
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrowave-assisted functionalization of the Aurivillius phase Bi2SrTa2O9: diol grafting and amine insertion vs. alcohol grafting†
Yanhui
Wang
a,
Maria
Nikolopoulou
a,
Emilie
Delahaye
 a,
Cédric
Leuvrey
a,
Fabrice
Leroux
b,
Pierre
Rabu
a and
Guillaume
Rogez
*a
a,
Cédric
Leuvrey
a,
Fabrice
Leroux
b,
Pierre
Rabu
a and
Guillaume
Rogez
*a
aUniversité de Strasbourg, CNRS, Institut de Physique et Chimie des Matériaux de Strasbourg, UMR 7504, 23 rue du Loess, BP 43, 67000 Strasbourg, France. E-mail: rogez@unistra.fr
bInstitut de Chimie de Clermont-Ferrand, Université Clermont Auvergne, UMR CNRS 6296, SIGMA Clermont, 24 Avenue des Landais, BP 80026, 63171 Aubière cedex, France
First published on 26th July 2018
Abstract
Microwave-assisted functionalization of the layered Aurivillius phase Bi2SrTa2O9 by alcohols is thoroughly investigated. The grafting of linear aliphatic and bulky alcohols is studied as a function of the starting material, underlining the importance of the prefunctionalization of the layered perovskite, for instance by butylamine. In addition, the functionalization by α,ω-alkanediols is explored. α,ω-alkanediols bearing long alkyl chains (nC > 3) adopt an unprecedented pillaring arrangement, whereas 1,3-propanediol and ethyleneglycol adopt a bilayer arrangement, only one out of the two hydroxyl groups being coordinated. Finally, the reactivities of alcohols and amines towards insertion are compared: the preferential reactivity of the two functional groups appears to be strongly dependent of the reaction conditions, and especially of the water content. This study is further extended to the case of amino-alcohol insertion. In this case, the amine group is preferentially bound, but it is possible to control the grafting of the alcohol moiety, thus going from a bilayer arrangement to a pillaring one. This work is of particular importance to be able to functionalize easily and rapidly layered oxides with elaborated molecules, bearing several different potentially reactive groups.
1 Introduction
Ion-exchangeable layered metal oxides have received growing attention because they can be functionalized under soft conditions, providing access to a very wide variety of kinetic metastable phases, inaccessible via classical solid-state reactions.1–3 Such new phases, very recently reviewed,4 range from complex new inorganic solids,5 organic–inorganic hybrid materials6 to 2D exfoliated nanosheets.7–9 They can present various interesting properties for instance in the field of energy (fuel cells, artificial photosynthesis, photovoltaics, batteries…) or in nanoelectronics (high-κ dielectrics, ferroelectrics, multiferroics…).4Among ion-exchangeable layered metal oxides, layered perovskites, Dion–Jacobson, Ruddlesden–Popper and Aurivillius phases, have probably been the most widely studied.1,10–15 Very recently, several reports have underlined the considerable progress offered by microwave activation for the functionalization of such layered perovskites, notably in terms of speed of the reactions.6,16–18 In addition, apart from classical insertion or grafting reactions,16–18 microwave-assisted post-synthesis modification19 and exfoliation20 have also been reported.
Nevertheless, despite the rapidity of the microwave approach compared to classical methods, the molecules which have been inserted up to now into the interlamellar space of layered perovskites remain essentially limited to relatively simple ones, which hence reduces the range of possible applications. As for amine insertion, we have recently shown that microwave-assisted reactions allowed to extend the variety of accessible amines, to aromatic or chiral ones for instance.18 As for grafting functionalization, either via classical heating, solvothermal or microwave-assisted reactions, a few Dion–Jacobson phases and Ruddlesden–Popper phases have been functionalized, essentially by simple alcohols (Table 1). To the best of our knowledge, only two examples describe the grafting of aliphatic linear polyols, namely (poly)ethylene glycols.13,21 In these reports, classical reactions are used, and only mono-grafting is observed. A more complicated alcohol (D-glucopyranose) has also been grafted into a Dion–Jacobson phase14 and into a Ruddlesden–Popper phase,22 and there again only one OH group (out of the five) reacts. Finally, only two examples of grafting with a molecule other than alcohol are reported (trifluoroacetic acid23 and linear phosphonic acids15). Until now, there is one single report of grafting reaction in the protonated form of an Aurivillius phase.19
| Phase | Protonated form | Intermediate | Organic phase | Solvent | Method | T | t | Ref. |
|---|---|---|---|---|---|---|---|---|
| Dion–Jacobson | HLaNb2O7 (HLN) | ∅ | Methanol | Water added | Stirring | R.T. | 1 d | 11 |
| ∅ | Ethanol | Water added | Stirring | R.T. | 7 d | 11 | ||
| ∅ | n-Alcohol, nC = 3–6, 8, 10, 12 | Water added | Classical heating | 80 °C | 7 d | 11 | ||
| n-Decoxyl-HLN | 2-Propanol, ethylene glycol | Water added | Solvothermal | 80 °C | 7 d | 13 | ||
| n-Decoxyl-HLN | tert-Butyl alcohol | Water added | Solvothermal | 80 °C | 14 d | 13 | ||
| n-Decoxyl-HLN | n-CnH2n+1PO(OH)2, 4 ≤ n ≤ 18 | 2-Butanone + 1% water | Classical heating | 80 °C | 2–3 d | 15 | ||
| n-Decoxyl-HLN | n-CH3(OCH2CH2)nOH, 1 ≤ n ≤ 4 | ∅ | Classical heating | 80 °C | 14 d | 12 | ||
| n-Decoxyl-HLN | CF3(CF2)7C2H4OH | 2-Butanone + 1% water | Classical heating | 80 °C | 7 d | 24 | ||
| n-Decoxyl-HLN | D-Glucopyranose | 2-Butanone + 10% water | Solvothermal | 70 °C | 3 d | 14 | ||
| Propoxyl-HLN | CF3COOH | ∅ | Solvothermal | 70 °C | 3 d | 25 | ||
| Propoxyl-HLN | 4-Penten-1-ol | ∅ | Solvothermal | 80 °C | 7 d | 23 | ||
| ∅ | Methanol or propanol | Water added | Microwave | 100 °C | 1 h | 17 | ||
| Methoxyl-HCN | n-Propanol | Water added | Microwave | 100 °C | 1 h | 17 | ||
| Methoxyl-HCN or propoxyl-HCN | n-Pentanol | Water added | Microwave | 120 °C | 1 h | 17 | ||
| Methoxyl-HCN or propoxyl-HCN | n-Decanol | Water added | Microwave | 150 °C | 0.5 h | 17 | ||
| HCa2Nb3O10 (HCaN) | ∅ | Methanol or ethanol | Water added | Solvothermal | 150 °C | 7 d | 26 | |
| Methoxyl-HCaN | n-Propanol | Water added | Solvothermal | 150 °C | 7 d | 26 | ||
| Propoxyl-HCaN | n-Alcohol, 4 ≤ nC ≤ 18 | Water added | Solvothermal | 150 °C | 7 d | 26 | ||
| HSr2Nb3O10 (HSN) | ∅ | Methanol | Water added | Microwave | Not-mentioned | 4 h | 16 | |
| Methoxyl-HSN | n-Propanol | Water added | Microwave | Not-mentioned | 4 h | 16 | ||
| Propoxyl-HSN | n-Hexanol | Water added | Microwave | Not-mentioned | 3.5 h | 16 | ||
| Ruddlesden–Popper | H2CaTa2O7 (HCT) | Methylamine-HCT | Methanol | Water added | Solvothermal | 100 °C | 3 d | 27 |
| Methoxyl-HCT | n-Propanol | Water added | Solvothermal | 80 °C | 7 d | 27 | ||
| Propoxyl-HCT | n-Alcohol, nC = 6, 10 | Water added | Solvothermal | 80 °C | 7 d | 27 | ||
| n-Decoxyl-HCT | D-Glucopyranose | 2-Butanone + 10% water | Solvothermal | 70 °C | 3 d | 22 | ||
| Propylamine-HCT | n-Propanol | Water added | Microwave | 110 °C | 1 h | 17 | ||
| Propoxyl-HCT | n-Alcohol, nC = 5, 10 | ∅ | Microwave | 120 °C | 1 h | 17 | ||
| H2La2Ti3O10 (HLT) | n-Butylamine-HLT | n-Propanol | Water added | Solvothermal | 180 °C | 5 d | 10 | |
| Propoxyl-HLT | n-Alcohol, nC = 4, 8, 10, 12 | Water added | Solvothermal | 150 °C | 5 d | 10 | ||
| Aurivillius | H2Bi0.1Sr0.85Ta2O7 (HST) | Ethylamine-HST | Ethanol | Water added | Microwave | 130 °C | 2 h | 19 |
| Ethoxyl-HST | 4-Pentyn-1-ol | Water added | Microwave | 110 °C | 2 h | 19 |
In this article, we explore the microwave-assisted grafting of alcohols into a protonated Aurivillius phase H2Bi0.1Sr0.85Ta2O7 (HST), derived from the Aurivillius phase Bi2SrTa2O9 (BST). We more particularly investigate the influence of the starting reactant (HST or pre-functionalized HST) on the grafting of alcohols, including bulky alcohols. We report for the first time the bi-grafting of long alkyl diols in pillaring arrangement, whereas for short diols (ethylene glycol and 1,3-propanediol) only mono-grafting is observed. Finally, the comparison of the reactivity of –NH2 and –OH groups towards HST is also investigated, depending on the water content in the reaction, either for an intermolecular competition (using butylamine and ethanol) or for an intramolecular competition (using 5-amino-pentan-1-ol). We describe in this article how the use of microwave-assisted reactions not only allows to speed up the insertion-grafting of aliphatic alcohols into a layered perovskite, but also provides new interesting tools to insert more complicated molecules (diols and amino alcohols) and to control the preferential reactivity of the different functional groups.
