
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA synergistic LUMO lowering strategy using Lewis acid catalysis in water to enable photoredox catalytic, functionalizing C–C cross-coupling of styrenes†
Elisabeth
Speckmeier
,
Patrick J. W.
Fuchs
and
Kirsten
Zeitler
 *
*
Institut für Organische Chemie, Universität Leipzig, Johannisallee 29, D-04103 Leipzig, Germany. E-mail: kzeitler@uni-leipzig.de
First published on 25th July 2018
Abstract
Easily available α-carbonyl acetates serve as convenient alkyl radical source for an efficient, photocatalytic cross-coupling with a great variety of styrenes. Activation of electronically different α-acetylated acetophenone derivatives could be effected via LUMO lowering catalysis using a superior, synergistic combination of water and (water-compatible) Lewis acids. Deliberate application of fac-Ir(ppy)3 as photocatalyst to enforce an oxidative quenching cycle is crucial to the success of this (umpolung type) transformation. Mechanistic particulars of this dual catalytic coupling reaction have been studied in detail using both Stern–Volmer and cyclic voltammetry experiments. As demonstrated in more than 30 examples, our water-assisted LA/photoredox catalytic activation strategy allows for excess-free, equimolar radical cross-coupling and subsequent formal Markovnikov hydroxylation to versatile 1,4-difunctionalized products in good to excellent yields.
Introduction
Visible light photocatalysis as a powerful tool for the selective generation of radicals has given rise to numerous unprecedented C–C-coupling reactions in recent years.1 Especially alkyl radicals2 have attracted great interest for the formation of highly functionalized natural products and pharmaceuticals.3 Back in 1985, Kellogg, one of the early pioneers of visible light photocatalysis, explored the reactivity of the electron deficient phenacyl radical generated by the photocatalytic single electron reduction of the corresponding phenacyl bromide.4 Since then, this electrophilic radical species has enabled the access to a variety of interesting radical trapping reactions for the implementation of new C–C-coupling transformations.5 However, phenacyl bromide is well known to promote radical polymerizations and also to be susceptible to nucleophilic substitutions (and hence enabling undesired polar reaction pathways) as for example, demonstrated for pyridines6 or under basic conditions.7 We hence assumed, that less activated α-acetoxy acetophenone could be a promising alternative for radical C–C coupling reactions avoiding undesired pathways.Especially, in addition, bromide itself cannot be considered as a photochemically innocent leaving group based on its redox properties: with an oxidation potential of only 0.66 V vs. SCE (Eox(Br˙/Br−) = 0.66 V) mesolytically cleaved bromide anions can easily get oxidized to the corresponding bromine radicals and hence potentially may also form elemental Br2 by most common photocatalysts.8–10 Although, as the reduction potential of the corresponding α-acetoxy acetophenone (Ered = −1.72 V vs. SCE) is more negative than the reduction potential of bromo acetophenone (Ered = −1.45 V vs. SCE), we considered based on our recent C–O-bond cleaving studies,11 that this reduced affinity to accept electrons could, in addition, be beneficially utilized as a promising, assistant tool to enhance reaction control and selectivity by deliberate lowering of the carbonyl LUMO.
Using synergistic photoredox catalysis with amino organocatalysis MacMillan and co-workers could already demonstrate that α-alkylation of aldehydes can be achieved with common α-bromo acetophenone12 and, very recently, as well as with significantly less reactive α-acetoxy derivatives as non-traditional radical precursors.13,14 We hence questioned, if such α-acetoxy derived radicals as “alkylation reagent” could provide the required selectivity for a photocatalytic sp3–sp2 C–C cross-coupling with styrenes followed by an oxidation to give the respective Markovnikov-type functionalized products (Scheme 1). This challenging photocatalytic/radical polar crossover reaction would offer unprecedented access to 1,4-functionalized synthons, such as γ-hydroxyketones, being versatile building blocks for the synthesis of a variety of bioactive compounds.16
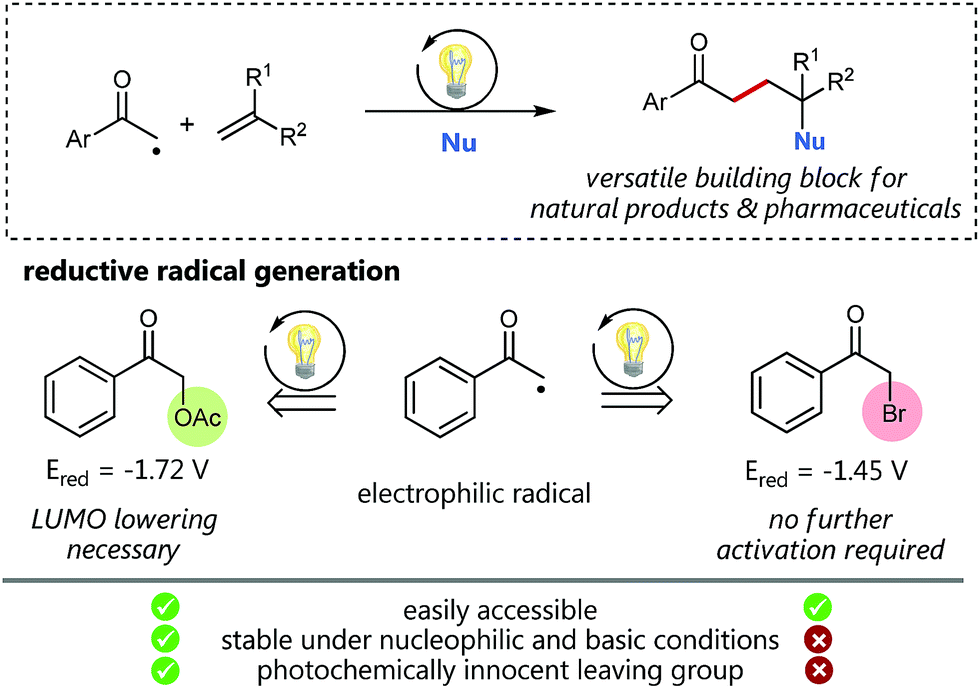 | ||
| Scheme 1 Design plan for a functionalizing sp3–sp2-photocatalytic cross-coupling reaction. | ||
Results and discussion
Based on our previous experience in C–O-bond activation,11 we approached our cross-coupling investigations right from the beginning with a focus on mechanism, especially considering the crucial role of the quenching cycles of the photocatalyst (Scheme 2).17 Initial test reactions employing simple oxidative (Scheme 2/left) and reductive quenching conditions (Scheme 2/right) for the coupling of α-acetoxy acetophenone (1) as radical precursor and 1,1-diphenylethylene (2) as well-established activated coupling partner,18 confirmed our hypothesis, that a reductive quenching cycle would be inappropriate for this intended reaction.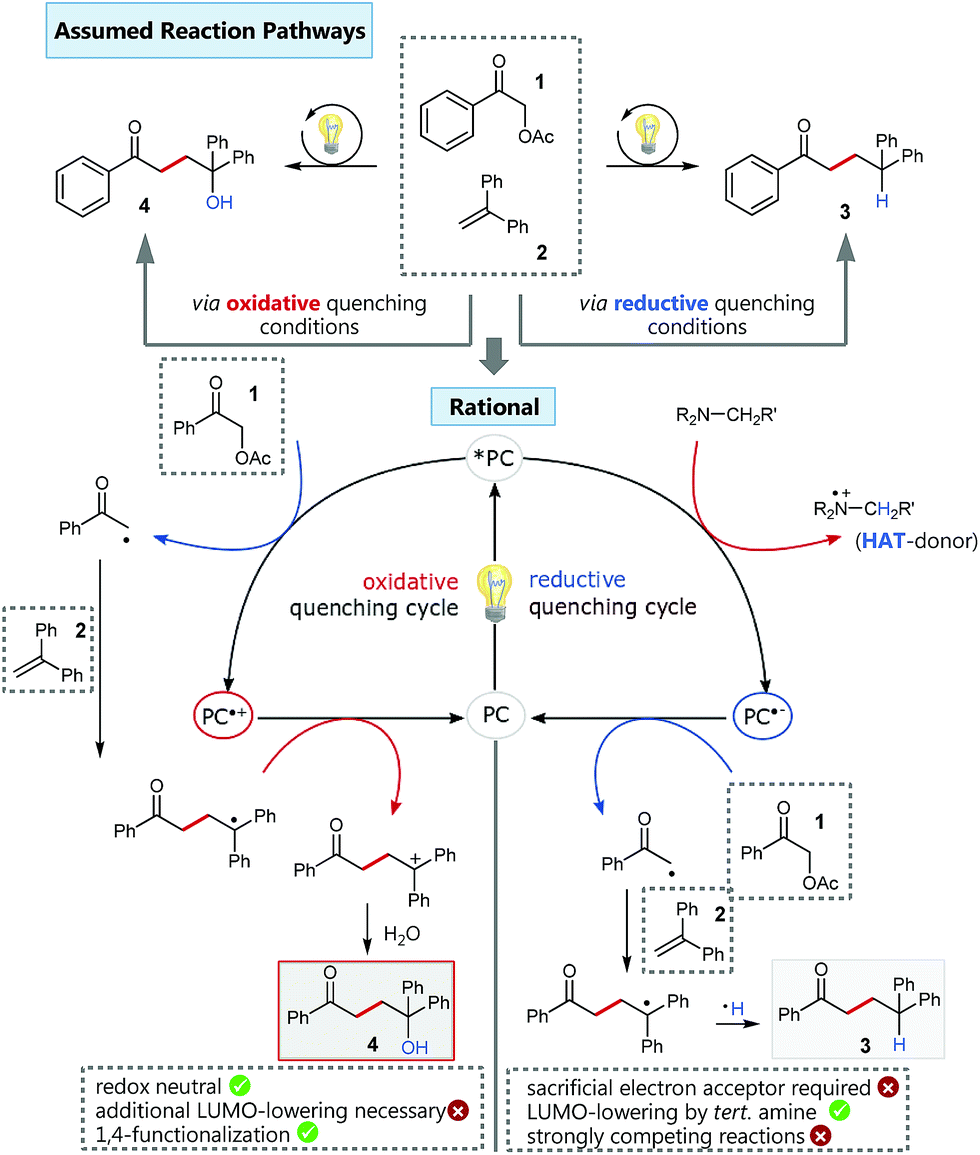 | ||
