
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancement of triplet-sensitized upconversion in rigid polymers via singlet exciton sink approach†
Steponas
Raišys
a,
Saulius
Juršėnas
a,
Yoan C.
Simon
 bc,
Christoph
Weder
b and
Karolis
Kazlauskas
*a
bc,
Christoph
Weder
b and
Karolis
Kazlauskas
*a
aInstitute of Photonics and Nanotechnology, Vilnius University, Saulėtekio av. 3, LT-10257 Vilnius, Lithuania. E-mail: karolis.kazlauskas@ff.vu.lt
bAdolphe Merkle Institute, University of Fribourg, Chemin des Verdiers 4, CH-1700 Fribourg, Switzerland
cSchool of Polymer Science and Engineering, University of Southern Mississippi, 118 College Drive #5050, Hattiesburg, Mississippi 39406, USA
First published on 13th July 2018
Abstract
To increase the practical usefulness of solid-state sensitized upconversion (UC) materials as components of solar energy harvesting systems, it is important to identify and suppress loss mechanisms, and increase the UC quantum yield (ΦUC). Here we focus on a benchmark UC system consisting of the emitter 9,10-diphenylanthracene (DPA) and the sensitizer platinum octaethylporphyrin (PtOEP) in a rigid poly(methyl methacrylate) (PMMA) matrix, and show that one of the major losses originates from Förster resonant energy transfer (FRET) from DPA back to PtOEP. Even though DPA emission lies within the PtOEP transparency window, the quantitative assessment of singlet exciton diffusion for samples with a high DPA content evidences that long-range FRET results in effective exciton trapping by PtOEP. A dramatic factor-of-6 reduction of the DPA emission quantum yield occurs even at PtOEP concentrations as low as 0.05 wt%. To alleviate this problem, we demonstrate a new concept based on the introduction of highly emissive sink sites to trap the singlet excitons produced upon annihilation prior to their quenching by the sensitizer. For DPA/PtOEP blends in PMMA, 1,6-bis-[2,5-di(dodecyloxyphenyl)ethynyl]pyrene is shown to be a useful sink, which results in 1.5-fold increase of the ΦUC. A maximum ΦUC of 2.7% was achieved, which is among the highest reported values for rigid sensitized UC polymers.
Introduction
Light upconversion (UC) relying on triplet–triplet annihilation (TTA) in organic molecules after energy transfer from a sensitizer is a useful process in the context of bioimaging,1 photocatalysis,2 oxygen sensing,3 drug delivery,4,5 and in particular solar energy harvesting.6–9 Namely, unexploited sub-bandgap photons are harvested and converted into a radiation of higher energy within the absorption region of a solar cell.10 UC proceeds via the triplet manifold of emitter molecules, which usually cannot be directly excited, and must therefore be populated via sensitization. The sensitizer serves a dual purpose: (i) it ensures the absorption of incident light as well as (ii) the rapid conversion of the photo-excited singlets into triplets via intersystem crossing. These processes are followed by triplet energy transfer to emitter molecules (see ESI Fig. S1† for detailed mechanistic information). When nonradiative decay pathways are unfavored, the population of these long-lived emitter (or annihilator) triplet states increases until TTA results in the formation of emitter singlet excited states, which relax radiatively.8 This process enables UC under weak (<10 mW cm−2) and noncoherent irradiation, which is readily available from the sun.11,12The most efficient sensitized TTA-UC attained so far are limited to solutions/liquids (ΦUC ≈ 30%),13 liquid nanodroplets encapsulated within rigid matrices (ΦUC = 15%)14 or ‘soft’ matrixes, e.g. elastomers,15 gels,16,17 and other materials18 in which the dye molecules enjoy a high degree of rotational and translational motion, and encounter of excited molecular species enable triplet energy transfer and TTA. In polymer matrices, high efficiency is thus readily observed in systems with low glass transition temperature (Tg), i.e. where T > Tg. For practical applications, rigid UC materials (i.e. with high Tg) are often preferable. In such systems, the positions of sensitizers and emitters are fixed, and TTA depends solely on the triplet exciton diffusion. Regrettably, here ΦUC is remarkably low and rarely exceeds 2%, and overcoming this limit proved challenging.19–24 Note that the theoretical maximum for TTA-UC process is defined as 50%, since only one photon can be emitted per two absorbed ones.25
Although a full understanding of the TTA-UC loss mechanisms is still lacking, the major losses in rigid solids are generally associated with poor triplet exciton diffusion.12,25–27 Nevertheless, we recently demonstrated that, in melt-processed UC polymer glasses at high emitter loadings (>25 wt%), exciton diffusion is not the limiting factor.20 Instead, diffusion-enhanced nonradiative decay of triplet emitter excitons was implicated as the dominant quenching mechanism. Another significant loss channel, which becomes increasingly important at high chromophore loadings (which are needed in rigid matrices to enable exciton diffusion), is related to back-transfer of upconverted emitter singlets to the sensitizer (ESI Fig. S1†).10,25,28–31 These losses are particularly severe in the case of recently introduced sensitizers such as semiconductor nanocrystals,10 thermally activated delayed fluorescence compounds,32,33 and direct triplet absorbers,34 which all feature broad absorption bands and lack a significant window of transparency.25 Obviously, long-range Förster resonant energy back-transfer and the diffusion of generated singlet excitons need to be properly managed to avoid diffusion-assisted trapping by the sensitizer and maximize ΦUC.