2 Results and discussion
Details of the experimental procedures and methods, as well as results of the elemental analyses are given in ESI.†2.1 Alcohol grafting and choice of the starting compound
It has been shown that some protonated forms of Dion–Jacobson phases, HLaNb2O7·xH2O, HCa2Nb3O10·xH2O and HSr2Nb3O10·xH2O, can react directly with n-alcohol to form grafted n-alkoxy derivatives.11,16,26 Yet, for Ruddlesden–Popper phases such a direct grafting of alcohol has been reported to be inefficient, either using classical heating10,27 or using microwave-assisted reactions.17HST (H2Bi0.1Sr0.85Ta2O7), the protonated phase derived from the Aurivillius phase Bi2SrTa2O9 (BST) can react directly with n-alkylamines to form inorganic–organic hybrids either using classical heating28 or using microwave-assisted reactions.18 Therefore it appeared worth trying to functionalize this phase by direct grafting of alcohol, but despite the use of drastic conditions in terms of temperature (up to 160 °C), duration (up to 3 days) and reaction conditions (classical heating, solvothermal or microwave activation), the direct grafting of ethanol into HST failed, providing only the unreacted starting compound. This result is not surprising since the same absence of reaction had already been reported for the structurally closely related Ruddlesden–Popper phases H2CaTa2O7 (ref. 17 and 27) and H2La2Ti3O10.10Yet, a solution to graft n-alcohols into the protonated double-layered perovskite H2CaTa2O7, has been reported.17,27 It consists in using an intermediate pre-intercalation of H2CaTa2O7 with an n-alkylamine. Such a pre-intercalation strategy, already used for various lamellar hybrid materials29–36 has been used more scarcely for layered oxides.15,17,18,27 The easy accessibility to HST functionalized by n-alkylamines using microwave-activated reactions18 offers extended possibilities for the subsequent interlayer surface modification of this Ta-based layered perovskite with n-alcohols.
Three different intermediates have been chosen as starting materials: NH3-HST (HST intercalated by ammonium), C2N-HST (HST intercalated by ethylamine) and C4N-HST (HST intercalated by butylamine). The microwave-assisted (130 °C, 2 h) grafting reactions were then performed using a large excess (more than 200 equivalents) of n-alcohols with respect to intermediates and ca. 1.3 mass% of water with respect to n-alcohols.
First, the intermediate NH3-HST has been used as a starting material to react with n-alcohols (CnOH, n = 1, 2, 3). Yet the XRD patterns and IR spectra of the reaction products show that no reaction occurs with ethanol and propanol (Fig. S1 and S2†). For methanol, the XRD pattern shows clearly a mixture of the starting material NH3-HST (interlamellar distance of 1.06 nm) and a new phase with an interlamellar distance of 1.23 nm, which corresponds to the effective grafting of methanol. This partial grafting is confirmed by IR spectroscopy (see ESI†). No improvement of these results could be obtained by changing the reaction conditions (temperature and duration), indicating that NH3-HST is not an ideal intermediate to perform grafting reactions with n-alcohols.
Fig. S3† shows the XRD patterns of the other intermediate tested, C2N-HST, and its reaction products with n-alcohols (CnOH, n = 1, 2, 3, 4, 7, 12). For reaction with alcohols possessing relatively short carbon chains (CnOH, n = 1, 2, 3, 4), the (00l) reflections of C2N-HST are no longer present in the final compounds, and new sets of (00l) reflections appear. This is a positive signal indicating the occurrence of grafting reactions between n-alcohols (CnOH, n = 1, 2, 3, 4) and HST. However, the compounds obtained after reactions with alcohols with longer carbon chains (CnOH, n = 7, 12) are multiphasic and with a very low crystallinity, whatever the reaction conditions used.
Infrared spectroscopy (Fig. S4†) shows for all compounds the appearance of signals of asymmetric and symmetric stretching vibrations of CH2 and CH3 groups in the region 2800–3000 cm−1. Yet, for the compounds obtained by reaction with C7OH and C12OH, these signals are particularly weaker than expected for such long alkyl chains. In addition, the band in the region 1100–1150 cm−1 corresponding to the formation of the C–O–Ta motif37 is visible for compounds with short alkyl chains but relatively weak for compound obtained with heptanol, or even absent for compound obtained with dodecanol.
It thus appears that the intermediate C2N-HST can be used to graft alcohols with relatively short carbon chains (CnOH, n = 1, 2, 3, 4), much better than the intermediate NH3-HST. Yet, for alcohols bearing longer carbon chain (n = 7, 12), no mono-phasic grafted products could be obtained.
Fig. 1a shows the XRD patterns of the last intermediate tested, C4N-HST and its reaction products with n-alcohols (CnOH, n = 1, 2, 3, 4, 7, 12). The grafted products are denoted as CnOH-HST (n represents the number of carbon atoms in n-alcohols). After reactions, the XRD patterns show that the (00l) reflections of C4N-HST disappear, and new sets of (00l) reflections appear. In contrast, the reflections, which are assigned to (100) and (110) in the XRD pattern of HST, are observed at the same positions (22.68° (3.92 Å) and 32.59° (2.74 Å) respectively, (CuKα1 = 0.1540598 nm)) in all the obtained products, which indicates the retention of the inorganic perovskite-like slab structure.
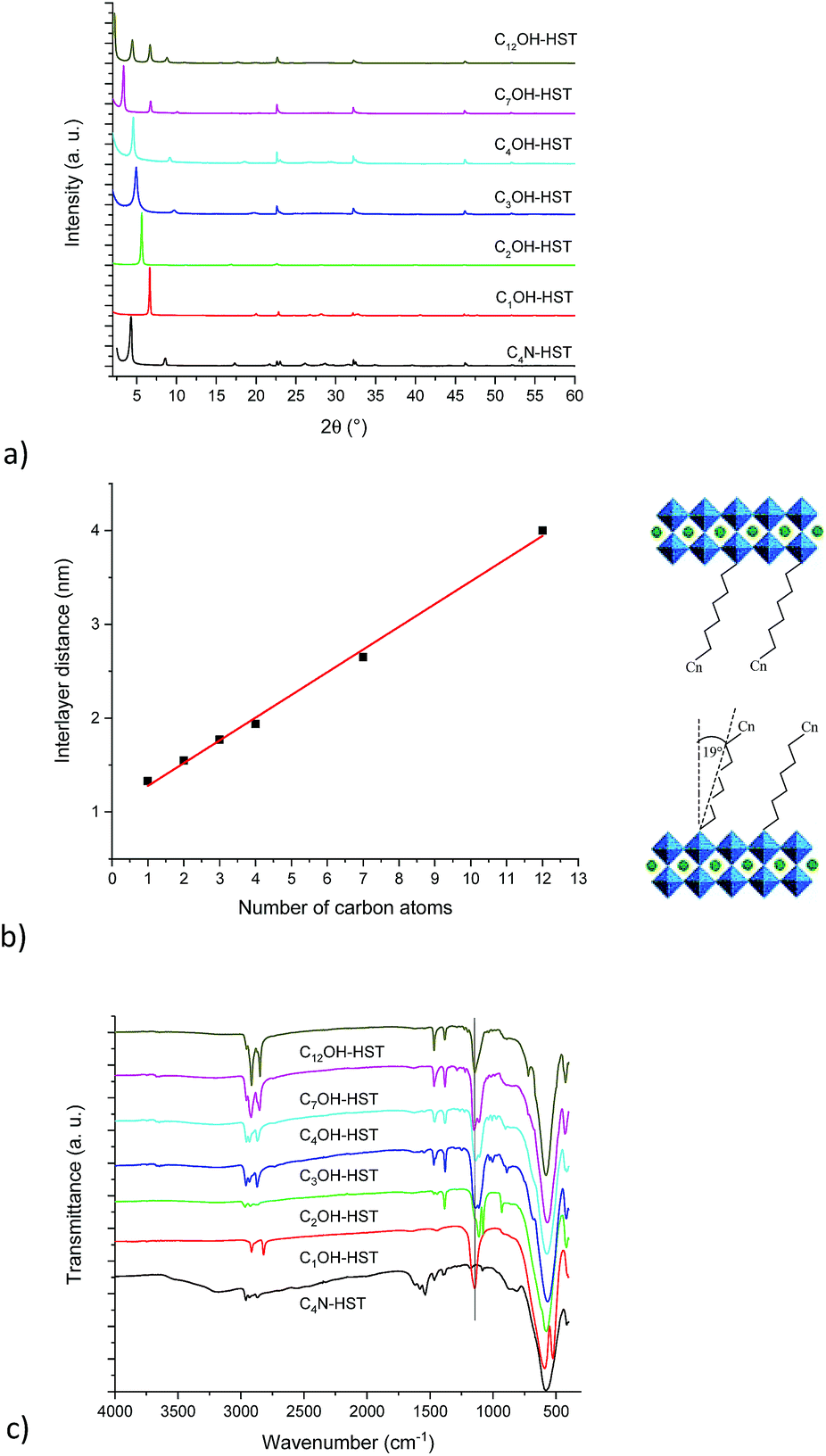 | ||
| Fig. 1 (a) PXRD patterns of the starting material C4N-HST and its reaction products with n-alcohols (CnOH, n = 1, 2, 3, 4, 7, 12). (b) Left: Relationship between the number of carbon atoms in the aliphatic chain of n-alcohols and the interlayer distance of the corresponding grafted products (full line corresponds to the best linear fit). Right: Scheme of the bilayer arrangement of the mono-n-alkoxy-HST derivatives. (c) Corresponding IR spectra. | ||
Fig. 1b shows a linear relationship between the interlayer distance and the number of carbon atoms in n-alcohols. This relationship can be expressed as follows: d001 = 0.24n + 1.04. This linear relationship indicates that the conformation of n-alkyl chains within interlayer spaces of the reaction products is similar. In addition, the slope, 0.24, is about twice the length of one CH2 group in the case of an all-trans ordered n-alkyl chain (0.127 nm/carbon atom) which indicates a bilayer arrangement.26 Similar results have been described for the intercalation of HST with n-alkylamines18,28 and for the grafting reactions of Ruddlesden–Popper phase H2CaTa2O7 (ref. 17 and 27) and of Dion–Jacobson phases HLaNb2O7·xH2O11,17 and HCa2Nb3O10·xH2O26 with n-alcohols.