Scheme 2 Oxidative vs. reductive quenching conditions for the C–C coupling of α-acetoxy acetophenone and 1,1-diphenylethylene and simplified comparison of the corresponding different reaction pathways.  conditions: fac-Ir(ppy)3, 0.5 mmol 1, 1.0 mmol 2 in MeCN/H2O 4 conditions: fac-Ir(ppy)3, 0.5 mmol 1, 1.0 mmol 2 in MeCN/H2O 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, irradiation with blue LEDs; yield of isolated product 4: 33%. :1, irradiation with blue LEDs; yield of isolated product 4: 33%.  conditions: 4CzIPN,15 1.5 equiv. NBu3, 0.5 mmol 1, 1.0 mmol 2 in MeCN/H2O 4:1, irradiation with blue LEDs; yield of isolated product 3: 83%. conditions: 4CzIPN,15 1.5 equiv. NBu3, 0.5 mmol 1, 1.0 mmol 2 in MeCN/H2O 4:1, irradiation with blue LEDs; yield of isolated product 3: 83%. | ||
Using tertiary amines as reductive quenchers typically offers the great advantage of an extensive LUMO-lowering of the carbonyl group via hydrogen bond/PCET19 activation or 2-center/3-electron interaction,20 respectively, effected by the tertiary amine's radical cation. However, we expected that such a reductive quenching cycle would impede the necessary oxidation of the benzylic radical to the corresponding benzylic cation being crucial for achieving the targeted, additional Markovnikov functionalization. Two possible pathways for this supposed limitation could be considered to be operating: apart from a competitive oxidation of the reductive quencher (e.g. R3N), well-established HAT (hydrogen atom transfer) from the tertiary amine's radical cation, being present in increasing concentrations during the progress of the reaction, could operate as an additional detrimental pathway. As anticipated, no Markovnikov product was obtained while conducting the reaction with tributylamine as electron donor, following a reductive quenching cycle (Scheme 2/right). Instead, hydrogen atom abstraction took place as subsequent reaction step after the catalytic cycle to provide the coupling product 3 in an isolated yield of 83%. However, our attempts to transfer these promising, “reductive quenching coupling conditions”, which albeit miss the targeted additional functionalization, to less reactive styrenes (instead of our initial test substrate 1,1-diphenylethylene 2) were not met with success. Further competitive reaction pathways (Scheme 3) impede the coupling with styrene, such as the direct abstraction of a hydrogen atom from the reductive quencher by the phenacyl radical (product 5) (HAT), as well as the radical addition onto an enamine species derived from the degradation of the amine quencher providing the alternative aldehyde coupling product 6via aminocatalysis (Scheme 3).
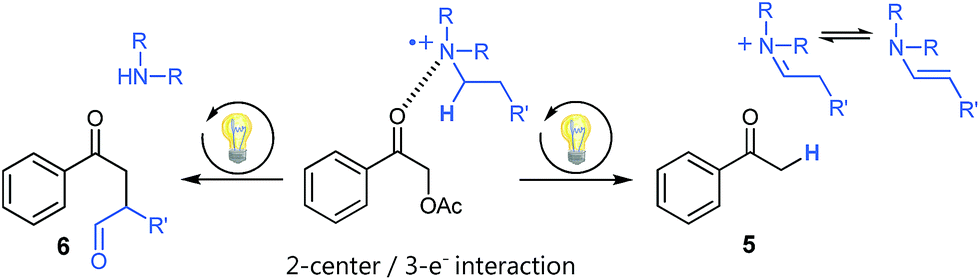 | ||
| Scheme 3 Undesired side products present in cross-coupling reactions following a reductive quenching cycle. | ||
Fortunately, employing fac-Ir(ppy)3 as photocatalyst in a redox neutral oxidative quenching cycle mediates the targeted functionalizing C–C coupling reaction to yield the desired 1,4-functionalized product 4. Remarkably, the α-acetoxy acetophenone (1) was reduced by fac-Ir(ppy)3 (E1/2(Ir(III)*/Ir(IV)) = −1.73 V vs. SCE)17b without further attempts to lower the carbonyl LUMO. Although the reduction potential of 1 seems to be too negative for a direct reduction (Ered = −1.72 V vs. SCE), we could already obtain 33% of the desired functionalized coupling product 4. Notably, this preliminary finding indicated, that the carbonyl group may here even be significantly activated for reduction, respectively for a PCET-type reduction with water as omnipresent hydrogen bond donor under our aqueous conditions.21
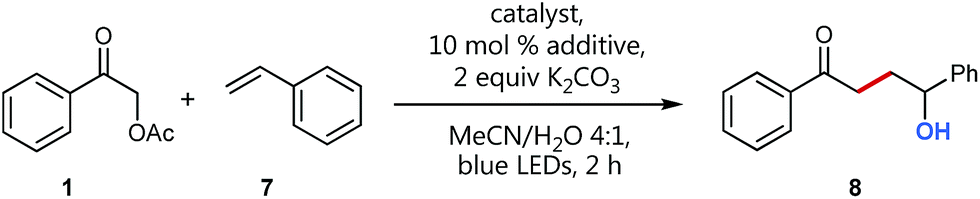
Having this in mind, we initiated a detailed screening, again using styrene (7) as a more challenging, less activated coupling partner (Table 1). Additional to water as H-bond donor present in our solvent mixture, we evaluated different activation modes for the LUMO lowering of carbonyl group,22 including Schreiner's thiourea,23 as well as different lanthanide-based, water-compatible Lewis acids.24,25 Interestingly, the choice of the activation mode (entries 2 and 3), as well as the choice of the lanthanide Lewis acid (entries 5–9) seems to be insignificant for the coupling product's yield.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additive |
1:7 |
Yielda |
|
a Conditions: catalyst, 10 mol% additive, 2 equiv. K2CO3, 0.25 mmol 1, 0.25 mmol 7 in 1 mL MeCN/H2O 4:1, degassed, irradiation with blue LEDs, 2 h, yield determined by GC-FID using mesitylene as internal standard.