To this end, detrimental Förster resonant energy transfer (FRET) from the emitter to the sensitizer was systematically investigated in a rigid poly(methyl methacrylate) (PMMA, Tg ≈ 105 °C) matrix by evaluating the singlet exciton diffusion in the presence and absence of sensitizer. Platinum octaethylporphyrin (PtOEP) and 9,10-diphenylanthracene (DPA) were chosen as a model sensitizer–emitter pair for this investigation. Upconverting films were melt-processed,35 to ensure homogeneous DPA distribution at high concentration and high ΦUC.20 The singlet exciton diffusion was quantified via a time-resolved photoluminescence bulk-quenching technique.36 Finally, a singlet exciton sink approach was tested to circumvent the FRET issue and enhance UC efficiency by trapping singlet excitons, at intentionally introduced, highly emissive sink sites, before they reached the sensitizer.
Results and discussion
Impact of FRET
Fig. 1a compares the fluorescence quantum yield (ΦFL) of DPA/PMMA and DPA/PtOEP/PMMA films and demonstrates the fluorescence quenching of DPA as a result of FRET to PtOEP. ΦFL was measured by directly exciting DPA at λex = 405 nm. The minute PtOEP content in DPA/PtOEP/PMMA films (several hundred times smaller than that of DPA) and the low extinction coefficient of PtOEP at λex = 405 nm ensured that sensitizer absorption was negligible. In the absence of sensitizer, ΦFL of DPA was found to be as high as ∼88%, decreasing down to 57% with increasing DPA concentration, due to self-quenching (Fig. 1a).20 In the presence of PtOEP, ΦFL of DPA was reduced to ∼15%, independently of the DPA content. The markedly reduced ΦFL of DPA clearly demonstrates considerable fluorescence quenching by the sensitizer, despite a concentration 300–800 times smaller than that of DPA (15–40 wt%). The quenching effect of the sensitizer is also reflected in fluorescence transients of DPA, which show accelerated decay and a significant drop in fluorescence lifetime from ∼9 to 3 ns upon incorporation of PtOEP (ESI Fig. S2†). DPA fluorescence quenching is explained by the overlap of the DPA emission and the PtOEP absorption (Fig. 1b). Even though DPA–PtOEP is considered to be a well-matched emitter–sensitizer pair, as DPA emission lies within the PtOEP “transparency window” (i.e. between the higher energy Soret-band and the lower energy Q-band), FRET-induced quenching is nevertheless significant and decreases ΦFL by 6 times at a DPA loading of 25 wt%. | ||
| Fig. 1 (a) Fluorescence quantum yield of DPA as a function of its concentration in the PMMA matrix without (circles) and with (squares) PtOEP (cPtOEP = 0.05 wt%). The excitation wavelength (λex = 405 nm) corresponds to the absorption maximum of DPA. Lines are guides to the eye. (b) Normalized absorption (dashed line) and emission spectra (solid line) of melt-processed PtOEP/PMMA (cPtOEP = 0.05 wt%) and DPA/PMMA (cDPA = 25 wt%) films, respectively. The shaded area indicates spectral overlap. | ||
Since the presence of the sensitizer, even in low concentration, was found to have a significant impact on the DPA ΦFL in DPA/PtOEP/PMMA films, the optical properties were systematically investigated as a function of the PtOEP concentration. Fig. 2 shows the decrease of ΦFL and ΦUC of DPA/PtOEP/PMMA films with increasing PtOEP concentrations. To map a broad parameter space, the experiments were performed at different DPA concentrations. Pictures of DPA/PtOEP/PMMA films show a visual increase in PtOEP phosphorescence as PtOEP concentration increases from 0.01 to 0.8 wt% (ESI Fig. S3†). For low PtOEP content and therefore lower absorbance at λex = 532 nm, the scattered green excitation from the laser is more apparent.
