
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic biology approaches and combinatorial biosynthesis towards heterologous lipopeptide production†
Fu
Yan‡
a,
Christian
Burgard‡
 a,
Alexander
Popoff
a,
Nestor
Zaburannyi
a,
Gregor
Zipf
b,
Josef
Maier
c,
Hubert S.
Bernauer
b,
Silke C.
Wenzel
a and
Rolf
Müller
*a
a,
Alexander
Popoff
a,
Nestor
Zaburannyi
a,
Gregor
Zipf
b,
Josef
Maier
c,
Hubert S.
Bernauer
b,
Silke C.
Wenzel
a and
Rolf
Müller
*a
aHelmholtz Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Centre for Infection Research and Department of Pharmacy at Saarland University, Saarland University Campus, Building E8.1, 66123 Saarbrücken, Germany. E-mail: rom@helmholtz-hzi.de
bATG:biosynthetics GmbH, Weberstraße 40, 79249 Merzhausen, Germany
cIStLS – Information Services to Life Sciences, Härlestraße 24/1, 78727 Oberndorf am Neckar/Boll, Germany
First published on 8th August 2018
Abstract
Synthetic biology techniques coupled with heterologous secondary metabolite production offer opportunities for the discovery and optimisation of natural products. Here we developed a new assembly strategy based on type IIS endonucleases and elaborate synthetic DNA platforms, which could be used to seamlessly assemble and engineer biosynthetic gene clusters (BGCs). By applying this versatile tool, we designed and assembled more than thirty different artificial myxochromide BGCs, each around 30 kb in size, and established heterologous expression platforms using a derivative of Myxococcus xanthus DK1622 as a host. In addition to the five native types of myxochromides (A, B, C, D and S), novel lipopeptide structures were produced by combinatorial exchange of nonribosomal peptide synthetase (NRPS) encoding genes from different myxochromide BGCs. Inspired by the evolutionary diversification of the native myxochromide megasynthetases, the ancestral A-type NRPS was engineered by inactivation, deletion, or duplication of catalytic domains and successfully converted into functional B-, C- and D-type megasynthetases. The constructional design approach applied in this study enables combinatorial engineering of complex synthetic BGCs and has great potential for the exploitation of other natural product biosynthetic pathways.
Introduction
Natural products and their derivatives contribute to a great number of clinical drugs.1 Many microbial natural products are synthesized by polyketide synthases (PKS) and/or nonribosomal peptide synthetases (NRPS).2 For many years, detailed analyses of the genetic basis of natural product biosynthesis revealed the important role of genetic alterations causing the diversity of chemical structures.3 In consequence, it has been widely recognized that “unnatural” natural products could be generated by refactoring genetic elements of biosynthetic pathways. Generally, natural product analogs could be produced by alterations at different biosynthetic stages, such as precursor-directed biosynthesis, engineering of the involved megasynthetases, and/or post-assembly modifications.4 As exemplified in the biosynthesis of lipopeptide antibiotics daptomycin and A54145, more than 120 non-natural derivatives were generated via engineering of biosynthetic machineries.5 In a recent study, over 10 novel peptides were generated by combination of NRPS subunits from Photorhabdus, Xenorhabdus and Bacillus.6 More information on recent progress regarding PKS and NRPS combinatorial biosynthesis can be found in recent reviews.7High-throughput sequencing of bacterial genomes has revealed great potential in the discovery of novel natural products.8 However, the translation of genomic sequences into novel natural products remains a challenging task.9 As most bacteria have not been cultured and many cultured species cannot be manipulated genetically,10 potential natural product BGCs of unknown function need to be cloned and transferred to suitable heterologous hosts for expression and engineering. Albeit the promising biosynthetic potential of bacterial genomes, BGC cloning and pathway engineering, especially in terms of the construction of combinatorial libraries, still remains a challenging task due to the complexity of bacterial genomes and the large sizes of BGCs. In addition, the production levels of natural products in heterologous hosts are often low. This is caused by numerous reasons, e.g. codon bias, inefficient transcription and incorrect protein folding, just to name a few.11 Recent developments in DNA synthesis provide opportunities to elaborately design and synthesize BGCs at will, and thus bear great potential in harnessing BGCs, identifying production-limiting factors and fostering combinatorial biosynthesis. As exemplified by work focusing on biosynthetic pathways of 6-deoxyerythronolide B, epothilone, spectinabilin and novobiocin, refactoring BGCs by using synthetic biology tools generated expected products.12 However, low production yields were observed and currently cannot be explained in detail.
Although the cost for DNA synthesis is on a rapid decline, synthesis of GC-rich DNA fragments larger than 5 kilobase pairs (kbp) is still a serious concern and expensive. Therefore, synthetic BGCs usually need to be assembled from relatively small DNA fragments. In recent years, DNA assembly technologies, such as recombination-based methods,13 PCR-based methods,14 Gibson assembly-like techniques,15 and type IIS restriction endonuclease (RE)-based techniques,16–19 have been developed and widely applied. Nevertheless, there are still significant limitations for the state-of-the-art techniques regarding scarless assembly of complex BGCs. Among these techniques, recombination-based methods exhibit high assembly efficiency. However, their potential limitation in the assembly of large synthetic BGCs with repetitive sequences is less investigated. Clearly, there is still a need to improve the fidelity of DNA sequences for PCR-based methods and Gibson assembly.20 The latest CRISPR-Cas9 technology coupled with homologous recombination or Gibson assembly showed prospective potential in gene cloning,21 however, it would require several rounds of complex manipulations for assembly of multiple synthetic DNA fragments. Type IIS restriction endonuclease-based tools, such as Golden Gate assembly,16,22 GoldenBraid,18 MoClo,17 and EcoFlex systems,19 revealed great advantages in DNA assembly, and bear potential for DNA shuffling and combinatorial biosynthesis.23 Unlike the most commonly used REs, type IIS REs recognize non-palindromic sequences and hydrolyze the DNA double strand outside of their recognition sites, thereby enabling the design of unique overhangs and the seamless ligation of two or more DNA fragments. However, their potential in genetic engineering of complex BGCs encoding multi-domain PKS/NRPS megasynthetases has not been investigated.