b 20 mol% additive.
c Without degassing.
d MeCN as solvent.
e Schreiner's thiourea catalyst.
|
||||
| 1b,c,d | 1 mol% fac-Ir(ppy)3 | Thioureae | 1:4 |
19% |
| 2b,c | 1 mol% fac-Ir(ppy)3 | Thioureae | 1:4 |
45% |
| 3b,c | 1 mol% fac-Ir(ppy)3 | Yb(OTf)3 | 1:4 |
46% |
| 4c | 1 mol% fac-Ir(ppy)3 | Yb(OTf)3 | 1:2 |
58% |
| 5 | 1 mol% fac-Ir(ppy)3 | Yb(OTf)3 | 1:2 |
82% |
| 6 | 1 mol% fac-Ir(ppy)3 | Er(OTf)3 | 1:2 |
85% |
| 7 | 1 mol% fac-Ir(ppy)3 | Gd(OTf)3 | 1:2 |
88% |
| 8 | 1 mol% fac-Ir(ppy)3 | Dy(OTf)3 | 1:2 |
90% |
| 9 | 1 mol% fac-Ir(ppy)3 | Nd(OTf)3 | 1:2 |
91% |
| 10 | 0.5 mol% fac-Ir(ppy)3 | Nd(OTf)3 | 1:1 |
92% |
However, Schreiner's thiourea catalyst proved to be rather unstable under these photocatalytic conditions. To avoid large catalyst loadings and additional side products formed via the degradation of the thiourea catalyst, we decided to continue our investigations with neodymium triflate as Lewis acid in a dual activation, synergistic catalysis.26 Furthermore, the yield of coupling product could be dramatically increased by degassing the reaction mixture (entries 4 and 5), to 91% (entry 9). Additionally, decreasing the styrene equivalents to an equimolar amount, as well as lowering the catalyst loading to 0.5 mol% had no negative influence nor on the yield neither on the reaction time (2 h) (entry 10 (92%)). Notably, these optimized reaction conditions allow for equimolar cross-coupling of radical precursor 1 and styrene 7 to the 1,4-functionalized product in excellent yield, avoiding typical side reactions, such as styrene polymerization or hydrogen abstraction of the α-acetylated acetophenone.
The control reactions (Table 2) confirmed the decisive role of both catalyst and visible light, as without each no reaction occurred during 48 h of elongated reaction time (entries 1 and 2). The omission of Lewis acid (entry 3) decreases the yield to only 64%. An experiment performed in acetonitrile without water, but offering MeOH as alternative nucleophile (entry 4), did not provide any of the corresponding methoxy adduct. In combination with entry 3 this clearly shows the synergistic interaction of both activation modes.21,26 Water can lower the carbonyl LUMO only by weak hydrogen bonds, however greatly contributes to the success of the reaction by creating a two-phase system, which additionally shifts the equilibrium to the cross-coupling process.
|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions | Yielda |
|
a Conditions: 0.5 mol% fac-Ir(ppy)3, 10 mol% Nd(OTf)3, 2 equiv. K2CO3, 0.25 mmol 1, 0.25 mmol 7 in 1 mL MeCN/H2O 4:1, degassed, irradiation with blue LEDs, 2 h, yield determined by GC-FID using mesitylene as internal standard.
b 48 h reaction time.
c Only minor conversion of 1; traces of 1,4-diphenylbut-3-en-1-one (corresp. alkene to 8) and dimerization product 1,4-diphenylbutane-1,4-dione were detected by GC/MS.
|
||
| 1b | No catalyst | 0% |
| 2b | No light | 0% |
| 3 | No Lewis acid | 64% |
| 4 | No water, but MeOH | 0%c |
| 5 | No base | 61% |
| 6 | Bromoacetophenone as radical source | 38% |
| 7 | Ir(dtbby)(ppy)2(PF6) as catalyst | 0% |
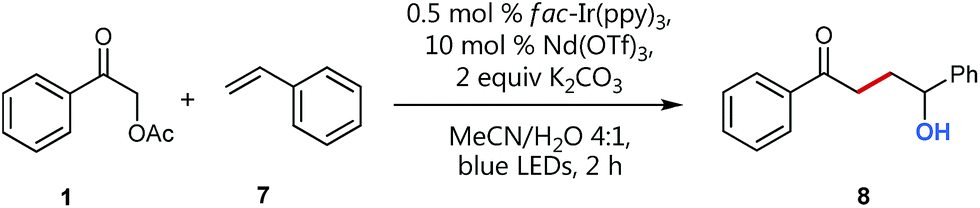
The catalytic amount of Lewis acid strongly enhances the effect on lowering the carbonyl's LUMO; this may also be in the context of the formation of Lewis acid water complexes to promote PCETs as well-known for SmI2-water systems.27,28 However, without the addition of water Nd(OTf)3 is almost insoluble in acetonitrile and therefore cannot effectively contribute to the carbonyl activation (entry 4). The exchange of the acetate leaving group with bromide (entry 6) significantly decreased the yield of the desired coupling product 8 to 38%. While full conversion of the bromo acetophenone was still reached after 2 h of irradiation, numerous, not further determined side products were formed, clearly proving the crucial role of the radical source for this cross-coupling/functionalization protocol. An additional test reaction using Ir(dtbbpy)(ppy)2(PF6) as strongly reductive catalyst for reductive quenching cycles, but weaker reductive power of its excited state (E(Ir(III)/Ir(II)) = −1.51 V vs. SCE; E(Ir(III)*/Ir(IV)) = −0.96 V vs. SCE)17b failed to provide any product and further confirms the oxidative quenching cycle of this cross-coupling.
We then set out to prove the synergistic combination of water and Lewis acid in a series of mechanistic studies. Initially, we wanted to “visualize” and somewhat quantify the LUMO lowering effect by cyclic voltammetry measurements29 to monitor the influence of different additives on the reduction potential (Fig. 1) and hence to gain a more detailed mechanistic understanding.