 | ||
| Fig. 2 Fluorescence (open symbols) and UC emission (filled symbols) quantum yield of DPA/PtOEP/PMMA films as a function of PtOEP concentration at different DPA concentrations (20, 25, 30 and 35 wt%). The excitation wavelengths for fluorescence and upconversion were λex,FL = 405 and λex,UC = 532 nm, respectively. Lines are guides to the eye. | ||
The ΦFL data indicate that the extent of fluorescence quenching increases with the PtOEP content, irrespective of DPA concentration. At cPtOEP < 10−3 wt%, ΦFL approached 80% but dropped by more than 2 orders of magnitude when the PtOEP content was increased to above 0.4 wt%. Interestingly, the ΦUC follows a similar trend, suggesting that the same emission quenching mechanism is at play, i.e. FRET from DPA to the PtOEP. Note however that a significant ΦUC (up to 1.8%) could be achieved by reducing the sensitizer concentration to 0.01 wt%. Unfortunately, a further reduction of the PtOEP content led to very low light absorption, and consequently a weak UC signal, making the ΦUC measurements unreliable. We stress that in all our experiments both ΦFL and ΦUC are defined in the same way, i.e. as the number of photons emitted per number of photons absorbed. Therefore, while for fluorescence maximal ΦFL is 100%, for UC theoretically attainable maximal ΦUC is only 50% (as TTA-UC process requires two absorbed photons per one emitted).25
To rule out the influence of possible sensitizer aggregation, the PtOEP phosphorescence quantum yield (ΦPh) was measured as a function of its concentration in PMMA (ESI Fig. S4†). ΦPh remained indeed constant up to a PtOEP content of 0.3 wt%, where we surmise aggregation starts to play a role. This finding indicates the absence of PtOEP aggregation in our films and points to FRET as the prime reason for emission quenching.
To quantify the FRET process, the singlet exciton diffusion length (LD) in the upconverting films was evaluated and compared to the average distance needed for the upconverted singlets to reach sensitizer molecules. In the DPA/PtOEP/PMMA films containing 0.05 wt% of PtOEP and 25 wt% of DPA, the average distance (d) between PtOEP molecules is estimated to be ∼14 nm (for a homogeneously dispersed film). Assuming that singlet excitons are generated via UC with equal probability in the entire volume of the film, LD must be at least 1/2 × d (= 7 nm) for the FRET to be effective. In the upconverting films, LD was evaluated via time-resolved fluorescence bulk-quenching measurements.36 For these measurements, films with an increasing concentration of quencher, phenyl-C61-butyric acid methyl ester (PCBM), were fabricated to estimate the relative fluorescence quenching efficiency and to determine the exciton diffusion by applying Stern–Volmer quenching analysis (see ESI Fig. S5†).37 Two sets of films, i.e. with and without PtOEP, were prepared to specifically address the FRET from DPA to the sensitizer.
The results revealed a steady increase of LD from 14 to 25 nm when the DPA concentration increased from 20 to 35 wt% in the absence of PtOEP (Fig. 3, squares). This behavior was found to be fully consistent with an enhanced exciton diffusivity on account of reduced intermolecular distances between DPA molecules. However, the values appear rather large taking into account that LD of singlet excitons in amorphous organic films is typically below 10 nm. The result can be explained on the basis of the work performed by Menke et al.,38 who reported a substantial extension of LD for chromophores that were diluted in a wide bandgap matrix. The dilution was shown to prolong fluorescence lifetime, increase ΦFL, and enlarge spectral overlap. A similar situation is likely occurring in the upconverting films studied in this work, viz. melt-processing ensures good dispersion and minimizes concentration quenching, thus maintaining high ΦFL and long fluorescence lifetime.
 | ||
| Fig. 3 Singlet exciton diffusion length as a function of DPA concentration in DPA/PMMA films with (circles, cPtOEP = 0.05 wt%) and without (squares) PtOEP sensitizer. Lines are guides to the eye. | ||
Conversely, in films with PtOEP, LD was almost independent of the DPA concentration and significantly shorter (∼8 nm) than that obtained for sensitizer-free films (Fig. 3, circles). Moreover, the estimated LD value nearly corresponds to 1/2 × d, confirming that the singlet excitons can readily diffuse to the sensitizer molecules. This result further demonstrates that the PtOEP molecules act as singlet exciton traps in the upconverting films, thereby severely limiting their LD and suppressing the intended radiative singlet decay in DPA. The findings validate the previously obtained ΦFL quenching amounting to ∼80% in DPA/PMMA films in the presence of the sensitizer (Fig. 1a).