Using the previous relationship, the tilt angle (with respect to the normal to the layers) can be estimated around 19°, which is the same as the one observed for the alkoxy derivatives of H2CaTa2O7.17,27 This tilt angle is larger than that of CnOH-H2La2Ti3O10 (15°) which can be attributed to larger lattice parameters.10,27 Finally, there is a considerable difference between the tilt angle of alkoxy derivatives of HST (CnOH-HST, 19°) and the one of the corresponding alkylamine intercalated HST (CnN-HST, 40°).18 This can be attributed to the different bonding modes, electrostatic in the case of amines, covalent in the case of alcohols.
Fig. 1c shows the IR spectra of the starting material C4N-HST and its reaction products with n-alcohols (CnOH, n = 1, 2, 3, 4, 7, 12). Comparing IR spectra of reaction products with that of the starting material, one notices the disappearance of the signals between 1530 and 1630 cm−1, which belong to the intermediate C4N-HST.18 The appearance of asymmetric and symmetric stretching bonds of CH2 or the stretching bonds of CH3 groups (2800–3000 cm−1) is clearly observed, and one can also notice an intense absorption around 1145 cm−1 attributed to the C–O vibration in the C–O–Ta group. Comparing with the corresponding n-alcohols (Fig. S5†), there is an obvious blue shift (ca. 95 cm−1) of the C–O band with respect to the corresponding free alcohol (ca.1050 cm−1). This blue shift is attributed to the coordination of the alcohol.37,38 In addition, the disappearance of stretching bonds of OH groups around 3300 cm−1 in the obtained products also indicates the success of the grafting reactions. The above analyses are in accordance with the removal of butylamine and the formation of a covalent bond C–O–Ta.
The major IR features are collected in Table 2. The asymmetric stretching bond of the CH3 groups is visible between 2950 and 2970 cm−1 for CnOH-HST (n = 2, 3, 4, 7 and 12) and hardly visible at around 2919 cm−1 for C1OH-HST.11,39 For all compounds, the CH3 symmetric stretching band is masked by the more intense CH2 stretching vibrations, except for C1OH-HST for which this CH3 symmetric stretching band is 2817 cm−1. The values of νas and νs for CH2 group observed for C7OH-HST and C12OH-HST correspond to all-trans conformation.40 When the length of the alkyl chains decreases, the frequencies of these bands increase as expected in the case of the presence of some gauche conformations,40 yet, the ratio of gauche conformation is too low to have an influence on the basal spacing, as evidenced by the linear variation of the basal spacing (Fig. 1b). The signals at around 1470 cm−1 and 1350 cm−1 are attributed to C–H bend and C–H rock, respectively. Ta–O elongation vibration is the dominant feature at 580 cm−1. This band is slightly shifted depending on the samples, without any clear correlation with the functionalization. Finally, no explanation was found for the presence of two signals (591 and 524 cm−1) in the Ta–O elongation vibration region for C1OH-HST.
| Product | ν as (CH2) (cm−1) | ν s (CH2) (cm−1) | ν as (CH3) (cm−1) | ν s (C–O) (cm−1) | ν s (Ta–O) (cm−1) |
|---|---|---|---|---|---|
| C1OH-HST | 2919 | 1147 | 591, 524 | ||
| C2OH-HST | 2926 | 2869 | 2968 | 1143 | 576 |
| C3OH-HST | 2933 | 2871 | 2960 | 1140 | 565 |
| C4OH-HST | 2931 | 2869 | 2957 | 1139 | 573 |
| C7OH-HST | 2919 | 2851 | 2955 | 1150 | 568 |
| C12OH-HST | 2916 | 2848 | 2957 | 1144 | 579 |
SEM observation for all compounds shows the same morphology of crystallites, inhomogeneous in size, with stratification typical of lamellar compounds (see Fig. S6† for representative examples).
In order to further demonstrate the formation of covalent bond C–O–Ta, solid state NMR spectroscopy was employed. As illustrations, the solid-state 13C CP/MAS NMR spectra of C1OH-HST and C4OH-HST are presented Fig. 2. The chemical shift of the α-carbon (–C–O–) of C1OH-HST and C4OH-HST are 60 and 74 ppm, respectively. Comparing with the chemical shift of the α-carbon of methanol and butanol in the liquid-state 13C NMR spectra (50.0 and 62.3 ppm, respectively41), there is a downfield shift of about 10 ppm. This downfield shift indicates unambiguously the formation of the covalent bond C–O–Ta in the reaction products.10,13,19,27
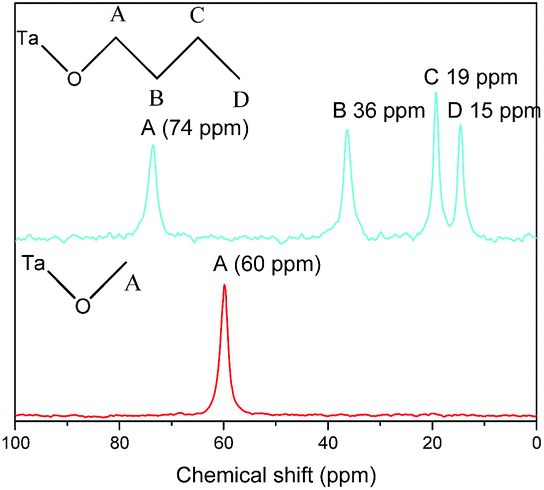 | ||
| Fig. 2 Solid-state 13C CP/MAS NMR spectra of C1H-HST (red) and C4OH-HST (cyan). | ||
The thermal behaviors of the grafted products have been studied with the help of thermo gravimetric and thermo differential analyses (TGA-TDA) (Fig. S7†). The TGA and TDA curves show small endothermic mass losses, of about 0.4–1.0%, before 120 °C, which are ascribed to the removal of water. It is worth noticing that there is no visible loss of free alcohol, which confirms the effective grafting of the alcohol molecules. Between 200 °C and 400 °C, the obvious mass losses, associated to exothermic peaks are ascribed to the decomposition of the n-alkoxy groups within the interlayer space of the layered perovskite. The XRD pattern of the compound obtained after heating at 450 °C (Fig. S8†) is very similar to the one of HST, suggesting that HST is recovered at this temperature. By analogy with what has been described on H2SrTa2O7, the XRD pattern at 800 °C indicates the formation of a three-dimensional perovskite of formula Bi0.1Sr0.85Ta2O6.42,43 It appears that the layered perovskite structure of HST is more thermally stable when the perovskite has been grafted by alcohols.
Combining the above analyses and the results of elemental analyses, the formulae of the grafted products are collected in Table S1.† Amounts of n-alkoxyl groups per HST are around 1 per Ta2 unit (except for methoxy), smaller than 2, the theoretical maximum value based on the chemical formula H2.0Bi0.1Sr0.85Ta2O7. This phenomenon has been well explained by steric reasons in a previous report concerning the insertion of amines into HST28 and the grafting of alcohols into the Ruddlesden–Popper phase H2La2Ti3O10.10 As a result, only half of the reaction sites can be occupied. However, the amount of CH3O– group per HST in C1O-HST is above this limit, around 1.5. It is noteworthy that this “excess” of methanol has already been reported for the grafting of alcohols into the Ruddlesden–Popper phase H2CaTa2O7.27 This dense arrangement of CH3O– group on the interlayer surface of the perovskite-like slabs may be ascribed to the smaller cross-section area of methanol (0.126 nm2),44 compared to the one all-trans aliphatic chains, estimated to be 0.186 nm2.45
The above study shows that the direct grafting reactions between HST and n-alcohols are totally inefficient and that the use of an intermediate is necessary. According to the work of van der Voort et al., primary amines are known as catalysts to promote the silylation of silica surface and formation of Si–O–Si linkage by acido-basic catalysis.46 But the fact that the intermediate NH3-HST cannot react with n-alcohols (except partially with methanol) proves that the only presence of NH3+ group is insufficient to promote the occurrence of grafting reactions in HST. n-alkylamine-HST hybrids are indeed efficient intermediates (provided the alkyl chain of the inserted amine is long enough). When using these intermediates (such as C4N-HST), the microwave-assisted reactions are indeed much faster than the classical solvothermal ones (Table 1). For instance, the grafting of aliphatic mono-alcohols into a Ruddlesden–Popper phase H2CaTa2O7 (very similar to the protonated Aurivillius phase HST, H2Bi0.1Sr0.85Ta2O7) lasts around 5 days using classical solvothermal approach.27 The general sketch of the synthetic procedure is given in Scheme 1.