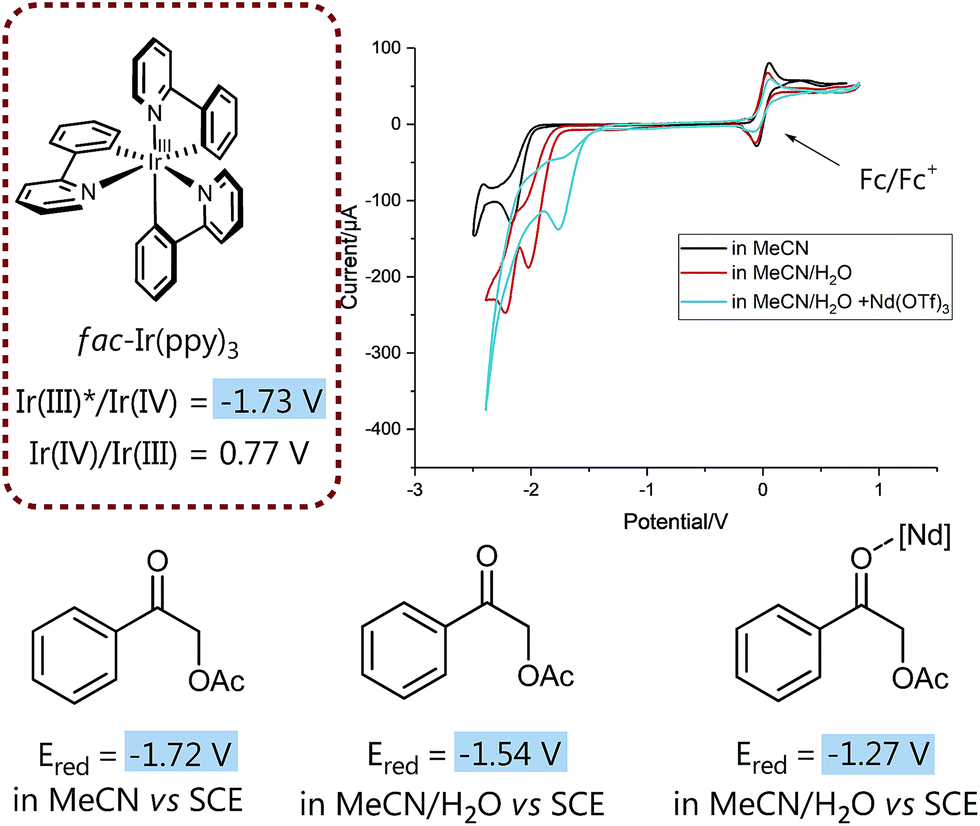 | ||
| Fig. 1 LUMO-lowering effects on reduction potential of 1 subject to different conditions. For details on electrodes, concentration and measurement conditions see ESI.† | ||
Insightfully, both the addition of water as well as of neodymium triflate results in a significant decrease of the measured reduction potential of α-acetoxy acetophenone 1. While without any additive the potential of 1 is slightly too high for a thermodynamically favorable reduction with fac-Ir(ppy)3, the solvent mixture (MeCN/water 4:1 (v/v)) already lowers the reduction potential sufficiently to enable the reduction of 1 and hence points to the often neglected, however crucial role of solvents, especially water, for (photo)redox processes.30
Notably, the combination of both activation modes, the aqueous solvent system plus a water-compatible Lewis acid,27 significantly lowers the potential by 0.5 V and hence contributes to an even more exergonic electron transfer process.
Furthermore, we could confirm these observations by Stern–Volmer experiments (Fig. 2), also corroborating our initial studies with diphenylethylene (see also Scheme 2).
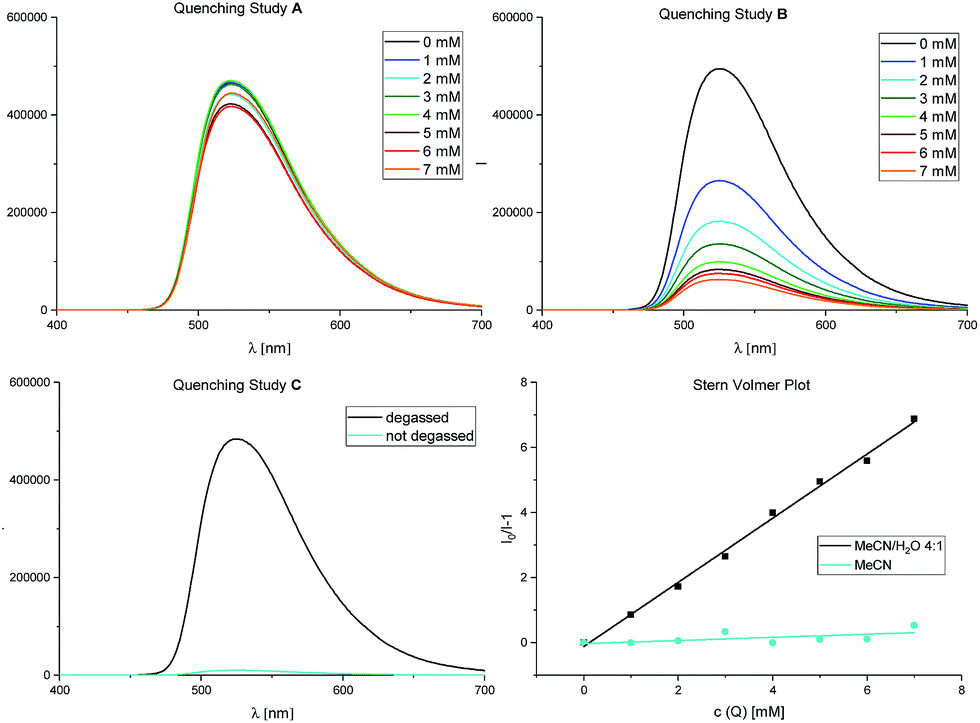 | ||
| Fig. 2 Stern–Volmer quenching studies. Fluorescence spectra for fac-Ir(ppy)3 (50 μM) with increasing amounts of α-acetoxy acetophenone 1 (0–7 mM) as quencher. Quenching study A: in MeCN; quenching study B: in MeCN/H2O 4:1; quenching study C: presence of oxygen. For further details, see ESI.† | ||
While quenching study A shows no relevant decrease in the photocatalyst's fluorescence with increasing concentrations of 1 in pure (dry) acetonitrile, only in quenching study B, performed in an acetonitrile/water mixture (4:1 (v/v)), the emission of fac-Ir(ppy)3 is clearly quenched by α-acetoxy acetophenone 1. We further proved the importance of the exclusion of oxygen (Table 1, entries 4 and 5) in this reaction: quenching study C shows energy transfer to O2 for the singlet oxygen production31 to be a strongly preferred quenching pathway of fac-Ir(ppy)3 in these solvent conditions.
Based on these studies and our experimental results, we propose the following mechanism for this functionalizing, radical C–C-coupling reaction (Scheme 4) starting with the excitation of the Ir catalyst followed by an oxidative quenching via single electron transfer to the activated carbonyl group of 1. The mesolytic cleavage of the C–O-bond leads to electron deficient radical 9, which can now attack the styrene. Subsequently, the newly created benzylic radical 10, which may be stabilized by intramolecular cyclization5g to radical 10cycl and hence suppress possible, undesired side reactions (polymerization etc.), can be oxidized by the Ir(IV) species. Upon regeneration of the photocatalyst this results in a radical polar crossover to benzylic cation 11, respectively its cyclized variant 11cycl. Final attack of water can then lead to the formation of the 1,4-ketohydroxy product.