Singlet exciton sink approach
Although a long triplet LD is essential to ensure efficient TTA in rigid UC materials, a long singlet LD is highly undesirable, as it can facilitate FRET to the sensitizer, and thus a reduction of ΦUC. A straightforward way to avoid FRET is to reduce the sensitizer concentration (see Fig. 2). Nevertheless, it will inevitably limit light absorption thereby diminishing the generation of triplet states and ultimately the TTA-UC process. In principle, this problem can be solved by sensitizer-free single-component organic TTA-UC systems based on direct triplet absorption induced by heavy atom effect.39 While the general feasibility of this approach has been demonstrated, such systems display very low UC efficiencies, and the low molar absorptivity of the S–T transition limits their usefulness.Here, we explore another solution to circumvent the FRET issue accompanying large singlet LD: a singlet exciton sink approach. The latter entails the intentional introduction of highly emissive exciton sinks, which trap singlets generated by TTA before they can even be quenched by a sensitizer molecule. Interestingly, Yanai and Kimizuka proposed a hypothetical, similar approach based on high-fluorescence-yield singlet energy collectors to improve the performance of TTA-UC systems through precisely controlled spatial organization.40 In contrast, our approach does not require a complex spatial distribution of the system components, but relies instead on homogeneously dispersed singlet sinks. This facile methodology is readily scalable and such materials were straightforwardly attained by the currently utilized melt-processing technique.
To be useful, the singlet sink must meet the following requirements: (i) display high ΦFL (desirably close to 100%), (ii) have a singlet energy that is (slightly) lower than that of the emitter to ensure efficient FRET to the sink (note however that the energy should not be too low to preserve anti-Stokes emission), (iii) the singlet lifetime should be shorter than that of the emitter to ensure rapid emission from the sink, and (iv) the triplet energy must be higher than that of the emitter to avoid triplet exciton depopulation in the emitter.
The pyreneethynylene derivative (PE, 1,6-bis-[2,5-di(dodecyloxyphenyl)ethynyl]pyrene, Fig. 4a) fulfills these requirements and was therefore chosen as a singlet sink for the DPA/PtOEP/PMMA blends presently investigated.41 PE exhibits a reasonably high ΦFL, 75% and 69% in dilute solution and melt-processed PMMA films, respectively, which is adequate for the demonstration of the approach. Moreover, the lowest energy and strongest absorption band of this compound at 435 nm overlaps well with the UC emission maximum of DPA (Fig. 4b). These conditions ensure efficient FRET from DPA to PE. The PE emission is redshifted by ∼12 nm relative to that of DPA, implying that the singlet energy is ∼75 meV lower, which suffices to guarantee exciton trapping at PE sites at ambient and even elevated temperature.
 | ||
| Fig. 4 (a) Chemical structure of the singlet exciton sink PE. (b) Normalized absorption and fluorescence spectra of the PE in PMMA (cPE = 0.01 wt%) and the UC emission spectrum of DPA/PtOEP/PMMA (cDPA = 25 wt%, cPtOEP = 0.01 wt%). The inset shows energy level alignment of DPA/PtOEP system with added singlet sink PE. (c) Fluorescence decays of PE (red points) and DPA (blue points) in PMMA (cPE = 0.01 wt%, cDPA = 25 wt%). Lines are single exponential fits. | ||
The fluorescence decays of PE and DPA in PMMA films are compared in Fig. 4c. Following a mono-exponential decay, PE has a 6-fold shorter fluorescence lifetime (∼1.8 ns) compared to DPA, which ensures much faster radiative relaxation of the trapped excitons and lower probability for their accumulation at the sink sites or back-transfer to DPA.
The triplet energy of PE, as determined from the onset of phosphorescence spectrum (ESI Fig. S7†), is 1.97 eV, which is ∼160 meV higher than that of DPA.42 This is important as it prevents triplet exciton transfer from DPA to PE. The energy level alignment of DPA/PtOEP system with added singlet sink PE is illustrated in the inset of Fig. 4b.
At first glance, the energetics regarding singlet sink may seem rather stringent. For the efficient FRET to the sink its singlet energy must to be lower than that of the emitter to ensure good spectral overlap between sink absorption and emitter emission. On the other hand, sink emission must not be to be too low in order to avoid absorption by the sensitizer's Q band. In part, this problem could be solved by utilizing sensitizers with lower Q band (larger energy separation between Soret- and Q-band). E.g. for NIR range, sensitizers based on metallonaphthalocyanines could be suitable.43 They have larger transparency window (400–700 nm) as compared to that of PtOEP (400–500 nm). On the other hand, finding a highly-emissive sink with desired energy levels should not be a big issue. A vast number of fluorophores (with ΦFL close to 100%) of desired wavelength with typically narrow absorption/emission bands and small Stokes shift are widely available. Semiconductor quantum dots, which emission/absorption wavelength can be finely tuned by varying their size, could be an attractive alternative to organic singlet sink compounds.
