 2015, 15, 7773–7778 CrossRef PubMed.
- Y. Wu, D. Wang, I. Willner, Y. Tian and L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 7790–7794 CrossRef PubMed.
- H. Meng and G. Li, Polymer, 2013, 54, 2199–2221 CrossRef.
- M. Yoshida, R. Langer, A. Lendlein and J. Lahann, Polym. Rev., 2006, 46, 347–375 Search PubMed.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef.
- Y. Hu, W. Guo, J. S. Kahn, M. A. Aleman-Garcia and I. Willner, Angew. Chem., Int. Ed., 2016, 55, 4210–4214 CrossRef PubMed.
- W. Guo, C. H. Lu, R. Orbach, F. Wang, X. J. Qi, A. Cecconello, D. Seliktar and I. Willner, Adv. Mater., 2014, 27, 73–78 CrossRef PubMed.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973–1985 RSC.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849–2853 CrossRef PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511–516 CrossRef PubMed.
- A. A. Kavitha and N. K. Singha, ACS Appl. Mater. Interfaces, 2009, 1, 1427–1436 CrossRef PubMed.
- W. M. Xu, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2016, 4, 10683–10690 RSC.
- T. V. Chirila, H. H. Lee, M. Oddon, M. M. L. Nieuwenhuizen, I. Blakey and T. M. Nicholson, J. Appl. Polym. Sci., 2014, 131, 1–12 CrossRef.
- J. Cui and A. del Campo, Chem. Commun., 2012, 48, 9302–9304 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02411a |
| This journal is © The Royal Society of Chemistry 2018 |