In a recent study on myxobacterial lipopeptide biosynthetic pathways we performed a comprehensive analysis of genetic changes in BGCs causing structural diversification of the lipopeptide products myxochromides, which seem to occur widespread among myxobacterial species.24 Previous studies indicate a physiological role of myxochromides in the developmental life cycle of myxobacteria,24 significant antimicrobial or cytotoxic activities were not detected in the applied testings.25,26 Until now, five different types of myxochromides (A, B, C, D and S) differing in the amino acid composition of their peptide cores, and the corresponding BGCs (mch gene clusters) have been identified (Fig. 1).24–27 The mch clusters are around 30 kb in size and contain four co-transcriptional genes. The gene mchA encodes a type I iterative PKS module, which synthesizes the polyunsaturated lipid chains. The NRPS genes mchB and mchC encode multimodular NRPS subunits, which direct the biosynthesis of myxochromide peptide cores. A short coding DNA sequence (CDS) located downstream of mchC was recently identified and designated as mchD, encoding a putative membrane protein of unknown function.24 Phylogenetic analysis suggested the A-type mch cluster as the common ancestral gene cluster of the other known mch clusters.24 The domain organizations in the megasynthetase subunits MchA and MchB among all pathways are identical, while significant differences have been found in MchC (Fig. 1). The observed evolutionary diversification of MchC by point mutations and recombination events led to the production of different types of myxochromides. In addition to studies with the native myxochromide producer strains, proficient heterologous expression systems using Myxococcus xanthus, Pseudomonas putida and Corallococcus macrosporus as hosts could be established,28,29 facilitating future efforts to further exploit this compound class.24
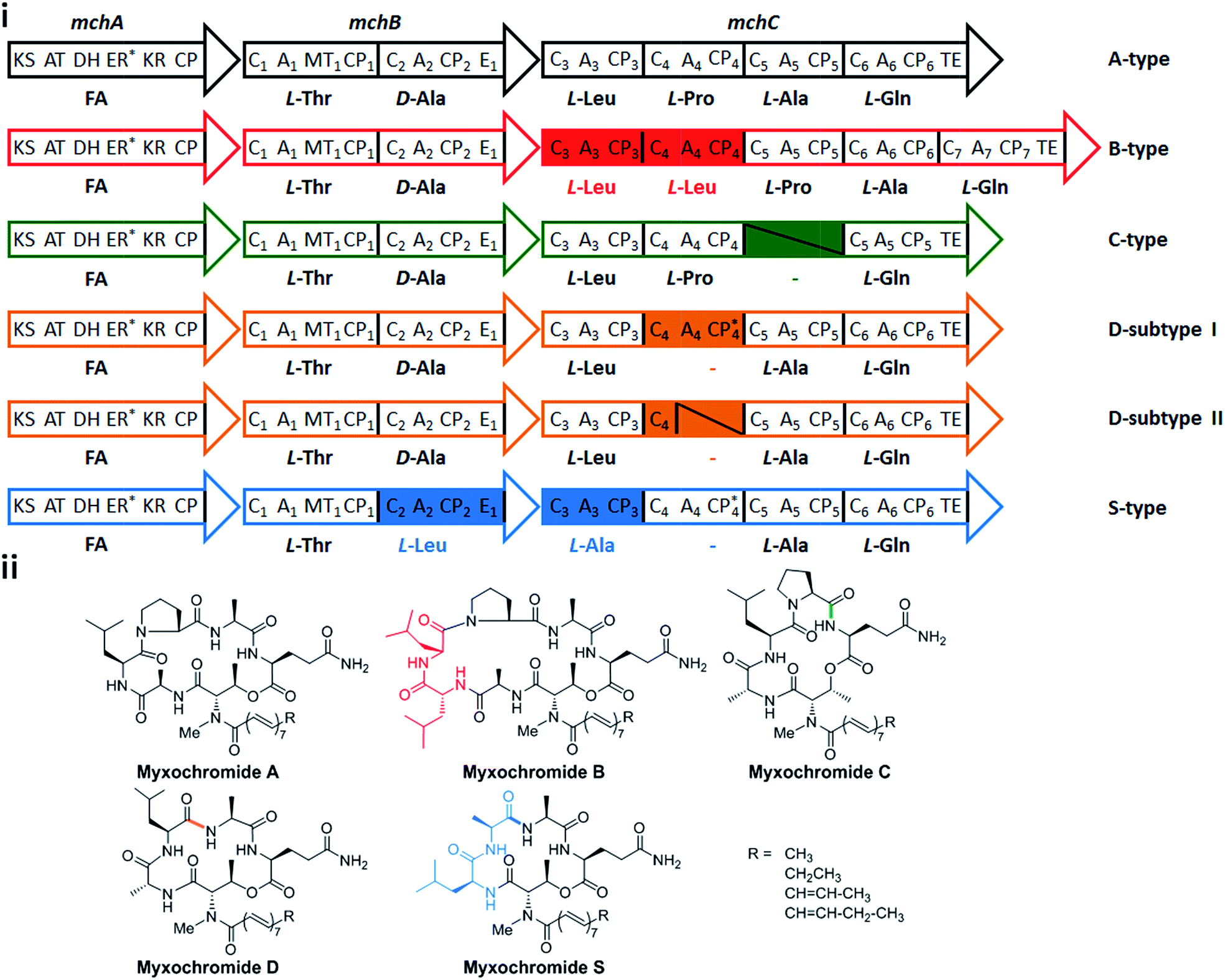 | ||
| Fig. 1 Myxochromide biosynthetic pathways and chemical structures. (i) The mch gene clusters consist of a pks gene (mchA), two nrps genes (mchB and mchC) and mchD (not shown in the figure). The catalytic domains of the encoded PKS/NRPS subunits are illustrated and abbreviated as follows: KS, ketosynthase; AT, acyltransferase; DH, dehydratase; ER, enoylreductase; KR, ketoreductase; CP, carrier protein; C, condensation domain; A, adenylation domain; MT, methyltransferase domain; E, epimerization domain; TE, thioesterase. In the case of NRPS encoding genes, domains are numbered according to the modular organization and the incorporated substrates are indicated. The color filled modules highlight the differences between the A-type and other pathway types. Module deletions are marked with slashes. Domains marked with an asterisk are supposed to be inactive. The figure is reproduced from ref. 24. (ii) Chemical structures of myxochromides. | ||
The divergent myxochromide pathways and lipopeptide structures provide an ideal system to apply synthetic biology approaches for combinatorial biosynthesis and evolutionary-guided pathway engineering. In this study we developed an efficient strategy for the assembly and versatile engineering of BGCs encoding multifunctional PKS/NRPS megasynthetases. Based on synthetic DNA, more than thirty artificial myxochromide BGCs were generated and heterologously expressed in a derivative of the myxobacterial model strain M. xanthus. Different engineering approaches were applied in order to expand the structural diversity of the myxochromide lipopeptide family.