To verify the proposed strategy of singlet exciton trapping, the best performing DPA/PtOEP/PMMA mixture (containing 25 wt% DPA and 0.01 wt% PtOEP) was used to create blends with PE. The PE concentration was varied from 0.001 to 1 wt%. For these compositions, ΦUC is shown as a function of PE concentration in Fig. 5. At PE concentrations below 2 × 10−3 wt%, almost no impact of the sink on ΦUC is noticed. However, when the PE content is increased to 0.01 wt%, a 1.5-fold enhancement of ΦUC and a maximum value of 2.7% is observed. Beyond this point ΦUC gradually decreased, perhaps due to PE aggregation and partially due to increased FRET from PE to PtOEP (the Q bands of PtOEP overlap rather well with PE emission). Nevertheless, maximal ΦUC of 2.7% achieved via singlet sink approach is among the highest ΦUC reported so far in rigid amorphous solids.21–23
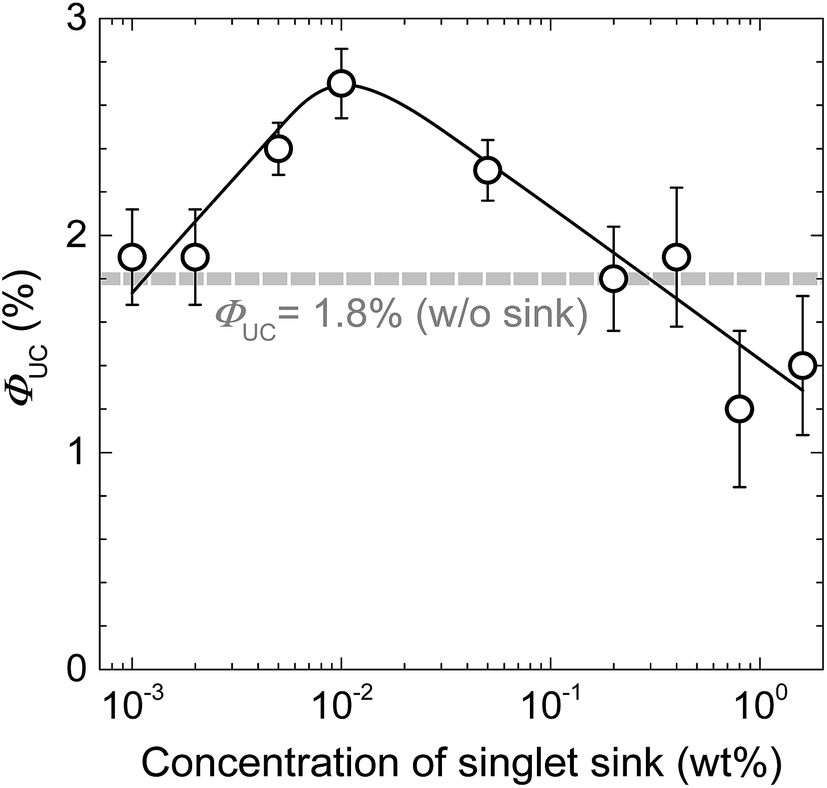 | ||
| Fig. 5 UC quantum yield of DPA/PtOEP/PE singlet sink/PMMA films (cDPA = 25 wt%, cPtOEP = 0.01 wt%) as a function of singlet sink concentration. The solid line is a guide to the eye. The dashed line indicates the maximum ΦUC of the corresponding material without the singlet sink. The error bars were derived by averaging data of three samples. | ||
To confirm that the FRET from PE to PtOEP in turn is responsible for ΦUC quenching observed at PE concentrations > 0.01 wt%, we have estimated and compared FRET rates for the DPA → PtOEP, DPA → PE, PE → DPA and PE → PtOEP processes at the fixed DPA and PtOEP concentrations of 25 wt% and 0.01 wt%, respectively (see ESI Table S2 and Fig. S6†). From this data it is clear that at low singlet sink (PE) concentrations excitons trapped by PE will eventually emit since FRET rates for PE → PtOEP are well below natural decay rate of PE (5 × 108 s−1). Before emission excitons can undergo numerous FRET cycles between PE and DPA due to the large transfer (∼1013 s−1) and back-transfer (∼3 × 1010 s−1) rates. With PE concentration well exceeding 0.01 wt%, FRET rate for PE → PtOEP becomes comparable to PE decay rate and thus can contribute to the observed ΦUC quenching.
Theoretically, since PtOEP (cPtOEP = 0.01 wt%) reduces ΦFL of DPA by a factor of 2.5, from 75% to 30% (Fig. 2), a maximal enhancement of ΦUC of up to 4.5% (1.8% × 2.5) should be attainable if all DPA singlets were collected by PE. The fact that a smaller enhancement of ΦUC is observed indicates that exciton trapping by the sink is incomplete.
A typical UC emission spectrum of the films with 0.01 wt% of PE is shown in Fig. 6. To elucidate its origin, the spectrum was superimposed with that obtained from the films without singlet sink and also with the fluorescence spectrum of a film of PE in PMMA. The comparison of these spectra clearly shows that the UC emission originates from both, the DPA and PE emitters. The quantitative spectral analysis (based on integrated intensities) reveals that the UC emission is composed of ∼2/3 DPA and ∼1/3 PE emission. This result confirms again that the collection of singlets by the sink is not fully effective.