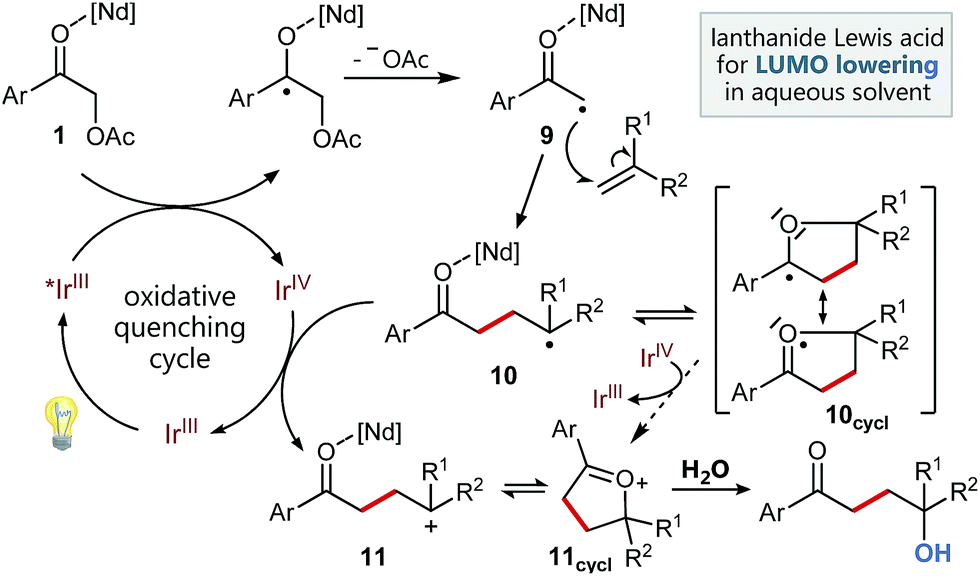 | ||
| Scheme 4 Proposed mechanism. | ||
Having optimized conditions and a mechanistic understanding at hand, we set out to examine the scope of the reaction starting with different styrenes (Scheme 5). Notably, all tested vinyl benzenes could successfully be coupled with the acetophenone radical. With exception of product 17, whose styrene precursor is known to be strongly prone to polymerization, all coupling products were obtained in good to excellent yields (up to 96%). Interestingly, there is no recognizable difference for the coupling of styrenes with electron withdrawing (12–15) and electron donating substituents (16, 18, 19). The direct comparison of the three different regioisomeric bromo styrenes (12–14) nicely shows the insensitivity of the transformation towards sterically hindrance being a typical advantage of radical coupling reactions.32
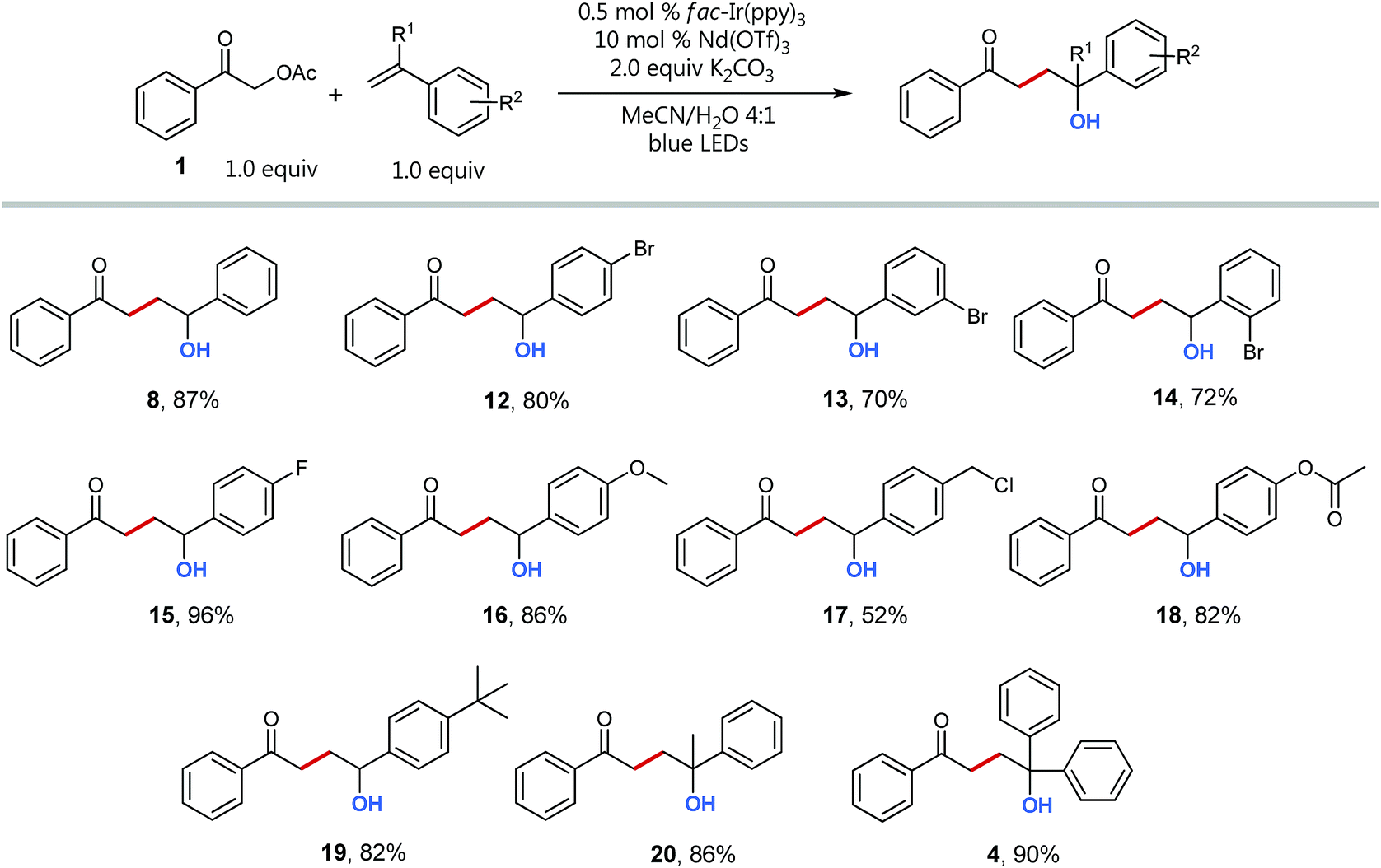 | ||
| Scheme 5 Substrate scope for C–C-coupling with styrene derivatives. Conditions: 0.5 mol% fac-Ir(ppy)3, 10 mol% Nd(OTf)3, 2 equiv. K2CO3, 0.5 mmol 1, 0.5 mmol styrene derivative in 2 mL MeCN/H2O 4:1, degassed, irradiation with blue LEDs; isolated yields. | ||
Even quaternary centers (e.g. tertiary alcohols) could be generated in excellent yields by either employing α-methylstyrene (product 20) or 1,1-diphenylethylene (product 4). To further elucidate the broad scope of our approach for radical cross-couplings with styrenes, we then investigated the trapping of radicals derived from different substituted α-acetoxy acetophenone precursors with a selection of four different styrenes (Scheme 6). Gratifyingly, we could obtain the desired products for all tested combinations within our “4 × 5 matrix”. While the yields for the combination of α-acetoxy acetophenone 1 with these styrenes are all consistently very good to excellent (82–96%; see Scheme 5 products: 8, 15, 16, 19) the difference in performance becomes more apparent for other, substituted α-acetylated acetophenones (Scheme 6: acetophenones I–V). The cross-coupling of styrenes with α-tetralone derivative I, as precursor for secondary radicals, tolerates styrenes bearing an electron withdrawing group (21) as well as a mesomeric electron donating effect (22) (77–89%). However, the yield decreases to 53% in combination with the sterically demanding and inductive electron donating tert-butyl substituent present (23). Interestingly, the electron donating methyl group (II) and the electron withdrawing chloro-substituent (III) did not show an opposing trend for their performance as coupling partners. According to this observed reactivity, the polarity match seems to be of minor importance for the success of this radical transformation. However, the mesomeric effect of the para-methoxy substituent (acetophenone IV) shows a notable decrease of all yields being best explained by the lowering of the electron deficiency of the attacking alkyl radical, respectively of the substrate's carbonyl group for the initial SET reduction.