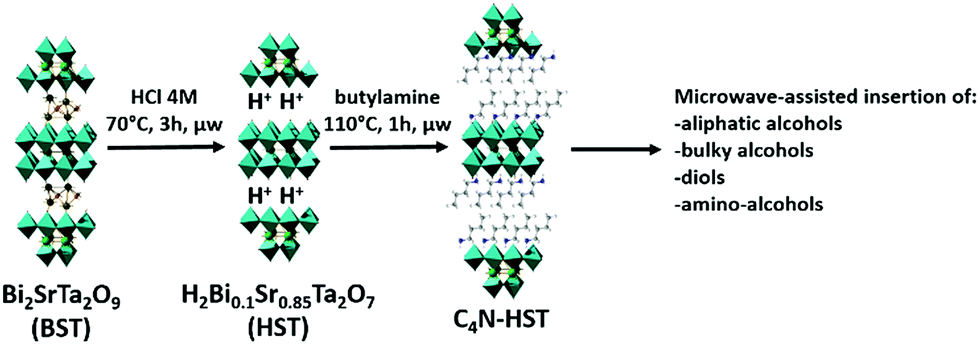 | ||
| Scheme 1 Scheme of the general synthetic procedure. | ||
2.2 Alcohol exchange reaction
Various alkoxy derivatives of HLaNb2O7·xH2O, HCa2Nb3O10·xH2O and H2La2Ti3O10 can be prepared by alcohol-exchange-type reactions with an n-alkoxy derivative as starting material to react with various alcohols.10,13,26 Therefore, following the results described above, it appears worth trying to prepare n-alkoxy derivatives of HST using microwave-assisted alcohol-exchange-type reactions.Among the obtained n-alkoxy derivatives, C2OH-HST was chosen as a starting material to react with some n-alcohols (CnOH, n = 3, 4, 7, 12) with the help of microwave irradiation (130 °C, 2 h). The grafting reactions were performed using a large excess (more than ×200) of n-alcohols with respect to the intermediate C2OH-HST, using a small amount of water (addition of ca. 1.3 mass% with respect to n-alcohols).
The XRD patterns and IR spectra of the reaction products of C2OH-HST with n-alcohols (CnOH, n = 3, 4, 7, 12) are identical to the ones obtained by using C4N-HST as an intermediate (Fig. S9†). The insertion-grafting rates are also similar to the ones determined above. Therefore, we can safely ascertain that n-alkoxy derivatives of HST can be prepared by alcohol exchange from C2OH-HST, and that the obtained compounds are identical to the ones obtained by using C4N-HST as an intermediate. Considering the observed crucial role of water (without water the exchange reaction described above are inefficient), the relatively easiness to hydrolyse alcohol-functionalized layered perovskites,13,19 and by analogy with what has been described in the literature for alcohol exchange in other layered perovskites,10,13,15,26 we propose a hydrolysis–etherification mechanism to account for alcohol exchange reactions.
2.3 Grafting of bulky alcohols
In order to further expand the scope of the microwave-assisted grafting reactions between HST and alcohols, we have tested some more bulky alcohols, other than linear aliphatic alcohols. As relatively bulky alcohols, propan-2-ol, tert-butanol and benzyl alcohol have been employed. The microwave assisted (130 °C, 2 h) grafting reactions were performed using a large excess (more than 200 times) of bulky alcohols, with respect to the intermediate C4N-HST, and a small amount of water (addition of ca. 1.3 mass% with respect to alcohols).After reaction with propan-2-ol, tert-butanol and benzyl alcohol, the interlayer distances change from that of C4N-HST (2.05 nm) to 1.57 nm, 1.72 nm and 2.06 nm, respectively (Fig. 3). In contrast, the reflections assigned to (100) and (110) are observed at the same positions (22.68° (3.92 Å) and 32.59° (2.74 Å) respectively, (CuKα1 = 0.1540598 nm)).
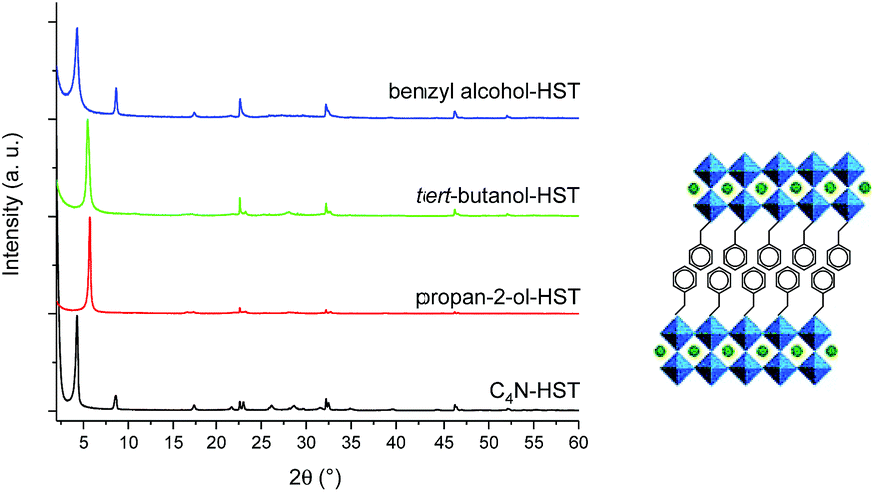 | ||
| Fig. 3 Left: PXRD patterns of C4N-HST and its reaction products with propan-2-ol, tert-butanol and benzyl alcohol. Right: As an example, proposition of arrangement of benzyl alcohol. | ||
Like for the insertion of aliphatic alcohols, IR spectra confirm the grafting of the bulky alcohols, essentially by the disappearance of the stretching OH vibration, and by the blue shift of the C–O vibration (see Fig. S10†). The formulae of the obtained compounds, established from elemental analyses and Thermo-Gravimetric Analyses (TGA) (Fig. S11†) are presented on Table S2.† The thermal behavior of the last compound, tert-butanol-HST is not reported here, because the results of elemental analyses show the significant presence of nitrogen (which is not the case for the other two compounds). It proves that in this case, the grafting is not complete, even using harder conditions (130 °C, 48 h, classical oven; 160 °C, 2 h, microwave). We attribute this difficulty to the bulkiness of tert-butanol, close to the anchoring function. On the contrary, elemental analyses for propan-2-ol-HST and benzyl alcohol-HST show no trace of nitrogen, confirming the complete removal of C4N.
It is noteworthy that, likely due to steric hindrance, the organic contents are 0.6 and 0.7 alcohol per Ta2 unit for 2-propanol-HST and benzyl alcohol-HST respectively, smaller than the value obtained for aliphatic linear n-alcohol-HST (ca. 1.0).
2.4 Grafting of α,ω-alkanediols
The reactivity of HST with α,ω-alkyldiamines has been demonstrated, using either classical heating47 or microwave-assisted reaction,18 but to the best of our knowledge, the reactivity of a layered perovskite towards diols has been described only twice, with the grafting of (poly)ethylene-glycols into a Dion–Jacobson phase HLaNb2O7.13,21 We thus explored the possibility to functionalize HST by α,ω-alkanediols, using microwave activated reactions. C4N-HST or C2N-HST can be used indifferently as a starting material to react with α,ω-alkanediols (HOCnH2nOH, n = 2, 3, 4, 7, 8, 9, 12).The microwave assisted (130 °C, 2 h) grafting reactions were performed using a large excess (more than ×200) of α,ω-alkanediols with respect to the intermediate C4N-HST, using a small amount of water (addition of ca. 13 mass% with respect to n-alcohols).
Fig. 4a shows the XRD patterns of C4N-HST and of the reaction products. After reacting with ethylene glycol, 1,3-propanediol and 1,4-butanediol, the interlamellar distances decrease from that of C4N-HST (2.05 nm) to 1.52 nm, 1.56 nm and 1.59 nm, respectively. For longer diols, the interlamellar distances of the grafted products increase regularly with the increase of the number of carbon atoms in alkane-diols.