Results and discussion
Strategy for gene cluster design and assembly
For the design of synthetic myxochromide BGCs and their expression in Myxococcus xanthus, we aimed at minimizing sequence changes within the CDS to reduce the risk of potential negative effects on pathway expression. Therefore, in contrast to our previous study on the construction of an artificial epothilone BGC,30 comprehensive sequence optimization approaches were not applied. The sequence design was rather focused on the development of a versatile assembly and engineering strategy for gene clusters encoding multifunctional PKS/NRPS megasynthetases. The template BGCs used to design artificial mch clusters are listed in Table S1.† During the design process, selected restriction sites used for the assembly of artificial myxochromide BGCs were engineered at the 5′ and 3′ ends of each synthetic fragment. These restriction sites were virtually removed from the mch cluster sequences by point mutations (Table S2 and S4†), and splitter elements (SEs) were introduced between the catalytic domain-encoding regions. SEs consist of a unique conventional type II restriction site flanked by two recognition sites of a type IIS restriction enzyme (Fig. S1 and Table S3†). The unique type II restriction sites allow for exchange of synthetic segments between two dedicated SEs on the stage of the mchA′/B′/C′_SE construct library (Fig. 2). After SE removal in the course of “desplitting” seamless and directed “rejoining” of mchA′/B′/C′ fragments is mediated via unique overhangs of the engineered type IIS restriction sites (Fig. 2, Table S3†). To prove our concept, we initially tested the assembly strategy on the A-type myxochromide BGC. Corresponding fragments of the biosynthesis genes mchAA, mchBA and mchCA, intergenic linker regions harboring the 3′/5′ ends of the genes, 3′mchA-5′mchB (3AA5BA) and 3′mchB-5′mchC (3BA5CA), as well as promoter/terminator fragments, promoter-5′mchA (P5mchAA) and the 3′mchC-mchD-terminator-rhlE (T3mchCA), were manufactured by DNA synthesis. Due to their large size the biosynthesis genes were subdivided into smaller fragments to synthesize mchAA and mchBA in two and mchCA in three parts. The putative ATP-dependent RNA helicase encoding gene (rhlE) downstream of mchD, which is part of the T3mchCA fragment, represents a homologous region for chromosomal integration in the heterologous host. Additionally, the cloning vector pSynbio1 as well as the expression vector pSynbio2 was designed and synthesized to facilitate cloning in E. coli (Fig. S2†). SEs were initially designed based on type IIS enzyme AarI. However, as the cleavage efficiency of AarI turned out to be very low in our assembly approach, we revised the sequence design after successful but tedious construction of the first pathway version by replacing AarI with BsaI. Following the concept illustrated in Fig. 2, the biosynthesis genes were first reconstituted from synthetic fragments by conventional restriction/ligation techniques. The generated constructs were subsequently hydrolyzed with BsaI (AarI in the first mchA BGC version) and the released SEs (∼30 bp) were removed by spin column purification of the gene fragments which are larger than 150 bp in size. The desplit gene fragments were then seamlessly “rejoined” by ligation to generate SE-free mchA′, mchB′ and mchC′ constructs. Using the promoter, linker and terminator fragments the entire mchA BGC (∼30 kb) was assembled by conventional restriction/ligation methods yielding the expression plasmids pSynMch1 (AarI-based desplitting) and pSynMch2 (BsaI-based desplitting) (Table 1, Fig. 3 and Table S7†). Both expression constructs were transferred into M. xanthus DK1622 ΔmchA-tet in which the native A-type myxochromide BGC was replaced by a tetracycline resistance gene.31 Chromosomal integration in the former mch gene cluster locus (upstream of rhlE) was achieved by homologous recombination via single cross-over (Fig. S3†). UPLC-MS analysis of crude extracts from the obtained mutant strains M. xanthus DK1622 ΔmchA-tet:pSynMch1/2 revealed the production of several compounds exhibiting retention times and MS/MS patterns similar to those of myxochromides A2–4 (shown for the pSynMch2 expression strain in Fig. S5†). Compared to the native A-type BGC, the synthetic version generated myxochromide A3 at equivalent levels based on quantification with authentic reference material (∼8 mg L−1; Fig. S6†). Other derivatives, myxochromides A2 and A4, were also detected in significant amounts indicating that the total myxochromide production was actually a bit higher with the synthetic BGC compared to the native pathway.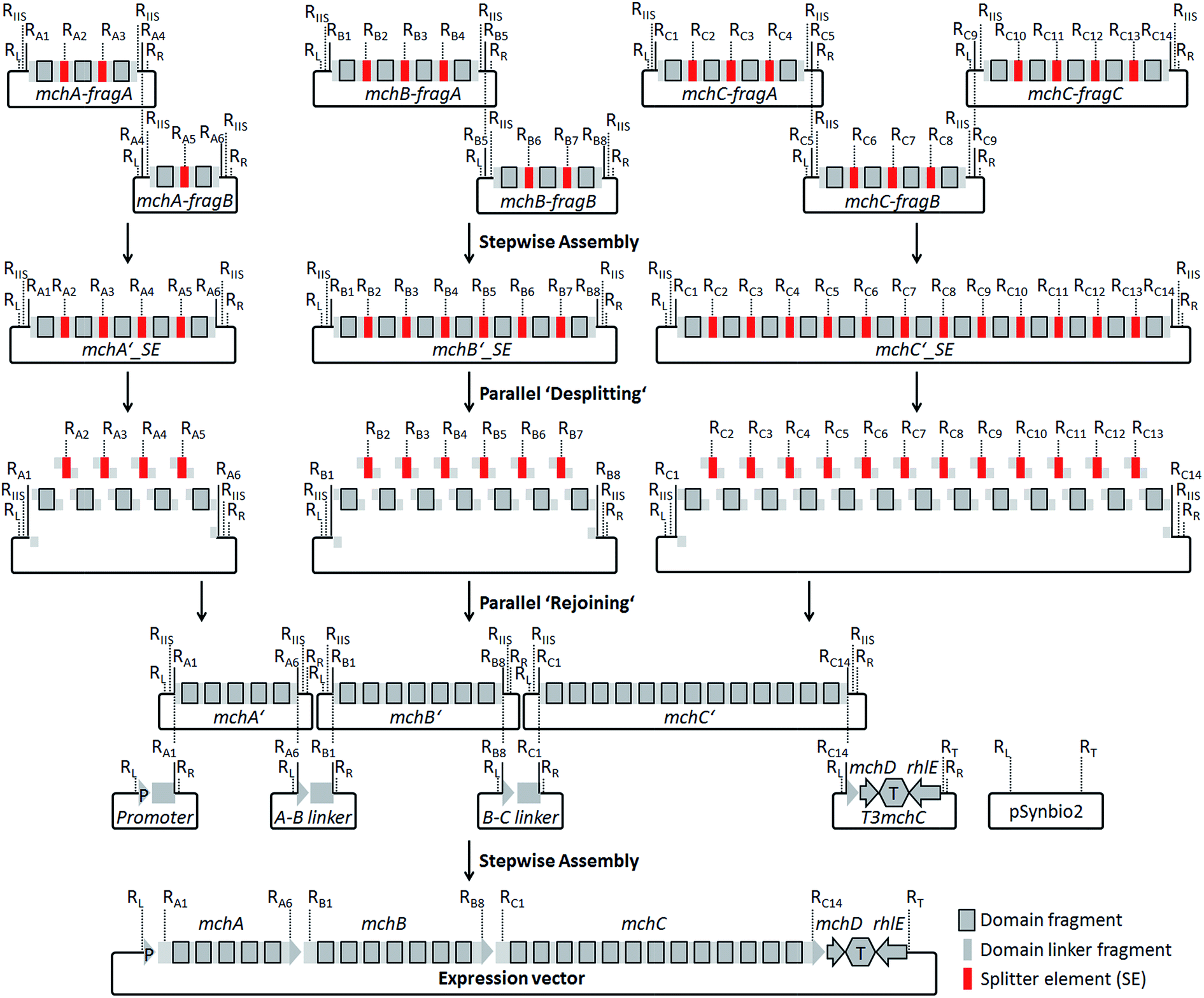 | ||
| Fig. 2 Assembly strategy for the generation of artificial myxochromide BGCs consisting of four steps: ligation of synthetic mch gene fragments on the cloning vector pSynbio1; ‘desplitting’ of gene constructs using type IIS restriction enzymes; ‘rejoining’ of the single domain encoding fragments after removal of splitter elements (SEs); reconstitution of the BGC by stepwise assembly of the promoter, 5′-/3′-truncated gene, intergenic linker and terminator fragments on the expression vector pSynbio2. Restriction sites (R) were introduced in SEs as well as at both termini of each gene synthesis fragment (RL, KpnI; RR, PmeI; RT, PvuI; other R-sites are listed in Table S3†). RIIS, type IIS R-site. Recognition sequences for IIS endonucleases used in the SEs (BsaI or AarI) were introduced in the flanking regions of mchA′/B′/C′ gene fragments for the desplitting process (Table S3†). | ||
| Plasmid | Descriptiona | GenBank Acc. No. |
|---|---|---|
| a mch clusters in pSynMch1-12 are under the control of the native promoter, while in pSynMch13-33 mch clusters the PTn5 promoter (PnptII) was used. The types of gene cluster in the corresponding synthetic segments are illustrated in lowercase; modified positions are indicated in lowercase and bold. b Design of A-type mch cluster from M. xanthus DK1622 (GenBank Acc. No. KX622595) based on AarI restriction sites. Except this construct, the design of all other mch clusters based on BsaI restriction sites. | ||
| pSynMch1 | Synthetic A-type mch clusterb (P5mchAA-mchAA-3AA5BA-mchBA-3BA5CA-mchCA-T3mchCA) | MG583853 |
| pSynMch2 | Synthetic A-type mch cluster (P5mchAA-mchAA-3AA5BA-mchBA-3BA5CA-mchCA-T3mchCA) | MG583854 |
| pSynMch3 | Synthetic B-type hybrid mch cluster (P5mchAA-mchAA-3AA5BA-mchBA-3BB5CB-mchCB-T3mchCB) | MG583855 |
| pSynMch4 | Synthetic C-type hybrid mch cluster (P5mchAA-mchAA-3AA5BA-mchBA-3BC5CC-mchCC-T3mchCC) | MG583856 |