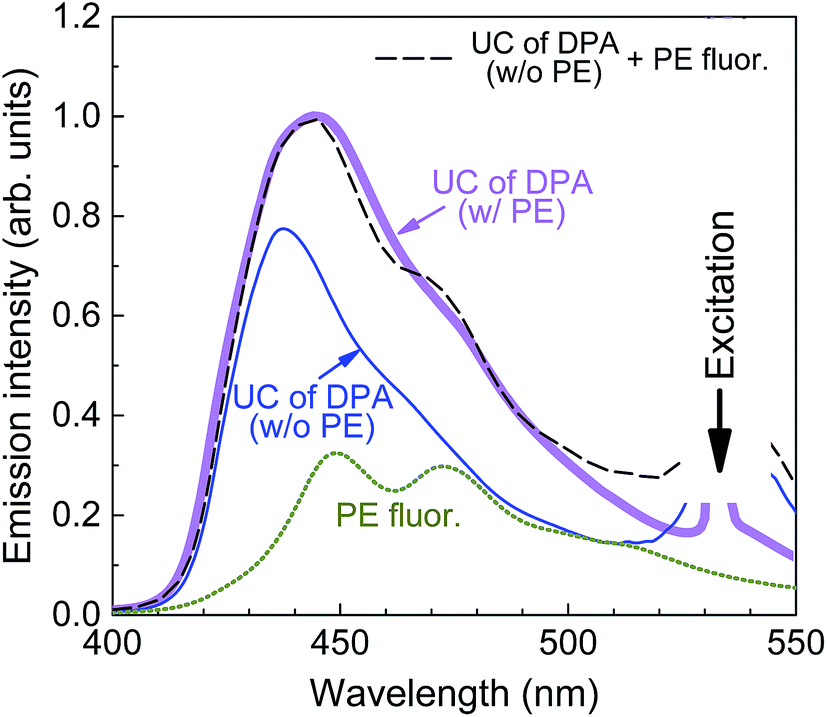 | ||
| Fig. 6 Fluorescence spectrum (dotted line) of the singlet sink PE in PMMA (cPE = 0.01 wt%), UC spectra of DPA/PtOEP/PMMA (cDPA = 25 wt%, cPtOEP = 0.01 wt%) without (thin line) and with (thick line) PE (cPE = 0.01 wt%), and a sum of the first two spectra (dashed line) at a ratio of 1/3 and 2/3, respectively. | ||
Taking into account that at PE concentration of 0.01 wt% FRET rate for PE → PtOEP is negligible as compared to the decay rate of excitons in PE, it is plausible to assume that all the exciton trapped by the sink will contribute to the emission. In other words, for the 1/3 part of UC emission associated with PE we can expect ΦUC to attain its maximum value, i.e. 4.5% (= 1.8% × 2.5), whereas for the rest 2/3 DPA-related part of UC emission only 1.8% as this part is subjected to quenching by PtOEP. Indeed this is confirmed by the sum of ΦUC of both parts 1/3 × 4.5% and 2/3 × 1.8%, which is equal to the overall ΦUC of DPA/PtOEP/PE/PMMA film (2.7%). This result indicates that there are no significant losses once transfer to PE is realized and the most important limitation in this system is DPA → PE transfer.
A plausible way to enhance the collection efficiency is to increase the sink concentration while avoiding its aggregation. Alternatively, the sink can be tethered to the DPA emitter in order to enhance efficient transfer.
In the DPA/PtOEP/PMMA films containing the PE singlet sink, the excitation density dependence of the UC emission intensity exhibited a typical quadratic behavior at lower excitation densities, which changed to linear at a threshold intensity (Ith) at higher power densities (ESI Fig. S8†).44,45Ith indicates the dominance of the TTA process over other decay pathways. It was estimated to be 39 mW cm−2, which is very close to the threshold for the best performing DPA/PtOEP/PMMA films (without the sink).20 Importantly, this invariance signifies that the singlet sink influences neither the TTA, nor the triplet exciton manifold of DPA, and only affects upconverted singlets (vide supra). Specifically, the involvement of PE triplet states would otherwise manifest itself through a reduced concentration of DPA triplets and a significant increase of Ith.
The singlet sink approach was successfully implemented to mitigate UC emission quenching by FRET in a DPA/PtOEP model system and should be readily generalizable to other solid state UC systems, provided that an adequate singlet sink meeting the aforementioned conditions be identified for each system. Certainly, suppressed detrimental energy transfer via introduction of a singlet sink implies some tradeoff between enhancing TTA-UC efficiency and reducing the energy of the harvested state. Therefore from a TTA-UC solar cell perspective, depending on the particular absorber used, a proper sensitizer–emitter and sink combination needs to be chosen to achieve the final improvement in photoconversion efficiency of the cell.