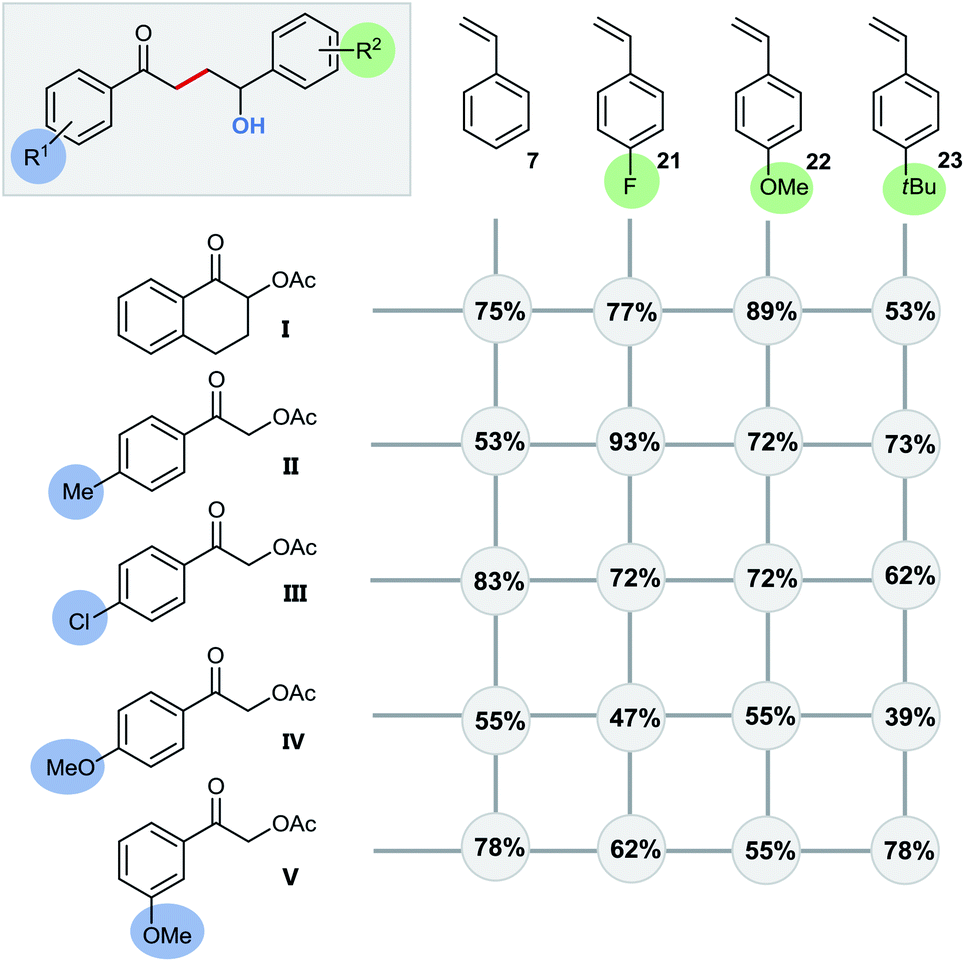 | ||
| Scheme 6 Substrate scope for functionalizing, radical polar cross-coupling reaction with styrenes. Conditions: 0.5 mol% fac-Ir(ppy)3, 10 mol% Nd(OTf)3, 2 equiv. K2CO3, 0.5 mmol acetoxy substrate, 0.5 mmol styrene derivative in 2 mL MeCN/H2O 4:1, degassed, irradiation with blue LEDs; isolated yields. | ||
Altering the position of the substituent further illustrates this effect with the observed increased yield for the coupling with meta-methoxy substituted radical source V (Scheme 6/line 5: –I effect only).
Conclusions
In summary, we have developed a novel equimolar cross-coupling reaction of α-acetoxy acetophenones and styrene derivatives with a subsequent Markovnikov functionalization to provide efficient access to versatile 1,4-substituted building blocks in good to excellent yields. The reaction was realized by the oxidative quenching of fac-Ir(ppy)3 in combination with effective synergistic activation of the carbonyl group with water and Nd(OTf)3. Detailed mechanistic investigations were performed, including cyclic voltammetry and Stern–Volmer experiments, to examine the LUMO-lowering effect in detail. Our mechanistic studies reveal the impact of radical source and choice of photocatalyst on both reactivity and selectivity and support the crucial importance of the deliberate application of fac-Ir(ppy)3 as an “oxidative quenching cycle”-only catalyst for this challenging functionalizing cross-coupling reaction. Moreover, our studies shed further light on the often neglected, but critical role of aqueous solvent systems in photoredox reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by the Deutsche Forschungsgemeinschaft (DFG, GRK 1626).Notes and references
- (a) D. Ravelli, S. Protti and M. Fagnoni, Carbon–Carbon Bond Forming Reactions via Photogenerated Intermediates, Chem. Rev., 2016, 116, 9850–9913 CrossRef PubMed; (b) M. H. Shaw, J. Twilton and D. W. C. MacMillan, Photoredox Catalysis in Organic Chemistry, J. Org. Chem., 2016, 81, 6898–6926 CrossRef PubMed.
- J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, Photoredox-Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development, ACS Catal., 2017, 7, 2563–2575 CrossRef PubMed.
- (a) J. J. Douglas, M. J. Sevrin and C. R. J. Stephenson, Visible Light Photocatalysis: Applications and New Disconnections in the Synthesis of Pharmaceutical Agents, Org. Process Res. Dev., 2016, 20, 1134–1147 CrossRef; (b) M. D. Kärkäs, J. A. Porco Jr and C. R. J. Stephenson, Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed; (c) T. P. Nicholls, D. Leonori and A. C. Bissember, Applications of Visible Light Photoredox Catalysis to the Synthesis of Natural Products and Related Compounds, Nat. Prod. Rep., 2016, 33, 1248–1254 RSC.
- A. H. Mashraqui and R. M. Kellogg, 3-Methyl-2,3-dihydrobenzothiazoles as Reducing Agent. Dye Enhanced Photoreaction, Tetrahedron Lett., 1985, 26, 1449–1452 CrossRef.