| pSynMch5 | Synthetic D-type hybrid mch cluster (P5mchAA-mchAA-3AA5BA-mchBD-3BD5CD-mchCD-T3mchCD) | MG583857 |
| pSynMch6 | Synthetic S-type hybrid mch cluster (P5mchAA-mchAA-3AA5BA-mchBS-3BS5CS-mchCS-T3mchCS) | MG583858 |
| pSynMch8 | Synthetic AS-type hybrid mch cluster (P5mchAA-mchAA-3AA5BA-mchBA-3BA5CA-mchCS-T3mchCS) | MG583859 |
| pSynMch9 | Synthetic SA-type hybrid mch cluster (P5mchAA-mchAA-3AA5BA-mchBS-3BA5CA-mchCA-T3mchCA) | MG583860 |
| pSynMch10 | Synthetic SC-type hybrid mch cluster (P5mchAA-mchAA-3AA5BA-mchBS-3BC5CC-mchCC-T3mchCC) | MG583861 |
| pSynMch11 | Synthetic SB-type hybrid mch cluster (P5mchAA-mchAA-3AA5BA-mchBS-3BB5CB-mchCB-T3mchCB) | MG583862 |
| pSynMch12 | Synthetic SD-type hybrid mch cluster (P5mchAA-mchAA-3AA5BA-mchBS-3BD5CD-mchCD-T3mchCD) | MG583863 |
| pSynMch13 | Synthetic A-type mch cluster (PTn5mchAA-mchAA-3AA5BA-mchBA-3BA5CA-mchCA-T3mchCA) | MG583864 |
| pSynMch14 | Synthetic SA-type hybrid mch cluster (PTn5mchAA-mchAA-3AA5BA-mchBS-3BA5CA-mchCA-T3mchCA) | MG583865 |
| pSynMch15 | Synthetic SB-type hybrid mch cluster (PTn5mchAA-mchAA-3AA5BA-mchBS-3BB5CB-mchCB-T3mchCB) | MG583866 |
| pSynMch16 | Synthetic SD-type hybrid mch cluster (PTn5mchAA-mchAA-3AA5BA-mchBS-3BD5CD-mchCD-T3mchCD) | MG583867 |
| pSynMch17 | Synthetic A-type mch cluster with inactivated CP1 domain (PTn5mchAA-mchAA-3AA5BA-mchBA-CP1*-3BA5CA-mchCA-T3mchCA) | MG583868 |
| pSynMch18 | Synthetic A-type mch cluster with inactivated CP2 domain (PTn5mchAA-mchAA-3AA5BA-mchBA-CP2*-3BA5CA-mchCA-T3mchCA) | MG583869 |
| pSynMch19 | Synthetic A-type mch cluster with inactivated CP3 domain (PTn5mchAA-mchAA-3AA5BA-mchBA-3BA5CA-mchCA-CP3*-T3mchCA) | MG583870 |
| pSynMch20 | Synthetic A-type mch cluster with inactivated CP4 domain (PTn5mchAA-mchAA-3AA5BA-mchBA-3BA5CA-mchCA-CP4.1*-T3mchCA) | MG583871 |
| pSynMch21 | Synthetic A-type mch cluster with inactivated CP4 domain (PTn5mchAA-mchAA-3AA5BA-mchBA-3BA5CA-mchCA-CP4.2*-T3mchCA) | MG583872 |
| pSynMch22 | Synthetic A-type mch cluster with inactivated CP5 domain (PTn5mchAA-mchAA-3AA5BA-mchBA-3BA5CA-mchCA-CP5*-T3mchCA) | MG583873 |
| pSynMch23 | Synthetic A-type mch cluster with inactivated CP6 domain (PTn5mchAA-mchAA-3AA5BA-mchBA-3BA5CA-mchCA-CP6*-T3mchCA) | MG583874 |
| pSynMch24 | Synthetic S-type mch cluster with reactivation of CP4 domain (P5mchAA-mchAA-3AA5BA-mchBS-3BS5CS-mchCS-CP4-T3mchCS) | MG583875 |
| pSynMch25 | Synthetic A-type mch cluster with duplicated module 1 (PTn5mchAA-mchAA-3AA5BA-mchBA[duplM1]-3BA5CA-mchCA-T3mchCA) | MG583876 |
| pSynMch26 | Synthetic A-type mch cluster with duplicated module 2 (PTn5mchAA-mchAA-3AA5BA-mchBA[duplM2]-3BA5CA-mchCA-T3mchCA) | MG583877 |
| pSynMch27 | Synthetic A-type mch cluster with duplicated module 3 (PTn5mchAA-mchAA-3AA5BA-mchBA-3BA5CA-mchCA[duplM3]-T3mchCA) | MG583878 |
| pSynMch28 | Synthetic A-type mch cluster with duplicated module 6 (PTn5mchAA-mchAA-3AA5BA-mchBA-3BA5CA-mchCA[duplM6]-T3mchCA) | MG583879 |
| pSynMch29 | Synthetic A-type mch cluster with deletion of module 1 (PTn5mchAA-mchAA-3AA5BA-mchBA[delM1]-3BA5CA-mchCA-T3mchCA) | MG583880 |
| pSynMch30 | Synthetic A-type mch cluster with deletion of module 2 (PTn5mchAA-mchAA-3AA5BA-mchBA[delM2]-3BA5CA-mchCA-T3mchCA) | MG583881 |
| pSynMch31 | Synthetic A-type mch cluster with deletion of module 3 (PTn5mchAA-mchAA-3AA5BA-mchBA-3BA5CA-mchCA[delM3]-T3mchCA) | MG583882 |
| pSynMch32 | Synthetic A-type mch cluster with deletion of module 4 (PTn5mchAA-mchAA-3AA5BA-mchBA-3BA5CA-mchCA[delM4]-T3mchCA) | MG583883 |
| pSynMch33 | Synthetic A-type mch cluster with deletion of module 5 (PTn5mchAA-mchAA-3AA5BA-mchBA-3BA5CA-mchCA[delM5]-T3mchCA) | MG583884 |
 | ||
| Fig. 3 Synthetic mch gene clusters and generated myxochromides. Synthetic mch clusters for production of native (i) and novel hybrid (iii) myxochromides. Abbreviations of the assembled gene cluster fragments are shown on the top. The lowercase letter indicates from which gene cluster type the fragment originates, which is also reflected by coloring (A-type, white; B-type, red; C-type, green; D-type, orange or S-type, blue). Simplified chemical structures of the produced native (ii) and hybrid (iv) myxochromides. The corresponding myxochromide type is indicated on the right; detailed structural information of the novel hybrid types (AS, SA, SB, SC, and SD) is given in ESI section 5.† FA, polyunsaturated fatty acid. | ||
Heterologous production of native myxochromides from synthetic hybrid pathways
After successful assembly and expression of the A-type myxochromide BGC, we applied the same constructional sequence design approach to establish heterologous expression platforms for the biosynthesis of the four other native myxochromide types (B, C, D and S-type). Considering the high similarity of the PKS-encoding mchA gene sequences and the generated polyunsaturated fatty acid side chains in the five known mch pathway types, the synthetic mchAA gene was also used for the construction of other synthetic mch BGC types. For the same reason, mchBA was used for the assembly of B-type and C-type mch BGCs. However, mchBD was synthesized separately taking into account the relatively low sequence similarity to the mchBA. Overall, six additional mch gene sequences (mchCB, mchCC, mchBD, mchCD, mchBS and mchCS) were generated in analogy to the design of mchBA/mchCA. The synthesized gene fragments were stitched and the resulting mchB′_SE and mchC′_SE constructs were desplit and rejoined as mentioned above. As this could not be accomplished for mchC′B (∼16 kb, 15 SEs), the gene was redesigned and assembled from four synthetic fragments with a total of three SEs. To maintain the native MchB/MchC subunit docking regions as well as the native C-terminus of MchC, respective mchB/mchC linker fragments (3BB5CB, 3BC5CC, 3BD5CD and 3BS5CS) and terminator fragments (T3mchCB, T3mchCC, T3mchCD and T3mchCS) were designed and synthesized. According to the established assembly strategy, expression constructs for the heterologous production of myxochromides B, C, D, and S were generated (pSynMch3, pSynMch4, pSynMch5 and pSynMch6, Table 1, Fig. 3i and Table S7†) and subsequently transferred into the heterologous host M. xanthus DK1622 ΔmchA-tet. The chromosomal integration of synthetic gene clusters was verified by colony PCR (Fig. S3 and S4†), while myxochromide production was analyzed by UPLC-MS. In all cases, the expected myxochromides were indeed produced in decent yield in M. xanthus (Fig. S5 and S6†), demonstrating the functionality of the artificial myxochromide BGCs. For the synthetic mch gene clusters under