Conclusions
In summary, this work addressed the serious FRET issue limiting radiative decay of the singlet emitter excitons (produced via TTA) through their back-transfer to the sensitizer. This issue is a challenging problem especially for next-generation sensitizers, such as semiconductor nanocrystals and direct triplet absorbers, which lack transparency window for UC emission. It causes substantial UC losses even in the well-studied DPA/PtOEP model system designed to reduce overlap of DPA emission and PtOEP absorption. Backed up by quantitative Stern–Volmer analysis of singlet exciton diffusion in highly DPA-loaded (15–40 wt%) rigid UC films, FRET induced quenching can decrease ΦFL of DPA to 1/6th of its original value for PtOEP concentrations as low as 0.05 wt%. To suppress FRET-assisted singlet exciton quenching by sensitizer molecules, we implemented a singlet exciton sink approach, which necessitated an alternative singlet transfer route to intentionally introduced, highly emissive traps (sinks). The utilization of a pyreneethynylene derivative as the sink for the current solid-state UC system confirmed the validity of our methodology and exhibited very promising results leading to a 1.5-fold increase of ΦUC, while keeping Ith almost unchanged. Although quantitative productive harvesting of upconverted singlets by intentionally incorporating singlet sinks could have improved ΦUC up to 2.5 times (up to 4.5%), the achieved maximal ΦUC of 2.7% remains one of the highest reported so far for rigid amorphous sensitized UC systems. These results obtained for DPA/PtOEP system are extremely encouraging. They suggest that the singlet sink approach can be extended to other UC systems suffering from FRET issues and open the door to a variety of novel energy transfer strategies to increase the efficiency and therefore the usefulness of upconverting systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research at Vilnius University was funded by a grant (No. S-MIP-17-77) from the Research Council of Lithuania.References
- O. S. Kwon, H. S. Song, J. Conde, H. Kim, N. Artzi and J.-H. Kim, ACS Nano, 2016, 10, 1512–1521 CrossRef PubMed.
- R. S. Khnayzer, J. Blumhoff, J. A. Harrington, A. Haefele, F. Deng and F. N. Castellano, Chem. Commun., 2012, 48, 209–211 RSC.
- M. Borisov Sergey, L. Christoph and K. Ingo, Adv. Funct. Mater., 2012, 22, 4360–4368 CrossRef.
- W. Wang, Q. Liu, C. Zhan, A. Barhoumi, T. Yang, R. G. Wylie, P. A. Armstrong and D. S. Kohane, Nano Lett., 2015, 15, 6332–6338 CrossRef PubMed.
- S. H. C. Askes, M. S. Meijer, T. Bouwens, I. Landman and S. Bonnet, Molecules, 2016, 21, 1460 CrossRef PubMed.
- S. Baluschev, T. Miteva, V. Yakutkin, G. Nelles, A. Yasuda and G. Wegner, Phys. Rev. Lett., 2006, 97, 143903 CrossRef PubMed.
- V. Gray, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, Phys. Chem. Chem. Phys., 2014, 16, 10345–10352 RSC.
- T. F. Schulze and T. W. Schmidt, Energy Environ. Sci., 2015, 8, 103–125 RSC.
- Y. Y. Cheng, A. Nattestad, T. F. Schulze, R. W. MacQueen, B. Fückel, K. Lips, G. G. Wallace, T. Khoury, M. J. Crossley and T. W. Schmidt, Chem. Sci., 2015, 7, 559–568 RSC.
- L. Frazer, J. K. Gallaher and T. W. Schmidt, ACS Energy Lett., 2017, 2, 1346–1354 CrossRef.
- A. Monguzzi, R. Tubino, S. Hoseinkhani, M. Campione and F. Meinardi, Phys. Chem. Chem. Phys., 2012, 14, 4322–4332 RSC.
- Y. C. Simon and C. Weder, J. Mater. Chem., 2012, 22, 20817–20830 RSC.
- T. Ogawa, N. Yanai, A. Monguzzi and N. Kimizuka, Sci. Rep., 2015, 5, 10882 CrossRef PubMed.
- R. Vadrucci, A. Monguzzi, F. Saenz, B. D. Wilts, Y. C. Simon and C. Weder, Adv. Mater., 2017, 29, 1702992 CrossRef PubMed.
- A. Monguzzi, F. Bianchi, A. Bianchi, M. Mauri, R. Simonutti, R. Ruffo, R. Tubino and F. Meinardi, Adv. Energy Mater., 2013, 3, 680–686 CrossRef.
- R. Vadrucci, C. Weder and Y. C. Simon, Mater. Horiz., 2014, 2, 120–124 RSC.
- P. Duan, N. Yanai, H. Nagatomi and N. Kimizuka, J. Am. Chem. Soc., 2015, 137, 1887–1894 CrossRef PubMed.
- D. C. Thévenaz, A. Monguzzi, D. Vanhecke, R. Vadrucci, F. Meinardi, Y. C. Simon and C. Weder, Mater. Horiz., 2016, 3, 602–607 RSC.