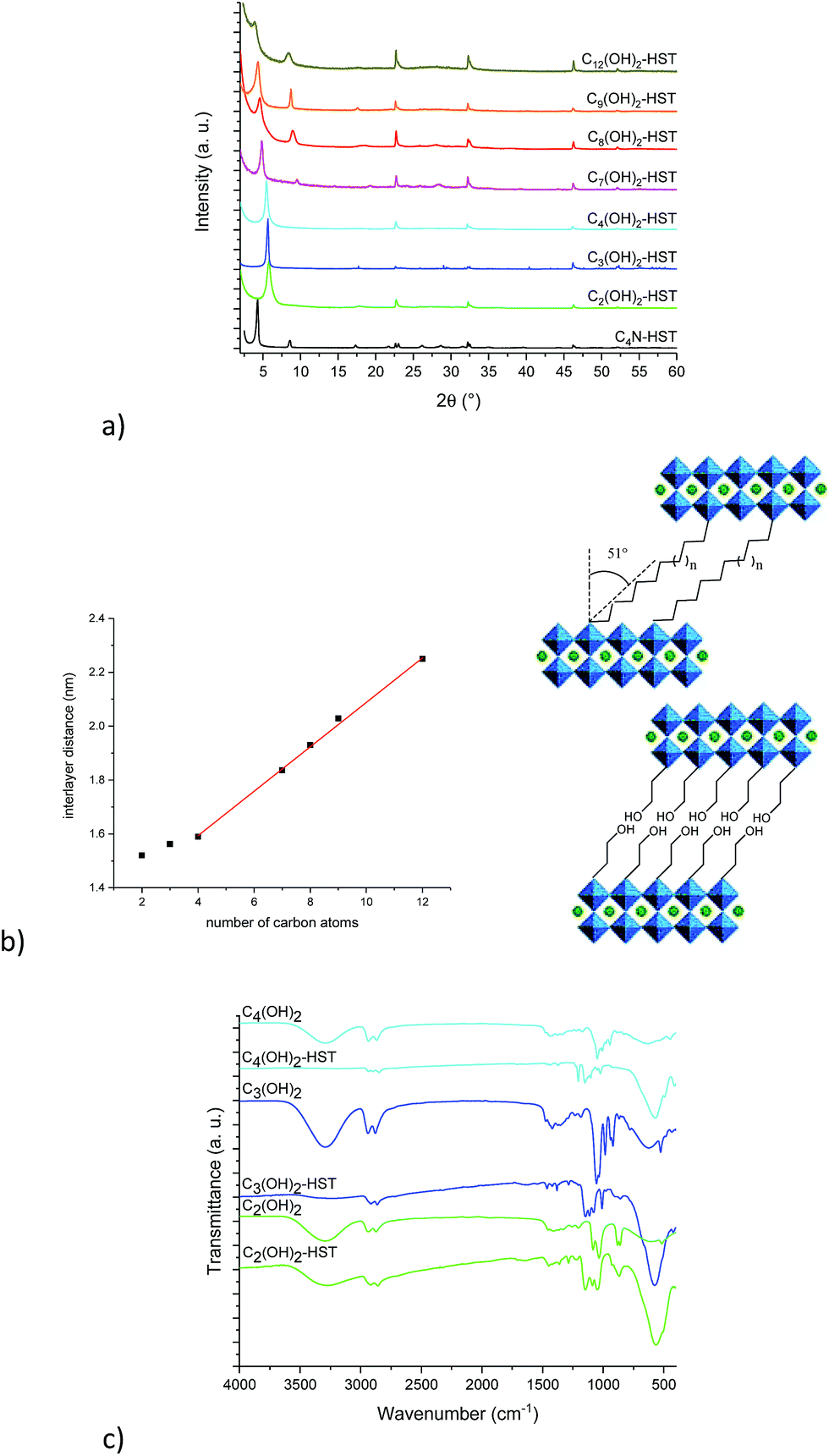 | ||
| Fig. 4 (a) PXRD patterns of the starting material C4N-HST and its reaction products with α,ω-alkanediols (HOCnH2nOH, n = 2, 3, 4, 7, 8, 9, 12). (b) Left: Relation between the number of carbon atoms in the aliphatic chain of α, ω-alkanediols and the interlayer distance of the corresponding grafted products (full line corresponds to the best linear fit for the longer diols (see text)). Right: Schemes of the pillaring arrangement of α, ω-alkanediols with long alkyl chains (top) and of the bilayer arrangement of ethyleneglycol or of 1,3-propanediol. (c) IR spectra of ethylene glycol, 1,3-propanediol, 1,4-butanediol, C2(OH)2-HST, C3(OH)2-HST and C4(OH)2-HST. | ||
The relation between the interlayer distance and the number of carbon atoms in aliphatic linear α,ω-alkanediols is plotted in Fig. 4b. For the longer diols (n = 4, 7, 8, 9 and 12), there is a clear linear relationship between the interlayer distance and the number of carbon atoms in the aliphatic chain of α,ω-alkanediols. The relation can be expressed as follows: d001 = 0.08n + 1.26. This linear relationship indicates that the conformation of n-alkyl chains in the interlayer space of the reaction products is identical for all compounds with n ≥ 4. The slope provides an estimation of the tilt angle of the alkyl chains with respect to the normal to the inorganic layers around 51° (Fig. 4b right). This tilt angle is much larger than the one obtained for aliphatic mono-alcohols (19°, see above) and larger than the one obtained for α,ω-alkanediamines (30°).18,47 Yet it is in line with the one observed for the grafting of aliphatic mono-alcohols into Nb-based layered Dion–Jacobson phases.11,17,26
It is worth noticing that C2(OH)2-HST and C3(OH)2-HST present an interlayer distance similar to that of C2OH-HST (1.52 nm vs. 1.55 nm) or slightly smaller than that of C3OH-HST (1.56 nm vs. 1.77 nm) respectively, whereas the interlamellar distance of hybrids with diols with longer alkyl chains (n ≥ 4) is significantly smaller than that of the corresponding mono-alcohols hybrids. For instance, the interlamellar distance C4(OH)2-HST (1.59 nm) is much smaller than the one of C4OH-HST (1.94 nm). Moreover, the interlamellar distance of C4(OH)2-HST is similar to that of C4N2-HST (1.56 nm), for which 1, 4-butanediamine has a pillaring arrangement.18,47
Fig. 4c shows IR spectra of ethylene glycol, 1,3-propanediol, 1,4-butanediol, C2(OH)2-HST, C3(OH)2-HST and C4(OH)2-HST. The disappearance of the signal of –CH3 group at 2962 cm−1 from the starting material C4N-HST is clearly observed, confirming the total removal of butylamine. The spectra of C2(OH)2-HST and C3(OH)2-HST present strong absorptions at the same positions as in ethylene glycol and propanediol respectively: 3296 cm−1, ascribed to free –OH group and 1086 cm−1 ascribed to C–O group. This indicates the existence of free –OH groups in C2(OH)2-HST and C3(OH)2-HST. In addition, the appearance of a new signal at 1149 cm−1 indicates the formation of covalent bond C–O–Ta between ethylene glycol and layered perovskite.37,38 On the contrary, the spectrum of C4(OH)2-HST shows the disappearance of the signal of the –OH group (visible at 3290 cm−1 in the spectrum of 1,4-butanediol). In addition, there is a blue shift of about 100 cm−1 of the signal of the C–O group (1149 cm−1 for the hybrid and 1048 cm−1 for 1,4-butanediol), without remaining signal at 1048 cm−1.37,38 The spectra of Cn(OH)2-HST (n = 7, 8, 9, 12) present the same characteristics (Fig. S12†).
The above analyses strongly suggest that C2(OH)2-HST and C3(OH)2-HST present a bilayer arrangement of the guest species, with only one side of the diol grafted to the perovskite-like slab, the other OH group remaining free. Cn(OH)2-HST (n ≥ 4) conversely, present a pillaring arrangement of guest species with both sides of the diols grafted to the perovskite-like slab.
This analysis is supported by solid state NMR spectroscopy (Fig. 5). The solid state 13C NMR spectrum of C2(OH)2-HST shows two signals at 75 and 64 ppm whereas the liquid state 13C NMR spectrum of ethylene glycol shows only one signal at 63.7 ppm.41 For C3(OH)2-HST, three signals are observed at 69, 59 and 37 ppm, whereas 1,3-propanediol presents only two signals at 61.7 and 34.1 ppm.41 Therefore, for both compounds, there is a downfield shift about 10 ppm for one α-carbon, while the other remains unchanged. This confirms that one alcohol group is coordinated to the perovskite layer, while the other remains free. The small signals at 36 and 14 ppm (marked with asterisks) are ascribed to residual C2N (for this peculiar experiment, C2N-HST was used as starting material instead of C4N-HST). For C4(OH)2-HST, only one signal for the α-carbon is observed, at 73 ppm, downfield shifted by about 10 ppm with respect to 1,4-butanediol (62.6 ppm), whereas the other signals (B and B′) remain unchanged.41 Therefore, both oxygen atoms of 1,4-butanediol are coordinated in C4(OH)2-HST. The small signal around 62 ppm (marked with asterisk) is ascribed to free –OH groups of 1,4-butanediol, coming from residual solvent, defects or 1,4-butanediol mono-grafted onto the surface of the crystallites.
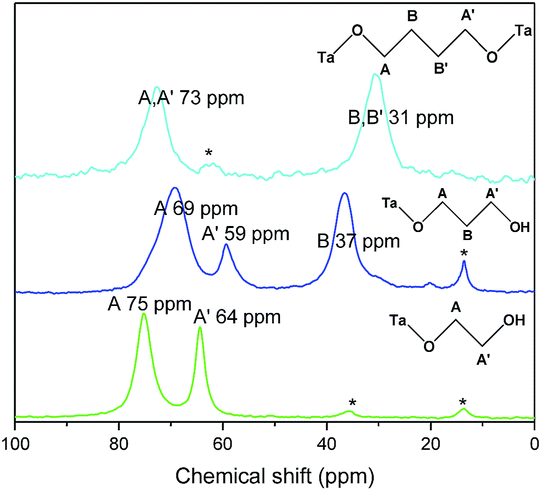 | ||
| Fig. 5 Solid-state 13C CP/MAS NMR spectra of C2(OH)2-HST (green), C3(OH)2-HST (blue) and of C4(OH)2-HST (cyan). | ||
According to proposed formulae, established from elemental analyses and Thermo-Gravimetric Analyses (TGA) (Table S3†), it is worth noticing that C2(OH)2-HST and C3(OH)2-HST contain about 0.8 and 0.6 diol respectively per formula unit, which, considering that the theoretical maximum number of hydroxyl group per Ta2 unit is 1,11,13 constitutes a further argument in favor of the monografting of ethylene glycol and propanediol.