control of the native promoter, the production titers of myxochromides D and S were similar to myxochromide A, while the production of myxochromides B and C was relatively higher (Fig. S5 and S6†). The production difference of the native myxochromides probably results from the different transcription efficiencies of the gene clusters.In the course of assembly of mch gene clusters, the efficiency of molecular cloning was mainly limited by the “rejoining” of the mchC gene. It seems that the efficiency of the cluster assembly is related to the size of the genes and the number of fragments. In our experiments, we were able to achieve 70–90% and 50–75% correct clones for the desplitting and assembly of mchA′ (∼6.2 kb with 4 splitter elements, 6 fragments in total) and mchB′ (∼9 kb with 6 splitter elements, 8 fragments in total), respectively, while the efficiency for the assembly of mchC′ (13–16 kb, 12–15 splitter elements, 14–17 fragments in total) was relatively low (10–30% correct clones). However, in the desplitting and assembly of mchC′B (16 kb, 3 splitter elements, 5 fragments in total), 75% clones were correct as judged by restriction analysis. Reducing the number of SEs or separation of large genes to several assembly units may improve the efficiency in the “rejoining” step. Notably, in contrast to the relatively high frequency of mutations shown for PCR-based assembly and Gibson assembly,20 the fidelity of the synthetic gene sequences was retained in our assembly process. In all of the sequenced constructs, point mutations occurred only rarely and are thought to be generated during plasmid replication in E. coli. Only three of the 32 sequenced constructs contained point mutations. Upon sequencing of a second clone, constructs free of mutations could be identified.
Production of hybrid myxochromides via recombination of NRPS subunits from different pathways
After establishing heterologous production platforms for native myxochromides, we aimed at extending the combinatorial gene cluster assembly approach in order to direct production towards novel myxochromide peptide cores. As the diversity of native myxochromide peptide cores originates from different substrate specificities or domain arrangements in the NRPS subunits MchB and MchC, we intended to generate novel myxochromides by recombining mchB and mchC genes from different pathway types (Fig. 3iii). Novel hybrid mch BGCs were generated via engineering of the A-type BGC expression construct, yielding pSynMch8 (AS-type BGC), pSynMch9 (SA-type BGC), pSynMch10 (SC-type BGC), pSynMch11 (SB-type BGC) and pSynMch12 (SD-type BGC) (Table 1, Fig. 3iii and Table S7†). For expression of the mch hybrid BGCs the constructs were transferred into M. xanthus DK1622 ΔmchA-tet. Subsequent UPLC-MS analysis revealed the production of several compounds with molecular masses corresponding to the expected novel hybrid myxochromides (Fig. 3iv and S7†). Interestingly, compared to myxochromide A, a significantly higher amount of myxochromide AS was produced after replacement of the mchCA′ of the A-type cluster with mchCS′. The reason for the higher production of hybrid myxochromide AS could not be identified at present but is worthwhile to be analyzed in future studies. The yield of myxochromides SA, SB and SC was comparable with myxochromide A, while the production of myxochromides SD was a bit lower. The yield of hybrid myxochromides was probably affected by the interplay between megasynthetase subunits.In order to ultimately validate the produced novel peptide cores, we cultivated the corresponding M. xanthus mutants in large scale and tried to isolate them for structure elucidation. Myxochromide AS was initially purified and structurally validated. Myxochromide SC was also purified, however, due to the low yield the purification process turned to be labor consuming. To facilitate the purification of sufficient compound material, we set out to increase the production titer of other hybrid myxochromides by means of promoter replacement. In a previous study, high production of myxochromide S was achieved by expression of the S-type mch gene cluster under control of the PTn5 (PnptII) constitutive promoter in the heterologous host M. xanthus.29 Therefore, we initially replaced the native promoter of the synthetic mchA cluster with the PTn5 promoter. The resulting construct pSynMch13 (Fig. 3 and Table 1) was transferred into M. xanthus DK1622 ΔmchA-tet and, expectedly, subsequent LC-MS analysis of the culture extracts revealed a remarkable increase (∼60 fold) for the production of myxochromide A (Fig. S5 and S6†). In analogy, the production titers of myxochromides SA, SB and SD were optimized by replacement of the native promoter of the corresponding hybrid myxochromide BGC with PTn5 promoter (pSynMch14, pSynMch15 and pSynMch16, Table 1), yielding sufficient compound material for structural elucidation. The planar structure of each representative of the purified hybrid myxochromides was unambiguously confirmed by NMR spectroscopy (ESI section 5†), while the absolute configurations of amino acids were elucidated by using Marfey's method and UPLC-MS analysis (ESI section 5†).32
In addition to the successful generation of novel lipopeptide structures our combinatorial approach uncovered the reason for the presence of an L-configured amino acid (leucine) in position two of the myxochromide S peptide core in contrast to other native myxochromides. The incorporation of D-leucine by hybrid assembly lines in which the MchBS subunit is recombined with MchC from A-, B-, C- or D-type pathways (Fig. 3) clearly shows that the E domain from MchBS is active and functional on a leucine residue. Therefore, the L-leucine residue in myxochromide S seems to (exclusively) result from the stereoselectivity of the downstream C3 domain of the MchCS subunit. This is underpinned by the incorporation of L- instead of D-configured alanine in position two of the myxochromide AS peptide core produced by the MchBA–MchCS hybrid NRPS. Taken together, our results show that the E domain from all MchB subunits is functional and the C domain of the downstream module (C3 from the MchC subunits) acts as a gatekeeper and key determinant for the observed stereochemistry. However, according to our previous studies C3 domains from all native myxochromide pathways (including the S-type pathway) were predicted to represent the DCL-type selecting for D-configured peptide substrates from the upstream module,24 indicating that methods for the in silico analysis of C domain specificities have to be refined.