- For selected examples, see: (a) H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L.-A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Asymmetric Photoredox Transition-Metal Catalysis Activated by Visible Light, Nature, 2014, 515, 100–103 CrossRef PubMed; (b) N. Dange, A. H. Jatoi, F. Robert and Y. Landais, Visible-Light-Mediated Addition of Phenacyl Bromides onto Cyclopropenes, Org. Lett., 2017, 19, 3652–3655 CrossRef PubMed; (c) W. Chen, Z. Liu, J. Tian, J. Li, J. Ma, X. Cheng and G. Li, Building Congested Ketone: Substituted Hantzsch Ester and Nitrile as Alkylation Reagents in Photoredox Catalysis, J. Am. Chem. Soc., 2016, 138, 12312–12315 CrossRef PubMed; (d) N. Esumi, K. Suzuki, Y. Nishimoto and M. Yasuda, Synthesis of 1,4-Dicarbonyl Compounds from Silyl Enol Ethers and Bromocarbonyls, Catalyzed by an Organic Dye under Visible-Light Irradiation with Perfect Selectivity for the Halide Moiety over the Carbonyl Group, Org. Lett., 2016, 18, 5704–5707 CrossRef PubMed; (e) J. Cheng, X. Deng, G. Wang, Y. Li, X. Cheng and G. Li, Intermolecular C–H Quaternary Alkylation of Aniline Derivatives Induced by Visible-Light Photoredox Catalysis, Org. Lett., 2016, 18, 4538–4541 CrossRef PubMed; (f) Q. Liu, H. Yi, J. Liu, Y. Yang, X. Zhang, Z. Zeng and A. Lei, Visible-Light Photocatalytic Radical Alkenylation of α-Carbonyl Alkyl Bromides and Benzyl Bromides, Chem.–Eur. J., 2013, 19, 5120–5126 CrossRef PubMed; (g) H. Jiang, A. Cheng, Y. Zhang and S. Yu, De Novo Synthesis of Polysubstituted Naphthols and Furans Using Photoredox Neutral Coupling of Alkynes with 2-Bromo-1,3-dicarbonyl Compounds, Org. Lett., 2013, 15, 4884–4887 CrossRef PubMed; (h) X. Zhang, J. Qin, X. Huang and E. Meggers, One-Pot Sequential Photoredox Chemistry and Asymmetric Transfer Hydrogenation with a Single Catalyst, Eur. J. Org. Chem., 2018, 571–577 CrossRef and references reported therein.
- (a) A. Fábián, F. Ruff and Ö. Farkas, Mechanism of Nucleophilic Substitutions at Phenacyl Bromides with Pyridines. A Computational Study of Intermediate and Transition State, J. Phys. Chem., 2008, 21, 988–996 Search PubMed. For an example of using 4-methoxy pyridine to supposedly improve leaving group properties of α-bromopropionates in a photocatalytic reactions, see ref. 5f and (b) Y. Su, L. Zhang and N. Jiao, Utilization of Natural Sunlight and Air in the Aerobic Oxidation of Benzyl Halides, Org. Lett., 2011, 13, 2168–2171 CrossRef PubMed.
- P. Zhang, H. Le, R. E. Kyne and J. P. Morken, Enantioselective Construction of All-Carbo Quarternary Centers by Branch-Selective Pd-Catalyzed Allyl–Allyl Cross-Coupling, J. Am. Chem. Soc., 2011, 133, 9716–9719 CrossRef PubMed.
- M. Sheridan, Y. Wang, D. Wang, L. Troian-Gautier, C. J. Dares, B. D. Sherman and T. J. Meyer, Light-Driven Water Splitting Mediated by Photogenerated Bromine, Angew. Chem., Int. Ed., 2018, 57, 3449–3453 CrossRef PubMed.
- For investigations on the direct UV light induced cleavage, see: J. Renaud and J. C. Scaiano, Hydrogen vs. Electron Transfer Mechanisms in the Chain Decomposition of Phenacyl Bromides. Use of Isotopic Labeling as a Mechanistic Probe, Can. J. Chem., 1996, 74, 1724–1730 CrossRef.
- See ESI† for the cyclovoltammetric detection of bromide and its oxidation during the analysis of phenacyl bromide (upon initial reductive measuring direction).
- (a) E. Speckmeier, C. Padié and K. Zeitler, Visible Light Mediated Reductive Cleavage of C–O Bonds Accessing α-Substituted Aryl Ketones, Org. Lett., 2015, 17, 4818–4821 CrossRef PubMed; (b) E. Speckmeier and K. Zeitler, Desyl and Phenacyl as Versatile, Photocatalytically Cleavable Protecting Group – A Classic Approach in a Different (Visible) Light, ACS Catal., 2017, 7, 6821–6826 CrossRef.
- D. A. Nicewicz and D. W. C. MacMillan, Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes, Science, 2008, 322, 77–80 CrossRef PubMed.
- E. D. Nacsa and D. W. C. MacMillan, Spin-Center Shift-Enabled Direct Enantioselective α-Benzylation of Aldehydes with Alcohols, J. Am. Chem. Soc., 2018, 140, 3322–3330 CrossRef PubMed.
- For a short overview on photocatalytic, defunctionalizing SET reduction of α-heteroatom carbonyl compounds, see: T. M. Monos, G. Magallanes, L. J. Sebren and C. R. J. Stephenson, Visible light mediated reductions of ethers, amines and sulfides, J. Photochem. Photobiol., A, 2016, 328, 240–248 CrossRef.
- (a) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence, Nature, 2012, 492, 234–238 CrossRef PubMed; (b) J. Luo and J. Zhang, Donor–Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(sp3)–C(sp2) Cross-Coupling, ACS Catal., 2016, 6, 873–877 CrossRef.
- A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Modern Advances in Heterocyclic Chemistry in Drug Discovery, Org. Biomol. Chem., 2016, 14, 6611–6637 RSC.
- For selected recent reviews, see: (a) Special issue “Photoredox Catalysis in Organic Chemistry”: Acc. Chem. Res. 2016, 49 Search PubMed; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef PubMed; (c) K. Zeitler, Photoredox Catalysis with Visible Light, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef PubMed.
- Selected recent examples: (a) W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, Organocatalysis in Cross-Coupling: DMEDA-Catalyzed Direct C–H Arylation of Unactivated Benzene, J. Am. Chem. Soc., 2010, 132, 16737–16740 CrossRef PubMed; (b) M. Nakajima, Q. Lefebvre and M. Rueping, Visible Light Photoredox-Catalysed Intermolecular Radical Addition of α-Halo Amides to Olefins, Chem. Commun., 2014, 50, 3619–3622 RSC. Review: (c) X.-W. Lan, N.-X. Wang and Y. Xing, Recent Advances in Radical Difunctionalization of Simple Alkenes, Eur. J. Org. Chem., 2018, 5821–5851 Search PubMed.
- (a) M. H. V. Huynh and T. J. Meyer, Proton-Coupled Electron Transfer, Chem. Rev., 2007, 107, 5004–5064 CrossRef PubMed; (b) D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. Fecenko Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. Granville McCafferty and T. J. Meyer, Proton-Coupled Electron Transfer, Chem. Rev., 2012, 112, 4016–4093 CrossRef PubMed; (c) K. E. C. Gentry and R. R. Knowles, Synthetic Applications of Proton-Coupled Electron Transfer, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef PubMed.
- M. Nakajima, E. Fava, S. Loeschner, Z. Jiang and M. Rueping, Photoredox-Catalyzed Reductive Coupling of Aldehydes, Ketones, and Imines with Visible Light, Angew. Chem., Int. Ed., 2015, 54, 8828–8832 CrossRef PubMed.