The bilayer arrangement of C2(OH)2 and C3(OH)2 in the interlayer space of HST is in good accordance with previous report of monografting of ethylene glycol on the Dion–Jacobson phase HLaNb2O7·xH2O,13 or of 1,2- and 1,3-propanediols in Kaolinite.48 In the latter example, when largely increasing the reaction times, the two OH groups of 1,3-propanediol are coordinated, but to the same inorganic layer. Yet, to the best of our knowledge, the pillaring arrangement of α,ω-alkanediols in the interlayer space of layered perovskite has never been described in literature. The modification of Kaolinite with butanediols shows no pillaring arrangement.49 The grafting reactions between HLaNb2O7 and polyethylene glycols (PEGs) with various molecular masses lead only to mono-grafted arrangements.21 Finally, the functionalization of HLaNb2O7 with D-glucopyranose has also been described as a mono-grafted arrangement.37
2.5 Alcohol grafting vs. amine insertion
Considering the above-described results and other reports in the literature concerning the microwave-assisted functionalization of layered perovskites by relatively simple amines and alcohols,6,16–19 it appears interesting to investigate the compared reactivity of amines and alcohols, and how the reaction conditions are able to change the preferential reactivity of HST towards –NH2 group or –OH group. This study is of great importance when times come for the insertion of very complicated molecules, bearing several functional groups. We focus here on the role of water.A series of reactions between an equimolar mixture of butylamine (5.1 mL, 3.7 g, 0.04 mol) and ethanol (2.9 mL, 2.3 g, 0.04 mol) and with HST (50 mg), using a variable amount of water were thus carried out (microwave, 130 °C, 2 h). The amount of distilled water is 0 mass%, 0.1 mass% and 1.0 mass% with respect to the total mass of the reactive mixture.
Fig. 6 shows the XRD patterns of HST and its reaction products depending on the reaction conditions. As expected, when no water is used, the reaction does not take place, neither with the amine, nor with ethanol. With a small amount of water (0.1 mass%), the interlamellar distance increases from 0.98 nm (HST) to 1.74 nm (which is slightly larger than that of C2OH-HST (1.54 nm)). When the amount of water increases further (1 mass%), the interlamellar distance increases to 2.07 nm, which corresponds to the one of C4N-HST.18
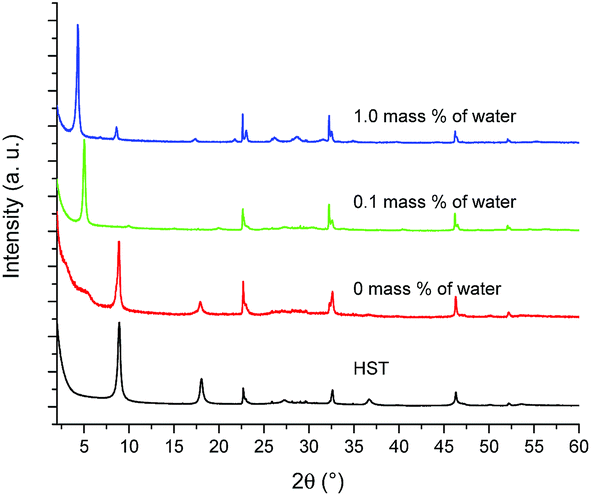 | ||
| Fig. 6 PXRD patterns of HST and its reaction products with an equimolar mixture of butylamine and ethanol and 0 mass%, 0.1 mass% and 1 mass% of distilled water. | ||
The corresponding IR spectra are shown in Fig. S13.† In the absence of water, the spectrum of HST is observed, as expected and in accordance with the corresponding XRD pattern. With 0.1 mass% of water, in addition to the signal coming from the alcohol essentially in the 1100 cm−1 region, the spectrum shows the antisymmetric and symmetric stretching bands of the CH2 group but also bands associated to the C–N group around 1500 cm−1. This strongly suggests the co-insertion of 1-butylamine and ethanol, which would then explain the interlamellar distance larger than that of C2OH-HST. When a larger amount of water is used, only the signals of CH2 group and the signals of C–N are observed and the signals of C–O band cannot be found which indicates only 1-butylamine is present in the interlayer space of HST.
Finally, the solid-state 13C CP/MAS NMR spectra of the reaction products with 0.1 mass% and 1.0 mass% of water further confirm the above observations (Fig. 7). When 1.0 mass% of water is used, only signals at 40, 30, 20 and 14 ppm were obtained, confirming the formation of C4N-HST. When a smaller amount of water is used, 0.1 mass%, the solid-state 13C CP/MAS NMR shows the signals which corresponds to the insertion of butylamine at 40, 30, 20 and 14 ppm, but also a signal at 68 ppm. This signal is attributed to the α-carbon (–C–O–) of ethanol, which is downfield shifted with respect to the corresponding signal for free ethanol in the liquid-state (57.8 ppm).41 This indicates the formation of the covalent bond C–O–Ta and confirms the co-insertion of ethanol and butylamine when only 0.1 mass% of water is used.
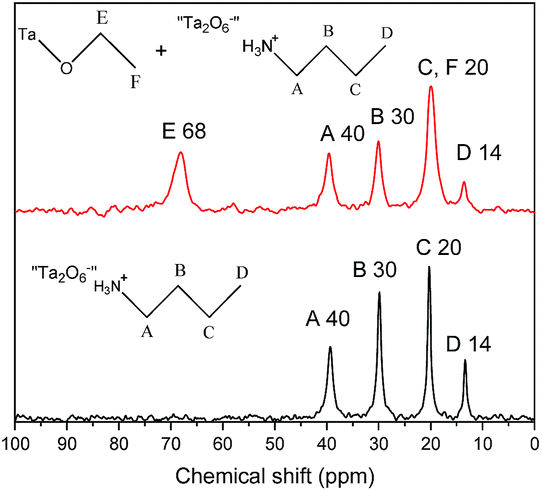 | ||
| Fig. 7 Solid-state 13C CP/MAS NMR spectra of the products resulting from the reaction between HST and an equimolar mixture of butylamine and ethanol and 0.1 mass% (red) and 1 mass% (black) of distilled water. | ||
As presented above, ethanol does not react with HST. Therefore, the fact that it is possible to graft ethanol into HST when a mixture of ethanol and amine is used indicates that butylamine serves as a catalyst. Depending on the conditions, C4N-HST is formed first, and then the amine is replaced by ethanol as described when C2N-HST or C4N-HST are used as starting compounds. Yet, when the amine is present in excess (and not only in the form of CnN-HST), the formation of a pure C2OH-HST phase is not possible. When the water content is high, the preferential reactivity of HST towards amines leads to the formation of a CnN-HST phase and no insertion-grafting of alcohol. One hypothesis to explain the inhibition of the reactivity of the alcohol moiety when the amount of water is high, is the reduction of the nucleophilic character of the hydroxyl group by solvation with water.
2.6 Amino-alcohol insertion
The above study of the role of water on the preferential reactivity of HST towards –NH2 group or –OH group, was further extended to the study of the reactivity of HST and related compounds (C2N-HST, C4N-HST and C2OH-HST) towards an aliphatic amino-alcohol, 5-amino-pentan-1-ol.Fig. 8 shows the XRD patterns of C2N-HST and its reaction products with 5-amino-1-pentanol, using different quantities of water. The interlayer distance of the obtained product increases from that of C2N-HST (1.57 nm) to 1.61 nm, when the volumetric ratio water/THF is 1%. This distance corresponds to the expected one in case of a pillaring arrangement (see Fig. 4a for pillaring diols and ref. 18 for pillaring diamines). When the volume of water is 2% or 3% the one of THF, the interlayer distance is around 1.75 nm. And when the volume of water is between 10% and 100% the volume of THF, the interlayer distance further increases to around 1.96 nm. The observed variation of the interlayer distances is attributed to the change of the arrangement of 5-amino-1-pentanol in the interlayer space, from pillaring arrangement to bilayer arrangement.
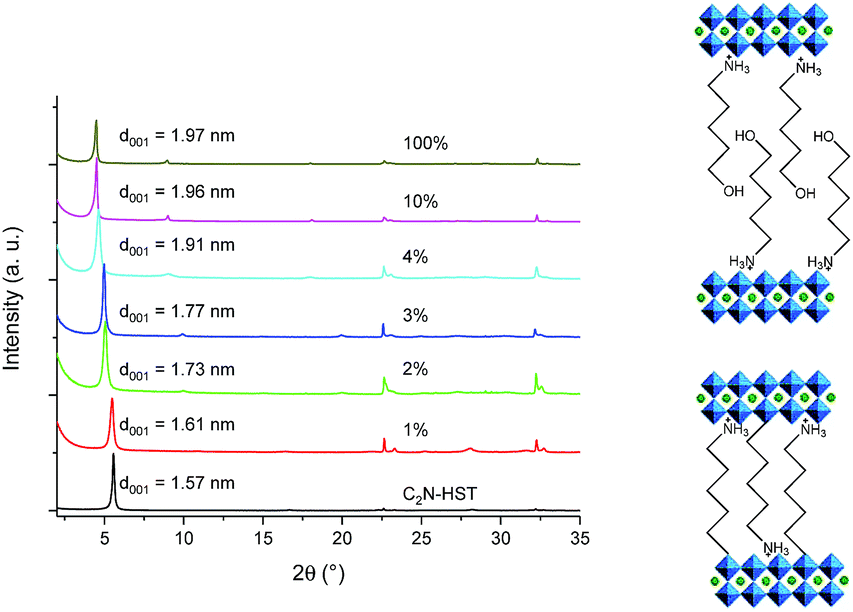 | ||
| Fig. 8 Left: XRD patterns of C2N-HST and its reaction products with 5-amino-1-pentanol using different quantities of water. Right: Scheme of the pillaring arrangement (bottom), and of the bilayer arrangement (top). | ||
These different arrangements are further confirmed and precised by IR spectroscopy (Fig. S14†). First it is worth noticing that the sharp strong bands at 3332 cm−1 and 3286 cm−1 due to NH stretching of free –NH2 group are greatly reduced in intensity in the spectra of the hybrid compounds compared to the spectrum of 5-amino-1-pentanol. In addition, new bands at 1578 cm−1 and 1545 cm−1 appear in the spectra of the hybrids, and can be attributed to –NH3+ groups deformation vibrations.50,51 This indicates that whatever the amount of water, the amino moiety is protonated and thus interacts with the inorganic negatively charged layers. Second, the signal coming from the stretching of the C–O bond appears at ca. 1060 cm−1 when the volume of water is more than 4% the one of THF, which indicates that the alcohol moiety is not coordinated, whereas for low water content (below 0.02 times the volume of THF), this band shifts to ca. 1130 cm−1, which indicates a grafting of the alcohol moiety to the inorganic layers and thus a pillaring arrangement of 5-amino-1-pentanol. When the volume of water is 2% and 4% the one of THF, the coexistence of signals around 1130 cm−1 and around 1060 cm−1 indicates the coexistence of the two arrangements. Unexpectedly, this coexistence of the two arrangements does not lead to a multiphasic compound (Fig. 8), but instead leads to the formation of compounds with an intermediate interlayer distance.