Evolutionary-guided engineering of the A-type myxochromide megasynthetase
To further broaden the application of the established assembly strategy and to expand the chemical diversity of myxochromides, we proceeded to engineer catalytic domains and modules of the NRPS. Assuming that the A-type pathway is the common ancestor of all myxochromide BGCs identified so far, we intended to mimic the evolutionary scenarios observed in nature by directed engineering of the synthetic A-type myxochromide pathway. We initially aimed at inducing “module skipping” processes observed in D-subtype I and S-type megasynthetases by inactivation of the CP domain in each NRPS module. Six CP-encoding fragments harboring a Ser → Ala mutation in the conserved CP core region, [GGHSL] for CPs from ordinary NRPS elongation modules and [GGDSI] for CPs from modules containing an additional E domain,33 were designed and synthesized (CP1inact1, CP2inact1, CP3inact1, CP4inact1, CP5inact1 and CP6inact1). The invariant serine residue is required for posttranslational activation by attachment of a 4′-phosphopantetheine cofactor that enables the binding of activated amino acids and growing peptide intermediates during biosynthesis.34 Our studies on the native myxochromide S biosynthetic pathway revealed that “module skipping” is caused by an inactive CP4 domain that differs in various positions in its core region from the described consensus motif, [GGNPS] instead of [GGHSL].25,35 Therefore, we generated an additional version of the CP4-encoding fragment from the A-type pathway featuring the corresponding three mutations (His → Asn, Ser → Pro and Leu → Ser; CP4inact2). The synthetic fragments were used to replace the respective CP-encoding regions in mchBA or mchCA. The resulting expression plasmids, pSynMch17–pSynMch23 (Table 1), were transferred into M. xanthus DK1622 ΔmchA-tet. Subsequent UPLC-MS analysis confirmed the biosynthesis of the expected product myxochromide D in M. xanthus DK1622 ΔmchA-tet:pSynMch20 and M. xanthus DK1622 ΔmchA-tet:pSynMch21 (Fig. S8†), in which CP4 domains were inactivated (Fig. 4). In the case of the other mutants expressing megasynthetases with an inactivated CP1, CP2, CP3, CP5 or CP6 domain, no myxochromide derivatives were detected indicating a complete abolishment of the biosynthesis. This might be explained by specific substrate requirements of downstream domains or structural features of the assembly line, e.g. including absent or nonfunctional domain interactions that might hamper skipping scenarios in the targeted positions. The retained functionality after CP4 inactivation demonstrates a certain degree of flexibility in this position in the present evolutionary stage of the A-type pathway. We also applied the reverse approach and aimed at reactivation of the CP4 domain in the S-type megasynthetase by restoring the [GGHSL] consensus motif (CP4react). The respective genetic modification in the synthetic S-type BGC abolished the production of myxochromide S, but also no other myxochromides including the expected novel lipohexapeptides could be detected. We assume that the CP4 domain was indeed reactivated and primed with proline, but biosynthesis is not accomplished probably due to substrate specificities of C4 and/or downstream domains. The biosynthesis seems to be blocked so that myxochromide S production via “module-skipping” is also prevented. | ||
| Fig. 4 Module modifications performed on the artificial A-type pathway. The upper side shows the artificial A-type cluster with the NRPS module encoding sequences indicated in different colors. The modified A-type clusters harboring corresponding module modifications are shown in the boxes, in which the respective engineered modules are highlighted in color. Structures of the generated myxochromides are presented below the corresponding modified gene cluster. | ||
Next, we aimed at modifying the artificial A-type BGC by engineering additional modules. In analogy to the duplicated domain region in the native myxochromide B megasynthetase,24 An-CPn-Cn+1 units were designed to target different positions of the A-type megasynthetase (Cn-An-CPn units for C-terminal subunit regions). The respective synthetic fragments were introduced into the mchBA or mchCA genes for subsequent modification of the expression construct pSynMch13. Due to difficulties in the “desplitting” process of the engineered mchC′A genes from plasmids pSyn1-MchC_A_duplM4_SE and pSyn1-MchC_A_duplM5_SE (Table S6†) constructs for duplications of A4-CP4-C5 (DuplM4) and A5-CP5-C6 (DuplM5) domain sets were not obtained. The four generated expression plasmids harboring duplicated domain sets, pSynMch25 (A1-MT1-CP1-C2; DuplM1), pSynMch26 (C2-A2-CP2; DuplM2), pSynMch27 (A3-CP3-C4; DuplM3) and pSynmch28 (C6-A6-CP6; DuplM6), were transferred into M. xanthus DK1622 ΔmchA-tet for expression and production analysis via UPLC-MS. As indicated in Fig. 4 myxochromides were detected in all four mutant strains. The duplication (DuplM3) revealed the expected lipoheptapeptide myxochromide B as the major product, while myxochromide A was produced as a minor derivative. Duplications of module 1, 2 or 6 (DuplM1, DuplM2 and DuplM6) in contrast did not result in the biosynthesis of lipoheptapeptides, but myxochromide A was still detected (Fig. S11†). In order to exclude a deletion of the duplicated module fragments by intramolecular recombination events, the presence of the respective fragments in the chromosomes of the M. xanthus expression strains was verified by Southern blot analysis, indicating that the BGC regions with the repetitive DNA sequences are indeed stable (Fig. S13†). The production of myxochromide A instead of lipoheptapeptides suggests that the duplicated domain set is skipped and that biosynthesis is achieved via the native domain/module interactions of the A-type megasynthetase, which is also observed as a “side reaction” in the DuplM3 experiment revealing myxochromide A as a minor product. The complete lack of lipoheptapeptide production with DuplM1, DuplM2 and DuplM6 might be due to the overall structural conformation of the engineered megasynthetase, dominant native interactions of the catalytic domains, non-functional fusion sites and/or restricted substrate specificities of downstream domains.36 Similar to the CP inactivation experiments, the module duplication approach was only successful in the position at which a natural duplication event was detected (DuplM3; see myxochromide B pathway).