- P. B. Merkel and J. P. Dinnocenzo, J. Lumin., 2009, 129, 303–306 CrossRef.
- S. Raišys, K. Kazlauskas, S. Juršėnas and Y. C. Simon, ACS Appl. Mater. Interfaces, 2016, 8, 15732–15740 CrossRef PubMed.
- A. Turshatov, D. Busko, N. Kiseleva, S. L. Grage, I. A. Howard and B. S. Richards, ACS Appl. Mater. Interfaces, 2017, 9, 8280–8286 CrossRef PubMed.
- A. Monguzzi, M. Mauri, M. Frigoli, J. Pedrini, R. Simonutti, C. Larpent, G. Vaccaro, M. Sassi and F. Meinardi, J. Phys. Chem. Lett., 2016, 7, 2779–2785 CrossRef PubMed.
- A. Monguzzi, M. Frigoli, C. Larpent, R. Tubino and F. Meinardi, Adv. Funct. Mater., 2012, 22, 139–143 CrossRef.
- M. Wu, D. N. Congreve, M. W. B. Wilson, J. Jean, N. Geva, M. Welborn, T. Van Voorhis, V. Bulović, M. G. Bawendi and M. A. Baldo, Nat. Photonics, 2016, 10, 31–34 CrossRef.
- V. Gray, K. Moth-Poulsen, B. Albinsson and M. Abrahamsson, Coord. Chem. Rev., 2018, 362, 54–71 CrossRef.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef.
- X. Li and M. L. Tang, Chem. Commun., 2017, 53, 4429–4440 RSC.
- S. P. Hill, T. Banerjee, T. Dilbeck and K. Hanson, J. Phys. Chem. Lett., 2015, 6, 4510–4517 CrossRef PubMed.
- N. Yanai and N. Kimizuka, Acc. Chem. Res., 2017, 50, 2487–2495 CrossRef PubMed.
- V. Gray, K. Börjesson, D. Dzebo, M. Abrahamsson, B. Albinsson and K. Moth-Poulsen, J. Phys. Chem. C, 2016, 120, 19018–19026 CrossRef.
- P. E. Keivanidis, S. Baluschev, G. Lieser and G. Wegner, ChemPhysChem, 2009, 10, 2316–2326 CrossRef PubMed.
- T. C. Wu, D. N. Congreve and M. A. Baldo, Appl. Phys. Lett., 2015, 107, 031103 CrossRef.
- N. Yanai, M. Kozue, S. Amemori, R. Kabe, C. Adachi and N. Kimizuka, J. Mater. Chem. C, 2016, 4, 6447–6451 RSC.
- Y. Sasaki, S. Amemori, H. Kouno, N. Yanai and N. Kimizuka, J. Mater. Chem. C, 2017, 5, 5063–5067 RSC.
- S. H. Lee, J. R. Lott, Y. C. Simon and C. Weder, J. Mater. Chem. C, 2013, 1, 5142–5148 RSC.
- J. D. A. Lin, O. V. Mikhnenko, J. Chen, Z. Masri, A. Ruseckas, A. Mikhailovsky, R. P. Raab, J. Liu, P. W. M. Blom, M. A. Loi, C. J. García-Cervera, I. D. W. Samuel and T.-Q. Nguyen, Mater. Horiz., 2014, 1, 280–285 RSC.
- H.-Y. Hsu, J. H. Vella, J. D. Myers, J. Xue and K. S. Schanze, J. Phys. Chem. C, 2014, 118, 24282–24289 CrossRef.
- S. M. Menke, W. A. Luhman and R. J. Holmes, Nat. Mater., 2013, 12, 152–157 CrossRef PubMed.
- K. Okumura, M. Matsuki, T. Yamada, N. Yanai and N. Kimizuka, ChemistrySelect, 2017, 2, 7597–7601 CrossRef.
- N. Yanai and N. Kimizuka, Chem. Commun., 2016, 52, 5354–5370 RSC.
- J. Gupta, S. Vadukumpully and S. Valiyaveettil, Polymer, 2010, 51, 5078–5086 CrossRef.
- H. Goudarzi and P. E. Keivanidis, ACS Appl. Mater. Interfaces, 2017, 9, 845–857 CrossRef PubMed.
- S. Amemori, N. Yanai and N. Kimizuka, Phys. Chem. Chem. Phys., 2015, 17, 22557–22560 RSC.
- A. Haefele, J. Blumhoff, R. S. Khnayzer and F. N. Castellano, J. Phys. Chem. Lett., 2012, 3, 299–303 CrossRef.
- A. Monguzzi, J. Mezyk, F. Scotognella, R. Tubino and F. Meinardi, Phys. Rev. B, 2008, 78, 195112 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02151a |
| This journal is © The Royal Society of Chemistry 2018 |