13C CP/MAS NMR spectra of different compounds (obtained with 1, 2 and 100% of water respectively) confirm the previous analyses (Fig. 9). Indeed, 13C NMR spectrum of free 5-amino-pentan-1-ol in solution in CDCl3 presents five signals at 61.6, 42.0, 33.3, 32.6 and 23.2 ppm.4113C CP/MAS NMR spectrum of the 5-amino-pentan-1-ol-HST hybrid obtained with 100% of water presents a signal at 62 ppm, with no evidence of a signal at lower field, indicating that the –OH group is not coordinated. On the contrary, for compounds obtained with 2 or 1% of water the signal of the C atom bearing the hydroxyl group is clearly down-field shifted with respect to free 5-amino-pentan-1-ol, indicating the deprotonation and coordination of the hydroxyl group.10,13,19,27 Yet a residual signal at 62 ppm shows the presence of a small amount of free hydroxyl groups (less pronounced when the quantity of water is decreased). The other signals are essentially unchanged, only slightly upfield shifted as expected due to amine protonation.18,47
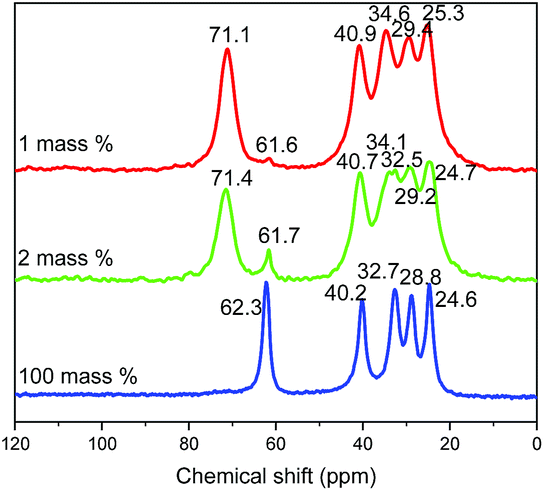 | ||
| Fig. 9 Solid-state 13C CP/MAS NMR spectra of the products resulting from the reaction between HST and 5-amino-pentan-1-ol with 1 mass% (red), 2 mass% (green) and 100 mass% (blue) of distilled water. | ||
Combining the above analyses and the results of elemental analyses, the formulae of the obtained products as a function of the amount of water used are collected in Table S4.†
The same results are obtained when C4N-HST is used instead of C2N-HST. With these starting materials, it is thus possible to finely control the interlayer spacing of the hybrids and the arrangement 5-amino-pentan-1-ol by playing with the water content in the reaction mixture.
On the contrary, when using C2OH-HST or HST are used as starting materials, only the bilayer arrangement, with an interlamellar distance around 1.96 nm, can be obtained. In addition, it is worth noticing that the insertion reaction of 5-amino-pentan-1-ol is far more difficult; complete disappearance of the starting materials C2OH-HST or HST requires a much larger quantity of water (Fig. S15–S18†).
2.7 Post-treatment: from bilayer to pillaring arrangement
The bilayer arrangement of 5-amino-pentan-1-ol leads to the presence of free –OH groups within the interlayer space of HST. These groups have the potential to undergo further grafting reaction to lead to pillaring arrangement. It is worth underlining that 5-amino-pentan-1-ol-HST in its bilayer form is stable during at least 10 days in ambient conditions (in air, around 20 °C and about 50% humidity). Yet, after a moderate heating (70 °C) during one day, the interlayer distance decreases from 1.98 nm to 1.70 nm and further decreases to 1.62 nm when the heating time is prolonged to two days. Longer heating times do not lead to any further change of the interlamellar distance (Fig. 10).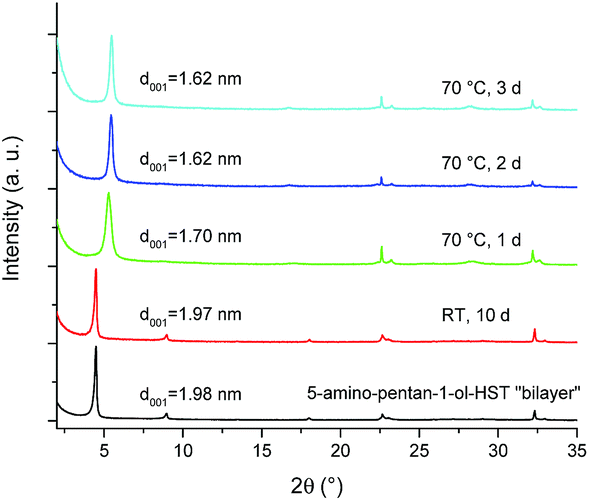 | ||
| Fig. 10 XRD patterns of 5-amino-pentan-1-ol-HST “bilayer” and of the products obtained after heating. | ||
It is noteworthy that the distance which is obtained after heating (1.62 nm) is the same as the one obtained by the reaction between C2N-HST and 5-amino-pentan-1-ol using a low quantity of water with respect to THF (Fig. 8) which corresponds to a pillaring arrangement of 5-amino-pentan-1-ol. The corresponding IR spectra further support the above hypothesis, of a temperature induced pillaring of 5-amino-pentan-1-ol (Fig. S19†). Notably, the C–O stretching vibration undergoes a clear blue shift from 1058 cm−1 (C–OH) to 1120 cm−1 (ascribed to C–OTa). Finally, the down-field shift of the solid-state 13C CP/MAS NMR signal attributed to the C bearing the hydroxyl group confirms the effective grafting of the alcohol moiety upon heating (Fig. 11). Nevertheless, a small peak at 61.7 ppm, corresponding to free –OH group, is still visible in the spectrum of the compound after heating. One hypothesis is that this peak is due to 5-amino-pentan-1-ol molecules linked to the surface of the crystallites via the ammonium group, and for which the terminal –OH group remains free. The broadening of the resonance lines is likely due to the more rigid structure induced by the pillaring arrangement of 5-amino-pentan-1-ol.
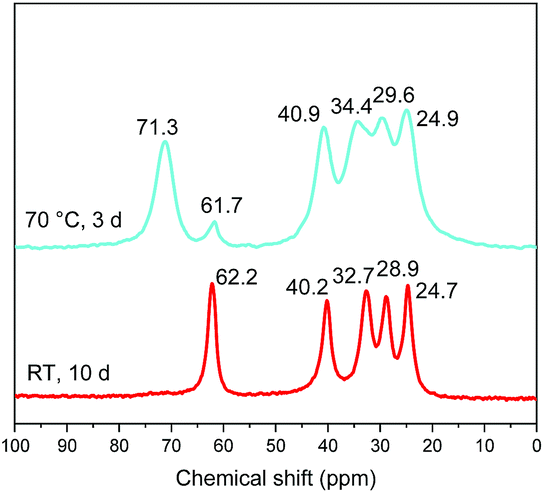 | ||
| Fig. 11 Solid-state 13C CP/MAS NMR spectra of 5-amino-pentan-1-ol-HST “bilayer” (red) and the product obtained after heating (cyan). | ||
3 Conclusions
The successful microwave-assisted interlayer surface modification of the protonated form of an Aurivillius phase, Bi2SrTa2O9 (BST) via grafting reactions with various alcohols (n-alcohols, linear diols and bulky alcohols) has been achieved. One marking result is the pillaring arrangement of α,ω-alkanediols with long alkyl chains in the interlamellar spacing of layered perovskite which is evidenced for the first time.Taking advantage of the rapidity of microwave assisted reaction, the influence of the reaction conditions has been investigated more precisely. The importance of the choice of the pre-intercalated material for the preparation of alkoxy derivatives the Aurivillius phase has thus been underlined. In addition, it has been shown that water plays a key role not only to allow grafting of alcohols, but also to drive the preferential reactivity of the protonated perovskite with amines or alcohols. In the example chosen, 5-amino-pentan-1-ol, the amine group is preferentially bound, but it is possible to control the grafting of the alcohol moiety either by adjusting the quantity of water in the reaction medium, or by a post-reaction moderate heating.
These results will be most useful when times come for the insertion of more complicated molecules, bearing several functional groups, necessary for the precise design of functional hybrids with specific structures and properties. Such precise synthetic approaches can for instance lead to new luminescent activators,52,53 vapour sensors54 or to a fine tuning of the ferroelectric properties of the starting BST phase.55
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the CNRS, the Université de Strasbourg, the Université de Clermont-Ferrand and the Agence Nationale de la Recherche (contract ANR-14-CE07-0004-01) for financial support, the Chemistry Department of the CNRS and the Région Alsace for the PhD grant of Yanhui Wang. The authors also acknowledge the support from the Labex NIE (ANR-11-LABX-0058_NIE within the Investissement d'Avenir program ANR-10-IDEX-0002-02). The present work is part of the research activity supported by the European COST action MP1202: HINT (Rational design of hybrid organic–inorganic interfaces: the next step towards advanced functional materials, http://www.cost-hint.cnrs.fr). The authors thank D. Burger and C. Kiefer for technical assistance. The help of C. Loth is also gratefully acknowledged.Notes and references
- R. E. Schaak and T. E. Mallouk, Chem. Mater., 2002, 14, 1455–1471 CrossRef.