The third strategy applied for myxochromide A pathway engineering was a module deletion approach. Inspired by recombination events proposed for myxochromide C BGC evolution,24 synthetic DNA fragments for deletion of An-CPn-Cn+1 units (including MT1 in the case of module 1) were generated. In order to maintain the native MchB/MchC subunit arrangement and docking regions we planned an A-CP didomain deletion for module 2 (similar to the A4-CP4 deletion during evolution of the D-subtype 2 megasynthetase).24 According to the established engineering procedure five expression plasmids with modified versions of the A-type BGC were constructed: pSynMch29 (ΔA1-MT1-CP1-C2; DelM1), pSynMch30 (ΔA2-CP2; DelM2), pSynMch31 (ΔA3-CP3-C4; DelM3), pSynMch32 (ΔA4-CP4-C5; DelM4) and pSynMch33 (ΔA5-CP5-C6; DelM5). The myxochromide production profiles of the transformed M. xanthus host strains were analyzed by UPLC-MS. The obtained data showed that mutants with DelM4 and DelM5 modifications produced the expected myxochromide types D and C, respectively (Fig. 4 and S12†). The modifications DelM1, DelM2 and DelM3, however, resulted in an abolishment of myxochromide production, indicating that the engineered myxochromide A megasynthetase is not functional most likely due to the significant structural changes. These results are in accordance with observations from native myxochromide pathways, for which An-CPn(-Cn+1) deletions were only detected for modules 4 and 5 so far.24
Based on our analyses and previous experiments the design for the duplications and deletions of myxochromide biosynthetic domain sets was based on An-CPn-Cn+1 (except DuplM2 and DuplM6) or An-CPn (for DelM2) units rather than functional modules (Cn-An-CPn). In a recent study on NRPS engineering, novel compounds were generated by combinatorial biosynthesis based on the same strategy of exchange units (An-CPn-Cn+1), and a consensus motif of flexible linker regions between C and A domains was used for unit-swapping.6 However, the C–A linker regions in myxochromide megasynthetases seem to be less conserved as in Photorhabdus and Xenorhabdus as mentioned by Bode and colleagues.6 The module engineering approach in this study was inspired by the natural module duplication/deletion events that occurred during mch BGC evolution. Therefore, instead of using the C–A linker, we used the fusion sites located at the N-terminus of the A domains (module duplications) and at the C-terminus of the C domains (module deletions) (Fig. S9 and S10†).24 Although no novel myxochromide derivative was generated by engineering of modules/domains, the regeneration of other myxochromides from the A-type pathway provides further evidence for the genetic interrelationship of myxochromide pathways as previously proposed.24
Conclusions
In this study, we developed a straightforward DNA assembly strategy for the construction and engineering of complex BGCs based on synthetic DNA. The described approach enables versatile modification and recombination of BGC elements and is suitable for the engineering of multifunctional PKS/NRPS megasynthetases, which was demonstrated by the construction of more than thirty artificial myxochromide BGCs. Using M. xanthus as the host, we were able to establish comparable expression systems for the five different myxochromide pathway types known from myxobacteria. Following a combinatorial biosynthesis approach novel lipopeptide structures, so-called hybrid myxochromides, could be generated via subunit recombination of the different myxochromide NRPS types. In addition, different scenarios for megasynthetase diversification (CP inactivation, module duplication/deletion) observed in the evolution of native myxochromide BGCs were broadly applied using the ancestral A-type pathway as the template. A switch from myxochromide A production to other types of myxochromides (myxochromides B, C and D) was achieved after targeting positions that are also altered in natural BGC evolution. In most other cases myxochromide biosynthesis was completely abolished or myxochromide A was still produced (in module duplication experiments) demonstrating that the interplay of the causative mutations, deletions or insertions with other features of the respective assembly line is of utmost importance for the functionality of the megasynthetase. In addition to targeted engineering approaches conducted in this study, the developed constructional BGC design enables the random generation of hybrid gene (cluster) libraries by e.g. simultaneous “one-pot” desplitting and rejoining of an initial gene set. The described approach can significantly accelerate the process of biosynthetic pathway engineering and thus offers potential to largely expand chemical diversity. The assembly strategy presented here can be applied on all types of BGCs, and represents not only an efficient method to exploit known pathways, but can e.g. also support genome-mining approaches.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by a grant from the German Federal Ministry of Education and Research (FKZ: 031A155). We thank ATG:biosynthetics GmbH and IStLS for supporting this study. We would like to thank PharmBioTec for support in this work.References
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629 CrossRef PubMed.
- (a) M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468 CrossRef PubMed; (b) J. L. Meier and M. D. Burkart, Chem. Soc. Rev., 2009, 38, 2012 RSC; (c) L. Katz and R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2016, 43, 155 CrossRef PubMed.
- C. T. Walsh and M. A. Fischbach, J. Am. Chem. Soc., 2010, 132, 2469 CrossRef PubMed.
- (a) H. Sun, Z. Liu, H. Zhao and E. L. Ang, Drug Des., Dev. Ther., 2015, 9, 823 Search PubMed; (b) C. C. Ladner and G. J. Williams, J. Ind. Microbiol. Biotechnol., 2016, 43, 371 CrossRef PubMed; (c) S. E. O'Connor, Annu. Rev. Genet., 2015, 49, 71 CrossRef PubMed.
- R. H. Baltz, ACS Synth. Biol., 2014, 3, 748 CrossRef PubMed.
- K. A. J. Bozhüyük, F. Fleischhacker, A. Linck, F. Wesche, A. Tietze, C.-P. Niesert and H. B. Bode, Nat. Chem., 2018, 10, 275 CrossRef PubMed.
- (a) K. J. Weissman, Nat. Prod. Rep., 2016, 33, 203 RSC; (b) M. Winn, J. K. Fyans, Y. Zhuo and J. Micklefield, Nat. Prod. Rep., 2016, 33, 317 RSC.
- N. Ziemert, M. Alanjary and T. Weber, Nat. Prod. Rep., 2016, 33, 988 RSC.
- S. E. Ongley, X. Bian, B. A. Neilan and R. Müller, Nat. Prod. Rep., 2013, 30, 1121 RSC.
- E. J. Stewart, J. Bacteriol., 2012, 194, 4151 CrossRef PubMed.
- D. C. Stevens, T. P. A. Hari and C. N. Boddy, Nat. Prod. Rep., 2013, 30, 1391 RSC.