- Please also see Stern–Volmer studies (Fig. 2 and ESI†) corroborating the pivotal role of water in the catalytic system.
- For a recent review, see: K. N. Lee and M.-Y. Ngai, Recent Developments in Transition-Metal Photoredox-Catalysed Reactions of Carbonyl Derivatives, Chem. Commun., 2017, 53, 13093–13112 RSC.
- (a) P. R. Schreiner, Metal-Free Organocatalysis through Explicit Hydrogen Bonding Interactions, Chem. Soc. Rev., 2003, 32, 289–296 RSC; (b) P. M. Pihko, Activation of Carbonyl Compounds by Double Hydrogen Bonding: An Emerging Tool in Asymmetric Catalysis, Angew. Chem., Int. Ed., 2004, 43, 2062–2064 CrossRef PubMed. For a seminal example of cooperative thiourea photoredox catalysis, see: (c) M. Neumann and K. Zeitler, A Cooperative Hydrogen-Bond-Promoted Organophotoredox Catalysis Strategy for Highly Diastereoselective, Reductive Enone Cyclization, Chem.–Eur. J., 2013, 19, 6950–6955 CrossRef PubMed.
- For other examples of successful carbonyl group activation with lanthanide-based triflates, also see: (a) J. Du, K. L. Skubi, D. M. Schultz and T. P. Yoon, A Dual-Catalysis Approach to Enantioselective [2+2] Photocycloadditions Using Visible Light, Science, 2014, 344, 392–396 CrossRef PubMed; (b) A. G. Amador, E. M. Sherbrook and T. P. Yoon, Enantioselective Photocatalytic [3+2] Cycloadditions of Aryl Cyclopropyl Ketones, J. Am. Chem. Soc., 2016, 138, 4722–4725 CrossRef PubMed; (c) T. P. Yoon, Photochemical Stereocontrol Using Tandem Photoredox–Chiral Lewis Acid Catalysis, Acc. Chem. Res., 2016, 49, 2307–2315 CrossRef PubMed.
- For a photoredox transformation with a lanthanide activation of alkenyl pyridines, see: K. N. Lee, Z. Lei and M.-Y. Ngai, β-Selective Reductive Coupling of Alkenylpyridines with Aldehydes and Imines via Synergistic Lewis Acid/Photoredox Catalysis, J. Am. Chem. Soc., 2017, 139, 5003–5006 CrossRef PubMed.
- For recent reviews covering dual activation photocatalysis, see: (a) M. Neumann and K. Zeitler, Synergistic Visible Light Photoredox Catalysis, in Chemical Photocatalysis, ed. B. König, deGruyter, Berlin, 2013, p. 151 Search PubMed; (b) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Dual Catalysis See the Light: Combining Photoredox with Organo-, Acid, and Transition-Metal Catalysis, Chem.–Eur. J., 2014, 20, 3874–3886 CrossRef PubMed; (c) K. L. Skubi, T. R. Blum and T. P. Yoon, Dual Catalysis Strategies in Photochemical Synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef PubMed; (d) J. Twilton, C. C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The Merger of Transition Metal and Photocatalysis, Nat. Rev. Chem., 2017, 1, 0052 CrossRef.
- For selected examples: (a) T. V. Chciuk, W. R. Anderson Jr and R. A. Flowers II, Proton-Coupled Electron Transfer in the Reduction of Carbonyls by Samarium Diiodide–Water Complexes, J. Am. Chem. Soc., 2016, 138, 8738–8741 CrossRef PubMed; (b) S. Shi, R. Szostak and M. Szostak, Proton-Coupled Electron Transfer in the Reduction of Carbonyls Using SmI2–H2O: Implications for the Reductive Coupling of Acyl-Type Ketyl Radicals with SmI2–H2O, Org. Biomol. Chem., 2016, 14, 9151–9157 RSC; (c) T. V. Chciuk, W. R. Anderson and R. A. Flowers II, High-Affinity Proton Donors Promote Proton-Coupled Electron Transfer by Samarium Diiodide, Angew. Chem., Int. Ed., 2016, 55, 6033–6036 CrossRef PubMed.
- The enhanced LUMO lowering effect by addition of lanthanide Lewis acids within the aqueous solvent system may also be considered as a case of “Lewis acid assisted Brønsted acid catalysis” (LBA) as classified by Yamamoto: H. Yamamoto and K. Futatsugi, “Designer Acids”: Combined Acid Catalysis for Asymmetric Synthesis, Angew. Chem., Int. Ed., 2005, 44, 1924–1942 CrossRef PubMed.
- (a) R. S. Nicholson, Theory and Application of Cyclic Voltammetry for Measurement of Electrode Reaction Kinetics, Anal. Chem., 1965, 37, 1351–1355 CrossRef; (b) A. Jutand, Contribution of Electrochemistry to Organometallic Catalysis, Chem. Rev., 2008, 108, 2300–2347 CrossRef PubMed; (c) For other mechanistic investigations by cyclic voltammetry see: T. Liedtke, P. Spannring, L. Riccardi and A. Gansäuer, Mechanism-Based Condition-Screening for Sustainable Catalysis in Single Electron Steps by Cyclic Voltammetry, Angew. Chem., Int. Ed., 2018, 57, 5006–5010 CrossRef PubMed.
- (a) See ref. 11b. For recent instructive examples, also see: ; (b) A. J. Boyington, M.-L. Y. Riu and N. T. Jui, Anti-Markovnikov Hydroarylation of Unactivated Olefins via Pyridyl Radical Intermediate, J. Am. Chem. Soc., 2017, 139, 6582–6585 CrossRef PubMed; (c) R. A. Aycock, H. Wang and N. T. Jui, A Mild Catalytic System for Radical Conjugate Addition of Nitrogen Heterocycles, Chem. Sci., 2017, 8, 3121–3125 RSC.
- (a) P. R. Ogilby, Singlet Oxygen: There Is Indeed Something New Under the Sun, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC; (b) A. Greer, Christopher Foote's Discovery of the Role of Singlet Oxygen [1O2 (1Δg)] in Photosensitized Oxidation Reactions, Acc. Chem. Res., 2006, 39, 797–804 CrossRef PubMed; (c) Q. G. Mulazzani, H. Sun, M. Z. Hoffman, W. E. Ford and M. A. J. Rodgers, Quenching of the Excited States of Ruthenium(II)–Diimine Complexes by Oxygen, J. Phys. Chem., 1994, 98, 1145–1150 CrossRef; (d) C. Tanielian, C. Wolff and M. Esch, Singlet Oxygen Production in Water: Aggregation and Charge-Transfer Effects, J. Phys. Chem., 1996, 100, 6555–6560 CrossRef.
- (a) G. L. Lackner, K. W. Quasdorf and L. E. Overman, Direct Construction of Quaternary Carbons from Tertiary Alcohols via Photoredox-Catalyzed Fragmentation of tert-Alkyl N-Phthalimioyl Oxalates, J. Am. Chem. Soc., 2013, 135, 15342–15345 CrossRef PubMed; (b) L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, Carboxylic Acids as A Traceless Activation Group for Conjugate Additions: A Three-Step Synthesis of (±)-Pregabalin, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef PubMed; (c) C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, Oxalates as Activating Groups for Alcohols in Visible Light Photoredox Catalysis: Formation of Quaternary Centers by Redox-Neutral Fragment Coupling, J. Am. Chem. Soc., 2015, 137, 11270–11273 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02106f |
| This journal is © The Royal Society of Chemistry 2018 |