- K. G. Sanjaya Ranmohotti, E. Josepha, J. Choi, J. Zhang and J. B. Wiley, Adv. Mater., 2011, 23, 442–460 CrossRef PubMed.
- J. Gopalakrishnan, T. Sivakumar, K. Ramesha, V. Thangadurai and G. N. Subbanna, J. Am. Chem. Soc., 2000, 122, 6237–6241 CrossRef.
- R. Uppuluri, A. S. Gupta, A. S. Rosas and T. E. Mallouk, Chem. Soc. Rev., 2018, 47, 2401–2430 RSC.
- D. Montasserasadi, D. Mohanty, A. Huq, L. Heroux, E. A. Payzant and J. B. Wiley, Inorg. Chem., 2014, 53, 1773–1778 CrossRef PubMed.
- S. Akbarian-Tefaghi and J. B. Wiley, Dalton Trans., 2018, 47, 2917–2924 RSC.
- L. Wang and T. Sasaki, Chem. Rev., 2014, 114, 9455–9486 CrossRef PubMed.
- B.-W. Li, M. Osada, Y. Ebina, S. Ueda and T. Sasaki, J. Am. Chem. Soc., 2016, 138, 7621–7625 CrossRef PubMed.
- R. Ma and T. Sasaki, Adv. Mater., 2010, 22, 5082–5104 CrossRef PubMed.
- S. Tahara, T. Ichikawa, G. Kajiwara and Y. Sugahara, Chem. Mater., 2007, 19, 2352–2358 CrossRef.
- S. Takahashi, T. Nakato, S. Hayashi, Y. Sugahara and K. Kuroda, Inorg. Chem., 1995, 34, 5065–5069 CrossRef.
- Y. Takeda, T. Momma, T. Osaka, K. Kuroda and Y. Sugahara, J. Mater. Chem., 2008, 18, 3581–3587 RSC.
- H. Suzuki, K. Notsu, Y. Takeda, W. Sugimoto and Y. Sugahara, Chem. Mater., 2003, 15, 636–641 CrossRef.
- C. Wang, K. Tang, D. Wang, Z. Liu, L. Wang, Y. Zhu and Y. Qian, J. Mater. Chem., 2012, 22, 11086–11092 RSC.
- A. Shimada, Y. Yoneyama, S. Tahara, P. H. Mutin and Y. Sugahara, Chem. Mater., 2009, 21, 4155–4162 CrossRef.
- J. R. Boykin and L. J. Smith, Inorg. Chem., 2015, 54, 4177–4179 CrossRef PubMed.
- S. Akbarian-Tefaghi, E. Teixeira Veiga, G. Amand and J. B. Wiley, Inorg. Chem., 2016, 55, 1604–1612 CrossRef PubMed.
- Y. Wang, E. Delahaye, C. Leuvrey, F. Leroux, P. Rabu and G. Rogez, Inorg. Chem., 2016, 55, 4039–4046 CrossRef PubMed.
- Y. Wang, E. Delahaye, C. Leuvrey, F. Leroux, P. Rabu and G. Rogez, Inorg. Chem., 2016, 55, 9790–9797 CrossRef PubMed.
- S. Akbarian-Tefaghi, T. Rostamzadeh, T. T. Brown, C. Davis-Wheeler and J. B. Wiley, ChemNanoMat, 2017, 3, 538–550 CrossRef.
- S. Hotta, N. Idota and Y. Sugahara, Key Eng. Mater., 2014, 616, 82–86 Search PubMed.
- Y. Wang, X. Zhu, X. Li, L. Wang, Y. Wang, Q. Hao and K. Tang, J. Mater. Chem. A, 2014, 2, 15590–15597 RSC.
- S. Yoshioka, Y. Takeda, Y. Uchimaru and Y. Sugahara, J. Organomet. Chem., 2003, 686, 145–150 CrossRef.
- Y. Asai, Y. Ariake, H. Saito, N. Idota, K. Matsukawa, T. Nishino and Y. Sugahara, RSC Adv., 2014, 4, 26932–26939 RSC.
- Y. Takeda, H. Suzuki, K. Notsu, W. Sugimoto and Y. Sugahara, Mater. Res. Bull., 2006, 41, 834–841 CrossRef.
- S. Tahara and Y. Sugahara, Langmuir, 2003, 19, 9473–9478 CrossRef.
- Y. Wang, C. Wang, L. Wang, Q. Hao, X. Zhu, X. Chen and K. Tang, RSC Adv., 2014, 4, 4047–4054 RSC.
- Y. Tsunoda, W. Sugimoto and Y. Sugahara, Chem. Mater., 2003, 15, 632–635 CrossRef.
- T. Coradin, R. Clément, P. G. Lacroix and K. Nakatani, Chem. Mater., 1996, 8, 2153–2158 CrossRef.
- P. G. Lacroix, R. Clément, K. Nakatani, J. Zyss and I. Ledoux, Science, 1994, 263, 658–660 CrossRef PubMed.
- J. L. Colon, C. Y. Yang, A. Clearfield and C. R. Martin, J. Phys. Chem., 1988, 92, 5777–5781 CrossRef.
- J. L. Colon, C. Y. Yang, A. Clearfield and C. R. Martin, J. Phys. Chem., 1990, 94, 874–882 CrossRef.
- Q. Wang, D. Yu, Y. Wang, J. Sun and J. Shen, Langmuir, 2008, 24, 11684–11690 CrossRef PubMed.
- G. Huang, S. Ma, X. Zhao, X. Yang and K. Ooi, Chem. Commun., 2009, 331–333 RSC.
- É. Delahaye, S. Eyele-Mezui, J.-F. Bardeau, C. Leuvrey, L. Mager, P. Rabu and G. Rogez, J. Mater. Chem., 2009, 19, 6106–6115 RSC.
- É. Delahaye, S. Eyele-Mezui, M. Diop, C. Leuvrey, P. Rabu and G. Rogez, Dalton Trans., 2010, 39, 10577–10580 RSC.
- C. Wang, K. Tang, D. Wang, Z. Liu, L. Wang, Y. Zhu and Y. Qian, J. Mater. Chem., 2012, 22, 11086–11092 RSC.
- M. V. Korolevich, R. G. Zhbankov and V. V. Sivchik, J. Mol. Struct., 1990, 220, 301–313 CrossRef.
- F. C. Cruz, A. Scalabrin, D. Pereira, P. A. M. Vazquez, Y. Hase and F. Strumia, J. Mol. Spectrosc., 1992, 156, 22–38 CrossRef.
- R. A. Vaia, R. K. Teukolsky and E. P. Giannelis, Chem. Mater., 1994, 6, 1017–1022 CrossRef.
- AIST, Spectral Database for Organic Compounds, SDBS, http://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi, (accessed April 13, 2018).
- M.-P. Crosnier-Lopez, F. L. Berre and J.-L. Fourquet, J. Mater. Chem., 2001, 11, 1146–1151 RSC.
- P. J. Ollivier and T. E. Mallouk, Chem. Mater., 1998, 10, 2585–2587 CrossRef.
- Y. Marcus, The properties of solvents, John Wiley & Sons Ltd., Chichester, 1998 Search PubMed.
- A. Goñi, J. Rius, M. Insausti, L. M. Lezama, J. L. Pizarro, M. I. Arriortua and T. Rojo, Chem. Mater., 1996, 8, 1052–1060 CrossRef.
- P. van der Voort and E. F. Vansant, J. Liq. Chromatogr. Relat. Technol., 1996, 19, 2723–2752 CrossRef.
- S. Tahara, T. Yamashita, G. Kajiwara and Y. Sugahara, Chem. Lett., 2006, 35, 1292–1293 CrossRef.
- T. Itagaki and K. Kuroda, J. Mater. Chem., 2003, 13, 1064–1068 RSC.
- J. Murakami, T. Itagaki and K. Kuroda, Solid State Ionics, 2004, 172, 279–282 CrossRef.
- K. Nakanishi, T. Goto and M. Ohashi, Bull. Chem. Soc. Jpn., 1957, 30, 403–408 CrossRef.
- R. A. Heacock and L. Marion, Can. J. Chem., 1956, 34, 1782–1795 CrossRef.
- T.-S. Chan, C.-L. Dong, Y.-H. Chen, Y.-R. Lu, S.-Y. Wu, Y.-R. Ma, C.-C. Lin, R.-S. Liu, J.-L. Chen, J. Guo, J.-F. Lee, H.-S. Sheu, C.-C. Yang and C.-L. Chen, J. Mater. Chem., 2011, 21, 17119–17127 RSC.
- S. Ida, C. Ogata, U. Unal, K. Izawa, T. Inoue, O. Altuntasoglu and Y. Matsumoto, J. Am. Chem. Soc., 2007, 129, 8956–8957 CrossRef PubMed.
- P. Ganter, L. M. Schoop, M. Däntl and B. V. Lotsch, Chem. Mater., 2018, 30, 2557–2565 CrossRef.
- N. C. Bristowe, J. Varignon, D. Fontaine, E. Bousquet and P. Ghosez, Nat. Commun., 2015, 6, 6677 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the experimental procedures, complete analyses and supplementary results. See DOI: 10.1039/c8sc01754a |
| This journal is © The Royal Society of Chemistry 2018 |