- (a) H. G. Menzella, S. J. Reisinger, M. Welch, J. T. Kealey, J. Kennedy, R. Reid, C. Q. Tran and D. V. Santi, J. Ind. Microbiol. Biotechnol., 2006, 33, 22 CrossRef PubMed; (b) S. C. Mutka, J. R. Carney, Y. Liu and J. Kennedy, Biochemistry, 2006, 45, 1321 CrossRef PubMed; (c) Z. Shao, G. Rao, C. Li, Z. Abil, Y. Luo and H. Zhao, ACS Synth. Biol., 2013, 2, 662 CrossRef PubMed; (d) P. Basitta, L. Westrich, M. Rösch, A. Kulik, B. Gust and A. K. Apel, ACS Synth. Biol., 2017, 6, 817 CrossRef PubMed.
- (a) M. Z. Li and S. J. Elledge, Nat. Methods, 2007, 4, 251 CrossRef PubMed; (b) Z. Shao, H. Zhao and H. Zhao, Nucleic Acids Res., 2009, 37, e16 CrossRef PubMed; (c) Y. Zhang, U. Werling and W. Edelmann, Nucleic Acids Res., 2012, 40, e55 CrossRef PubMed.
- (a) H. E. Klock, E. J. Koesema, M. W. Knuth and S. A. Lesley, Proteins, 2008, 71, 982 CrossRef PubMed; (b) J. Quan and J. Tian, PLoS One, 2009, 4, e6441 CrossRef PubMed; (c) S. Kadkhodaei, H. R. Memari, S. Abbasiliasi, M. A. Rezaei, A. Movahedi, T. J. Shun and A. B. Ariff, RSC Adv., 2016, 6, 66682 RSC; (d) J. Bitinaite, M. Rubino, K. H. Varma, I. Schildkraut, R. Vaisvila and R. Vaiskunaite, Nucleic Acids Res., 2007, 35, 1992 CrossRef PubMed.
- (a) D. G. Gibson, L. Young, R.-Y. Chuang, J. C. Venter, C. A. Hutchison and H. O. Smith, Nat. Methods, 2009, 6, 343 CrossRef PubMed; (b) C. Fu, W. P. Donovan, O. Shikapwashya-Hasser, X. Ye and R. H. Cole, PLoS One, 2014, 9, e115318 CrossRef PubMed.
- C. Engler, R. Kandzia and S. Marillonnet, PLoS One, 2008, 3, e3647 CrossRef PubMed.
- E. Weber, C. Engler, R. Gruetzner, S. Werner, S. Marillonnet and J. Peccoud, PLoS One, 2011, 6, e16765 CrossRef PubMed.
- A. Sarrion-Perdigones, E. E. Falconi, S. I. Zandalinas, P. Juárez, A. Fernández-del-Carmen, A. Granell and D. Orzaez, PLoS One, 2011, 6, e21622 CrossRef PubMed.
- S. J. Moore, H.-E. Lai, R. J. R. Kelwick, S. M. Chee, D. J. Bell, K. M. Polizzi and P. S. Freemont, ACS Synth. Biol., 2016, 5, 1059 CrossRef PubMed.
- J. Liang, Z. Liu, X. Z. Low, E. L. Ang and H. Zhao, Nucleic Acids Res., 2017, 45, e94 CrossRef PubMed.
- (a) C. Ronda, L. E. Pedersen, M. O. A. Sommer and A. T. Nielsen, Sci. Rep., 2016, 6, 19452 CrossRef PubMed; (b) N. C. O. Lee, V. Larionov and N. Kouprina, Nucleic Acids Res., 2015, 43, e55 CrossRef PubMed; (c) J.-W. Wang, A. Wang, K. Li, B. Wang, S. Jin, M. Reiser and R. F. Lockey, BioTechniques, 2015, 58, 161 CrossRef PubMed; (d) H. Wang, Z. Li, R. Jia, J. Yin, A. Li, L. Xia, Y. Yin, R. Müller, J. Fu, A. F. Stewart and Y. Zhang, Nucleic Acids Res., 2018, 46, e28 CrossRef PubMed.
- S. Werner, C. Engler, E. Weber, R. Gruetzner and S. Marillonnet, Bioeng. Bugs, 2012, 3, 38 Search PubMed.
- (a) C. Engler, R. Gruetzner, R. Kandzia and S. Marillonnet, PLoS One, 2009, 4, e5553 CrossRef PubMed; (b) C. Engler and S. Marillonnet, Methods Mol. Biol., 2013, 1073, 141 CrossRef PubMed.
- C. Burgard, N. Zaburannyi, S. Nadmid, J. Maier, H. Jenke-Kodama, E. Luxenburger, H. S. Bernauer and S. C. Wenzel, ACS Chem. Biol., 2017, 12, 779 CrossRef PubMed.
- S. C. Wenzel, B. Kunze, G. Höfle, B. Silakowski, M. Scharfe, H. Blöcker and R. Müller, ChemBioChem, 2005, 6, 375 CrossRef PubMed.
- B. Ohlendorf, S. Kehraus and G. M. König, J. Nat. Prod., 2008, 71, 1708 CrossRef PubMed.
- (a) W. Trowitzsch-Kienast, K. Gerth, V. Wray, H. Reichenbach and G. Höfle, Liebigs Ann. Chem., 1993, 1993, 1233 CrossRef; (b) S. C. Wenzel, P. Meiser, T. M. Binz, T. Mahmud and R. Müller, Angew. Chem., 2006, 118, 2354 CrossRef.
- (a) S. C. Wenzel, F. Gross, Y. Zhang, J. Fu, A. F. Stewart and R. Müller, Chem. Biol., 2005, 12, 349 CrossRef PubMed; (b) O. Perlova, K. Gerth, S. Kuhlmann, Y. Zhang and R. Müller, Microb. Cell Fact., 2009, 8, 1 CrossRef PubMed.
- J. Fu, S. C. Wenzel, O. Perlova, J. Wang, F. Gross, Z. Tang, Y. Yin, A. F. Stewart, R. Müller and Y. Zhang, Nucleic Acids Res., 2008, 36, e113 CrossRef PubMed.
- C. Osswald, G. Zipf, G. Schmidt, J. Maier, H. S. Bernauer, R. Müller and S. C. Wenzel, ACS Synth. Biol., 2014, 3, 759 CrossRef PubMed.
- H. Sucipto, D. Pogorevc, E. Luxenburger, S. C. Wenzel and R. Müller, Metab. Eng., 2017, 44, 160 CrossRef PubMed.
- R. Bhushan and H. Brückner, Amino Acids, 2004, 27, 231 CrossRef PubMed.
- U. Linne, S. Doekel and M. A. Marahiel, Biochemistry, 2001, 40, 15824 CrossRef PubMed.
- J. Crosby and M. P. Crump, Nat. Prod. Rep., 2012, 29, 1111 RSC.
- S. C. Wenzel, P. Meiser, T. M. Binz, T. Mahmud and R. Müller, Angew. Chem., Int. Ed., 2006, 45, 2296 CrossRef PubMed.
- K. J. Weissman, Nat. Chem. Biol., 2015, 11, 660 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, design of artificial gene cluster, genetic manipulation, LC-MS analysis and structure elucidation. See DOI: 10.1039/c8sc02046a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |