
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of iron binding on uranyl(V) stability†
Radmila
Faizova
,
Sarah
White
,
Rosario
Scopelliti
and
Marinella
Mazzanti
 *
*
Institut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
First published on 14th August 2018
Abstract
Here we report the effect of UO2+⋯Fe2+ cation–cation interactions on the redox properties of uranyl(V) complexes and on their stability with respect to proton induced disproportionation. The tripodal heptadentate Schiff base trensal3− ligand allowed the synthesis and characterization of the uranyl(VI) complexes [UO2(trensal)K], 1 and [UO2(Htrensal)], 2 and of uranyl(V) complexes presenting UO2+⋯K+ or UO2+⋯Fe2+ cation–cation interactions ([UO2(trensal)K]K, 3, [UO2(trensal)] [K(2.2.2crypt)][K(2.2.2crypt)], 4, [UO2(trensal)Fe(py)3], 6). The uranyl(V) complexes show similar stability in pyridine solution, but the presence of Fe2+ bound to the uranyl(V) oxygen leads to increased stability with respect to proton induced disproportionation through the formation of a stable Fe2+–UO2+–U4+ intermediate ([UO2(trensal)Fe(py)3U(trensal)]I, 7) upon addition of 2 eq. of PyHCl to 6. The addition of 2 eq. of PyHCl to 3 results in the immediate formation of U(IV) and UO22+ compounds. The presence of an additional UO2+ bound Fe2+ in [(UO2(trensal)Fe(py)3)2Fe(py)3]I2, 8, does not lead to increased stability. Redox reactivity and cyclic voltammetry studies also show an increased range of stability of the uranyl(V) species in the presence of Fe2+ with respect both to oxidation and reduction reactions, while the presence of a proton in complex 2 results in a smaller stability range for the uranyl(V) species. Cyclic voltammetry studies also show that the presence of a Fe2+ cation bound through one trensal3− arm in the trinuclear complex [{UO2(trensal)}2Fe], 5 does not lead to increased redox stability of the uranyl(V) showing the important role of UO2+⋯Fe2+ cation–cation interactions in increasing the stability of uranyl(V). These results provide an important insight into the role that iron binding may play in stabilizing uranyl(V) compounds in the environmental mineral-mediated reduction of uranium(VI).
Introduction
Uranyl(V)1 has been proposed as an important transient intermediate in the biological or abiotic mineral-mediated transformation of soluble uranyl(VI) compounds into the insoluble uranium(IV) dioxide (UO2). These processes provide a convenient strategy to sequester uranium in the environment and, as such, are very important for ground-water remediation. In particular, stable adsorbed or incorporated uranyl(V) species have been reported to form during the U(VI) reduction by Fe(II)-bearing minerals such as mica1e or magnetite ([Fe2+(Fe3+)2O4])2,3 and the presence of iron as the second nearest neighbour has been identified.4 UO2+ species have low stability in aqueous media and they quickly disproportionate to uranyl(VI) and U(IV),5 but the incorporation into iron minerals may prevent disproportionation or further reduction of U(V) to U(IV) and thus lead to long-term immobilization of U(V). However, the role of iron binding to uranyl(V) species and their stabilization remains ambiguous in spite of its importance for the correct speciation of uranium in the environment.Dinuclear or polynuclear complexes of uranyl(V) built from the interaction of a uranyl(V) oxo group with the uranium centre from a UO2+ moiety (UO2+⋯UO2+), also known as cation–cation interaction (CCI),6 have been proposed as intermediates in the proton promoted disproportionation of uranyl(V) to afford UO22+ and U(IV) species.1c,7 The subsequent addition of protons to these polynuclear uranyl(V) intermediates leads to complete electron transfer followed by dissociation of the resulting U(VI)⋯U(IV) complex. In aprotic media stable polynuclear UO2+⋯UO2+ complexes have been isolated.8 We showed that the addition of protons (PyHCl) to a pyridine solution of stable tetrameric UO2+⋯UO2+ complexes leads to the immediate disproportionation of the uranyl(V) species affording uranyl(VI) and U(IV) complexes and water.8b Disproportionation of polynuclear cation–cation complexes was also observed in the absence of protons upon addition of strong Lewis acids (Li+ or U4+)8b,9 to stable uranyl(V) Schiff base complexes and was found to lead to complex mixtures of soluble mixed-valent U(IV)/U(V) uranium oxo clusters. It was also reported that the binding of strong Lewis acids or Group 1 metals to the uranyl(VI) oxo group renders more favourable the reduction of U(VI) to U(V).10
Moreover, it has been demonstrated that the binding of strong Lewis acids such as B(C6F5)3 to the uranyl(V) oxo groups renders more accessible the reduction of U(V) to U(IV).10d,11,12 A fewer studies have been directed to investigate the effect of the interaction of uranyl(V) with 3d transition metals on the stability and redox reactivity of uranyl(V) species. Moreover, in spite of the fact that several uranyl(V) complexes stable in organic solution have been isolated in recent years,9,13 only a few examples of heteropolymetallic complexes presenting a UO2+⋯M interaction, where M is a 3d transition metal, have been prepared.14 The few reported UO2+⋯3d complexes have shown interesting single-molecule magnetic properties.14
The addition of FeI2 to an unstable putative uranyl(V) dipotassium complex of a macrocyclic Schiff base Pacman ligand was reported to result in a higher stability of the uranyl(V) Pacman complexes which was corroborated by the isolation of the corresponding heterobimetallic UO2+⋯Fe2+ CC complex. However, the effect of the interaction UO2+⋯Fe2+ on the stability of these uranyl(V) complexes was not further investigated.14c
Here we report two new stable complexes of uranyl(V) supported by the tripodal Schiff base ligand H3trensal (2,2′,2′′-tris(salicylideneimino)triethylamine): the UO2+⋯K+ [UO2(trensal)K]K, 3, and the heterobimetallic UO2+⋯Fe2+ complex [UO2(trensal)Fe(Py)3], 6. The reactivity of these complexes toward protons and their redox properties were compared and these studies unambiguously show the increased stability of the iron bound complexes.
Results and discussion
Uranyl(VI) and uranyl(V) complexes of trensal3−
The reaction of K3trensal with the nitrate salt of uranyl (VI) leads to the isolation of the uranyl(VI) complex [UO2(trensal)K], 1 in 59% yield. The broad 1H NMR spectrum of 1 in pyridine suggests the presence of fluxional solution species. A higher resolution of the 1H NMR spectrum is observed in deuterated THF and a well resolved 1H NMR spectrum could be obtained in CD3OD solution (Fig. S2†).X-ray quality crystals of 1 could not be obtained, but the addition of one equivalent of PyHCl to a pyridine solution of 1 led to the isolation of X-ray quality crystals of the neutral complex [UO2(Htrensal)], 2, in 60% yield. The 1H NMR spectrum of 2 in pyridine shows the presence of 15 overlapping narrow signals in agreement with the presence of C2 symmetric solution species (Fig. S3†).
The X-ray crystal structure of this complex is presented in Fig. 1 and shows that the uranium atom is heptacoordinated, with a slightly distorted pentagonal bipyramidal coordination geometry, by two uranyl oxygen atoms in the axial position and five donor atoms of the trensal3− ligand in the equatorial plane. The third protonated arm of the trensal3− ligand is not coordinated to the uranyl cation and the phenol proton is hydrogen-bonded with the Schiff base nitrogen N4. The values of the U(VI)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond lengths lie in the range of those typically observed for uranyl(VI) complexes (U–O3 = 1.783(3) Å and U–O4 = 1.787(3) Å).8a,d,9,15 The average U–Ophenoxide (2.231 Å) and the average U–Nimine (2.612 Å bond) lengths are also in the range of those found in other reported Schiff base complexes of uranyl(VI).8a,d,9,15
O bond lengths lie in the range of those typically observed for uranyl(VI) complexes (U–O3 = 1.783(3) Å and U–O4 = 1.787(3) Å).8a,d,9,15 The average U–Ophenoxide (2.231 Å) and the average U–Nimine (2.612 Å bond) lengths are also in the range of those found in other reported Schiff base complexes of uranyl(VI).8a,d,9,15
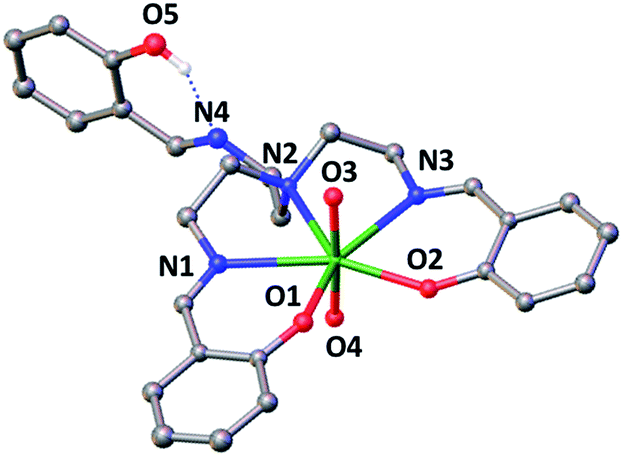 | ||
| Fig. 1 Ellipsoid plot at 50% probability of 2 (co-crystallised pyridine molecule and hydrogen atoms were omitted for clarity, C atoms are represented in grey, O in red, N in blue and U in green). Selected distances (Å) U(1)–O(1) 2.222(3), U(1)–O(2) 2.240(3), U(1)–O(3) 1.783(3), U(1)–O(4) 1.787(3), U(1)–N(1) 2.584(3), U(1)–N(2) 2.625(3), U(1)–N(3) 2.599(3), O(5)⋯N(4) 2.619(5) Å. | ||
In the attempt to reduce the uranyl(VI) complex 2 we added 1 eq. of decamethyl cobaltocene (Cp*2Co) to pyridine solutions of 2. The 1H NMR spectrum of the resulting reaction mixture immediately after addition shows the presence of a large number of signals in the −45 to 45 ppm range suggesting that a putative uranyl(V) intermediate complex undergoes rapid disproportionation (Fig. S4†). This suggests that the phenol arm protonates the more basic uranyl(V) (compared to uranyl(VI)) oxo group resulting in proton induced disproportionation.
In contrast, the uranyl(V) complex [UO2(trensal)K]K, 3, is conveniently prepared in 70% yield from the salt metathesis reaction between K3trensal and [(UO2Py5) (KI2Py2)]n in pyridine (Scheme 1).
 | ||
| Scheme 1 Synthesis of the complexes 3 and 4. | ||
The 1H NMR spectrum of 3 in deuterated pyridine showed the presence of fluxional species with signals in the paramagnetic region (−11 to 15 ppm) characteristic of U(V). Cooling down or heating up the NMR sample did not lead to a better resolution of the spectrum (Fig. S5†).
The addition of stoichiometric amounts of 2.2.2 cryptand (4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane) to complex 3 resulted in a well resolved 1H NMR spectrum (Fig. S6†). This suggests that fluxional potassium binding to the uranyl oxygen is the cause of the broad features in the 1H NMR spectrum of 3.
The complex [UO2(trensal)] [K(2.2.2crypt)][K(2.2.2crypt)], 4, was obtained analytically pure as a green solid in 62% yield. The solid-state structure of 4 was determined by X-ray diffraction studies and is presented in Fig. 2. The overall quality of the crystal structure of compound 4 is rather poor (very weakly diffracting sample) but its connectivity is well determined.
 | ||
| Fig. 2 Ellipsoid plot at 50% probability of the anion [UO2(trensal)] [K(2.2.2crypt)]− in 4 (H and [K(2.2.2crypt)]+ were omitted for clarity, C atoms are represented in grey, O in red, N in blue, K in light blue and U in green). Selected distances (Å) U(1)–O(3) 1.865(16), U(1)–O(4) 1.824(15). | ||
The coordination environment around the uranium centre is similar to that found in complex 2. In 4 the uranium atom is heptacoordinated in a pentagonal bipyramidal coordination geometry. Five donor atoms of the trensal ligand (two oxygen and three nitrogen atoms) occupy the equatorial plane of the uranium ion, while the third arm of the trensal3− ligand does not interact with any cation and one [K(cryptand)] cation is found as an isolated ion in the unit cell of 4. The bipyramid axial positions in 4 are occupied by two oxo ligands with U–O distances (1.824(15) and 1.865(16) Å) significantly longer than those found in the uranyl(VI) complex 2 (1.785(3) Å). These distances are in the range of those found in previously reported complexes of uranyl(V).13a,e,13f
The 1H NMR spectrum of 4 in pyridine shows the presence of 12 narrow signals in agreement with the presence of C2 symmetric solution species (Fig. S6†). This indicates that the molecular anionic fragment [UO2(trensal)] [K(2.2.2crypt)]− found in the X-ray structure of 4 dissociates in pyridine solution and the [K(2.2.2crypt)]+ cation is not bound to the uranyl(V) oxo group in pyridine solution.
The solid-state X-band EPR spectra measured at 298 K and 10 K revealed that the complex 3 is EPR silent.
In contrast, the solid-state X-band (9.40 GHz) EPR spectrum of 4 shows an intense signal at 10 K with a fitted rhombic set of g-values (g1 = 2.44; g2 = 1.10; g3 < 0.6), confirming the presence of uranium in the oxidation state +5 (Fig. S29†). Notably, encapsulation of potassium enables us to obtain an EPR signal from the otherwise EPR silent complex 3. The likely presence of two potassium binding both uranyl(V) oxo groups results in a different electronic structure of 3 compared to 4 (where only one potassium cation is bound) which results in the absence of the EPR signal.13e
Complex 4 is stable up to one month in the solid state and in pyridine and THF solutions. In order to assess the stability of these uranyl(V) complexes with respect to proton induced disproportionation, we have investigated the reaction of 3 and 4 with protons. After addition of 1 eq. of PyHCl to complex 3, partial disproportionation of the uranyl(V) complex was observed by 1H NMR spectroscopy. The addition of 2 eq. of PyHCl resulted in the complete disproportionation of the uranyl(V) to afford the uranyl(VI) complex 2 and unidentified U(IV) products as indicated by 1H NMR spectroscopy (Fig. S7†) and single crystal X-ray diffraction of the isolated crystal of the complex 2. The U(IV) compounds formed in the disproportionation were identified as the product of the hydrolysis of the [U(trensal)]Cl complex as confirmed by the 1H NMR spectrum of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 mixture of [UO2(Htrensal)], [U(trensal)]X (X = I, Cl) and H2O (Fig. S8†).
:1:2 mixture of [UO2(Htrensal)], [U(trensal)]X (X = I, Cl) and H2O (Fig. S8†).
On the other hand, addition of PyHCl to 4 initially resulted in the formation of NMR silent species, but after 3 days the 1H NMR spectrum shows the formation of the same disproportionation products as those found in the reaction of 3 with 2 eq. of PyHCl (Fig. S9†).
Iron binding to uranyl(V) complexes
In view of the potential important role of iron binding in the abiotic reduction of uranyl(VI) as well as in the stabilization of uranyl(V) at iron mineral surfaces we have investigated the reactivity of complexes 1 and 3 with iron salts.The reaction of 1 with FeI2 affords the trinuclear complex [{UO2(trensal)}2Fe], 5, in 93% yield according to Scheme 2.
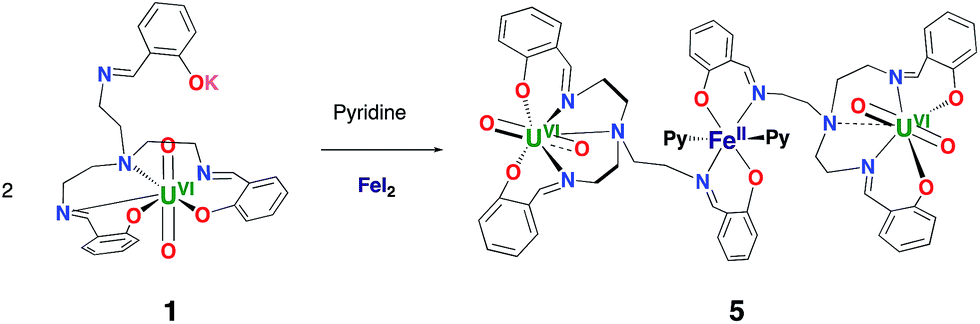 | ||
| Scheme 2 Synthesis of [(UO2(trensal))2Fe(Py)2], 5. | ||
The solid state structure of 5 (Fig. 3) shows the presence of a neutral trinuclear complex where two [UO2(trensal)] moieties are held together by a Fe(II) cation bound by two trensal O, N donor atoms not involved in the coordination of the uranyl cation. Thus, the replacement of the potassium cation in 1 with a Fe(II) cation leads to formation of a trinuclear structure.
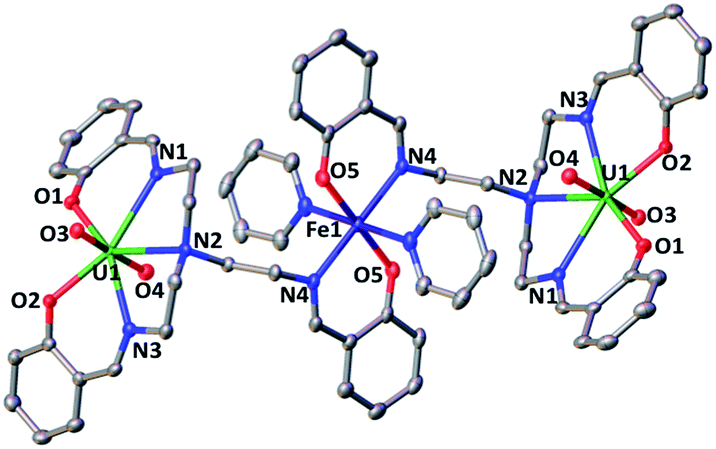 | ||
| Fig. 3 Ellipsoid plot at 50% probability of 5 (co-crystallised pyridine molecule and H were omitted for clarity, C atoms are represented in grey, O in red, N in blue, Fe in dark blue and U in green). Selected distances (Å) U(1)–O(1) 2.2321(18), U(1)–O(2) 2.223(2), U(1)–O(3) 1.7892(18), U(1)–O(4) 1.7851(18), U(1)–N(1) 2.595(2), U(1)–N(2) 2.604(3), U(1)–N(3) 2.558(2), Fe(1)-N(4) 2.140(2), Fe(1)–O(5) 2.0165(18). | ||
In order to prepare a trinuclear uranyl(V) analogue we allowed 5 to react with Cp*2Co. The 1H NMR spectrum after addition of 1 eq. of Cp*2Co to complex 5 revealed the formation of a complex reaction mixture. One of the products could be identified by X-ray diffraction studies, revealing the formation of the dinuclear heterobimetallic complex [UO2(trensal)Fe(py)3], 6. Addition of 2.5 eq. of Cp*2Co to a pyridine solution of 5 led to an intractable reaction mixture from which none of the components could be identified (Fig. S11c†).
Complex 6 can be conveniently prepared in 81% yield from the reaction of FeI2 with complex 3 in pyridine in a 1:1 ratio (Scheme 3).
 | ||
| Scheme 3 Synthesis of [UO2 (trensal)Fe(py)3], 6. | ||
The solid-state structure of 6, represented in Fig. 4, shows the presence of a neutral dinuclear complex where a [UVO2(trensal)] dianion binds a Fe2+ cation through a UO2+⋯Fe2+ CCI. The Fe2+ cation is hexacoordinated, with a slightly distorted octahedral geometry, by two donor atoms of the trensal3− ligand, one uranyl oxo group and three pyridine molecules. The uranium cation is heptacoordinate with a slightly distorted pentagonal bipyramid geometry, by two uranyl oxygen atoms and five donor atoms of the trensal3− ligand in the equatorial plane. The mean U–O bond lengths lie in the range of values typically observed for uranyl(V) complexes,1a,8c,13a,e,13f,14f with the UO2+⋯Fe2+ interaction resulting in a slight lengthening of the bond (U1–O4 = 1.930 (2) Å and U1–O3 = 1.837(3) Å). The value of the Fe–O bond length (2.018(3) Å) falls in the range of those found in the only two previously reported examples of uranyl(V) complexes presenting a cation–cation interaction with a Fe2+ cation (1.946(4) –2.132(4) Å).14c,f Similar Fe–O bond lengths ranging from 1.935(4) to 2.058(4) Å were reported for uranyl(VI) complexes bridged to Fe(III) via a hydroxo group.16
 | ||
| Fig. 4 Ellipsoid plot at 50% probability of complex 6 (co-crystallised pyridine molecule and H were omitted for clarity, C atoms are represented in grey, O in red, N in blue, Fe in dark blue and U in green). Selected distances (Å) U(1)–O(1) 2.300(3), U(1)–O(2) 2.301(3), U(1)–O(3) 1.837(3), U(1)–O(4) 1.930(2), U(1)–N(1) 2.595(4), U(1)–N(2) 2.715(3), U(1)–N(3) 2.598(3), Fe(1)–N(4) 2.147(3), Fe(1)–O(5) 2.023(3), Fe(1)–O(4) 2.018(3). | ||
The 1H NMR spectrum of 6 in pyridine shows the presence of 12 signals over a broad range of chemical shifts (−30 to + 51 ppm). The large shift of the 1H NMR signals observed for 6 compared to complex 4 indicates that the UO2+⋯Fe2+ CCI is present in pyridine solution (Fig. S12†). The ESI/MS spectrum ({UO2(trensal)Fe(Py)+}: m/z = 859.83) of 6 also indicates the presence of the heterobimetallic complex in pyridine solution (Fig. S26†).
The stability and reactivity of 6 were then investigated and compared with those found for 3 and 4 in order to elucidate the effect of the Fe2+ ion. The addition of 1 eq. of PyHCl to a solution of 6 in pyridine results in the partial disproportionation of the uranyl(V) complex (Scheme 4) with a 2:1 ratio of 6 to the disproportionation product [UO2(trensal)Fe(py)3U(trensal)]Cl 7b (Fig. S20†). The addition of 2 equivalents of PyHCl to 6 led to the complete disappearance of the signals of complex 6 in the 1H NMR spectrum (Fig. S13†) and to an increased intensity of the signals assigned to 7b. The presence of the uranyl(VI) complex [UO2(Htrensal)] as the second disproportionation product was also identified by 1H NMR spectroscopy.
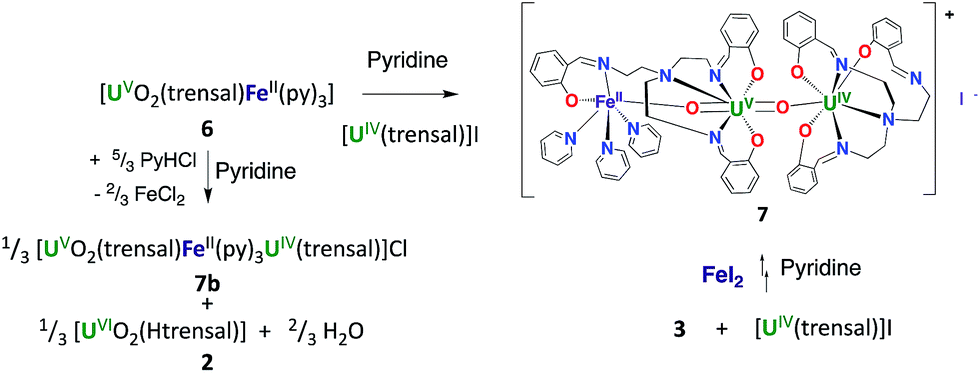 | ||
| Scheme 4 Addition of PyHCl to the complex 6 and synthesis of 7. | ||
However, in both cases the disproportionation was not complete. Notably, the trinuclear cation–cation complex 7b contains unreacted uranyl(V) (Scheme 4).
The formation of the [UO2(Htrensal)] by-product prevented the synthesis of 7b from the reaction of 6 with PyHCl (Scheme 4).
However, the iodide analogue [UO2(trensal)Fe(py)3U(trensal)]I, 7 was prepared in 80% yield from the reaction of complex 6 with 1 eq. of the [U(trensal)]I complex in pyridine (Scheme 4). This complex is a rare example of an actinide-functionalized uranyl complex and only the third example of a uranyl(V) complex presenting a CCI between the uranyl(V) oxo group and a U(IV) cation.9,13l
The structure of complex 7 (Fig. 5) shows the presence of a cationic trinuclear complex built via CCI between the U(IV) center from the [U(trensal)]+ complex and the oxo group of the uranyl(V) [UO2(trensal)Fe(py)3] fragment. The three metal ions adopt a close to linear arrangement with a Fe–O–U angle of 170.3(3)° and a U–O–U angle of 171.2(3)°. The UO bond distance for the uranyl(V) oxo group bound to the Fe2+ remains unchanged at 1.922(6) Å compared to complex 6, but a significant lengthening of the UO bond is observed upon binding of the U(IV) cation in 7 (1.960(6) Å). The UO2+⋯U(IV) distance (2.317(6) Å) is comparable to those found in the only two other complexes reported to have a UO2+⋯U(IV) CCI (2.198(13) and 2.245(3) Å).9,13l
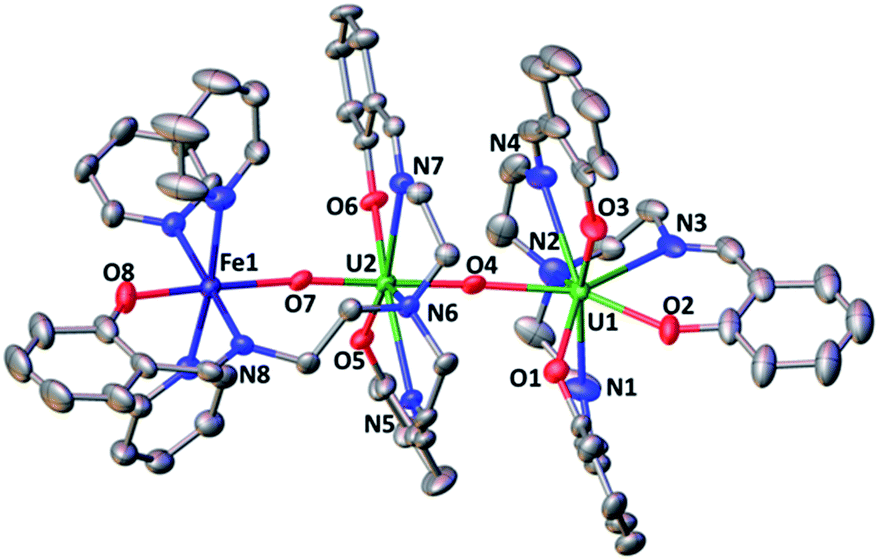 | ||
| Fig. 5 Ellipsoid plot at 50% probability of the cation [UO2(trensal)Fe(py)3U(trensal)]+ in 7 (co-crystallized pyridine molecule and H were omitted for clarity, C atoms are represented in grey, O in red, N in blue, Fe in dark blue and U in green). Selected distances (Å) U(1)–O(2) 2.216(6), U(1)–O(4) 2.317(6), U(1)–N(2) 2.713(8) U(2)–O(4) 1.960(6), U(2)–O(5) 2.267(6), U(2)–O(7) 1.922(6), U(2)–N(6) 2.653(7), Fe(1)–O(7) 2.144(6), Fe(1)–O(8) 2.033(6), Fe(1)–N(8) 2.159(7). | ||
The UO2+⋯Fe2+ distance (2.144(6) Å) is slightly longer than in 6 but is in the range of those found in the two previously reported complexes presenting a UO2+⋯Fe2+ interaction (1.946(4) Å–2.132(4) Å).14c,f
These results indicate that the presence of Fe2+ increases the stability of uranyl(V) in 6 with respect to proton induced disproportionation. Notably the addition of 2 eq. of PyHCl led to full disproportionation of the complexes 3 and 4 while it resulted only in the partial disproportionation of 6 and the formation of [UO2(Htrensal)] and of the Fe–U(V)–U(IV) trimer. The addition of five equivalents of pyridinium chloride is required for the full disproportionation of complex 6 to occur affording the same uranyl(VI) and U(IV) diproportionation products as observed after addition of acid to 3. This indicates that the iron bound uranyl(V) complex 6 displays an increased stability towards the proton induced disproportionation compared to the potassium bound uranyl(V) complexes 3 and 4 (Fig. S14†).
The binding of U(IV) to the uranyl(V) oxo group was previously reported to promote partial disproportionation and formation of multimetallic U(IV)–U(V) oxo-bridged complexes.9 The stabilizing effect of Fe2+ compared to U4+ can be explained in terms of the lower Lewis acidity of Fe2+ compared to U4+. This results in the formation of stable UO2+⋯Fe2+ adducts where the uranyl oxo group becomes less accessible to protonation by the Brønsted acid H+. In contrast, complex 7 is stable in pyridine solution over one month period. 1H NMR studies show that the addition of [U(trensal)]I to complex 3 also leads to the formation of a stable unidentified compound (Fig. S15†). The subsequent addition of FeI2 to this compound led to the formation of complex 7. These results suggest that stable U(IV)–U(V) complexes also form in the absence of iron bound to uranyl(V) oxo group. However, the formation of these compounds is not observed during the addition of PyHCl to 3, which undergoes complete disproportionation after the addition of 2 eq. of PyHCl. Moreover, the addition of 2 eq. of PyHCl to the U(IV)–U(V) adduct results in full disproportionation, as indicated by the 1H NMR spectrum, suggesting that the binding of U(IV) to the uranyl(V) oxo does not lead to increased stability (Fig. S15†). This further confirms the stabilizing role of Fe(II) binding with respect to proton induced disproportionation of uranyl(V).
The 1H NMR spectrum of 7 in pyridine shows the presence of 45 signals over a large range of chemical shifts (−35 to + 53 ppm) in agreement with the presence of the trimeric complex 7 in solution. (Fig. S16†). Additionally, the ESI/MS spectrum of 7 in pyridine solution {(UO2(trensal)Fe3U(trensal)+} m/z = 1474.42) indicated that the complex 7 retains its trinuclear structure in the pyridine solution (Fig. S27†).
The addition of 1 eq. of pyridinium chloride to 7 results in partial disproportionation with a 3:1 ratio of complex 7 to the disproportionation products as shown by 1H NMR spectroscopy (Fig. S17†). The complete disproportionation of complex 7 requires the addition of 4 eq. of PyHCl. The coordination of U(IV) does not increase the stability of the uranyl(V) species in 7 with respect to 6.
In view of the increased stability of 7 and 6 compared to 3 towards proton induced disproportionation, we set out to investigate how the coordination of a second Fe2+ cation to complex 6 would affect the structure and reactivity of the U(V) centre.
The 1H NMR of the reaction mixture resulting from the addition of 0.5 equivalents of iron(II) iodide to 6 in pyridine indicated the formation of a new species (Fig. S18†). X-ray quality crystals of [(UO2(trensal)Fe(py)3)2Fe(py)3]I2, 8, were obtained in 65% yield from this reaction (Scheme 5). The solid-state structure (Fig. 6) of 8 shows the presence of a pentametallic structure where a Fe(Py)3 moiety bridges two iron-bound uranyl(V) [UO2(trensal)Fe(py)3] moieties. Overall, this results in the presence of UO2+⋯Fe2+ CCIs for both uranyl(V) oxo-groups. The central Fe2+ cation is penta-coordinated by one oxo atom from each of the two uranyl(V) groups and three pyridine molecules. The mean Fe(2)–O(oxo) bond lengths is 1.988 Å. The [UO2(trensal)Fe(py)3] moieties of the crystal structure possess the same geometry found in the mononuclear complex 6, but the additional uranyl–iron interaction results in a slight lengthening of the UO2+⋯Fe2+ bonds compared to 6 (2.061(4) Å vs. 2.018(3) Å). The arrangement of the 5 metal ions is not linear, with a 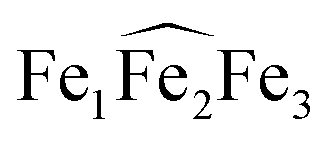 angle of 136°.
angle of 136°.
 | ||
| Scheme 5 Synthesis of [(UO2(trensal)Fe(py)3)2Fe(py)3]I2, 8. | ||
 | ||
| Fig. 6 Molecular structure of the dication [{UO2(trensal)Fe(py)3} 2Fe(py)3]2+ in 8 (H atoms were omitted for clarity, C atoms are represented in grey, O in red, Fe in dark blue, N in blue, I in purple and U in green). Selected distances U1–O2 = 1.920 (4) Å, U1–O5 = 1.935 (4) Å, U2–O6 = 1.927 (4) Å and U2–O9 = 1.927 (5) Å. | ||
The ESI/MS spectrum of 8 (Fig. S28†) did not show the presence of a pentanuclear architecture in pyridine solution but showed only the peaks corresponding to the trinuclear {Fe–U–Fe} and dinuclear {Fe–U} complexes. The addition of 1 eq. of the complex [UO2(trensal)Fe(py)3] to a solution of 8 in pyridine resulted only in a slight broadening of the 1H NMR signals of 8 suggesting the presence in solution of a fast exchange between the [UO2(trensal)Fe(py)3] moiety and 8 (Fig. S19†).
The labile binding of the central Fe(Py)32+cation in 8 does not lead to an increased stability of 8 towards proton induced disproportionation compared to 6. Notably, the 1H NMR indicated a 2:1 ratio between the starting complex 8 and the disproportionation products upon addition of 1 eq. of H+ per uranyl(V) which is identical to the ratio observed for the complex 6 (Fig. S20†).
Redox reactivity
Iron binding to the uranyl(V) oxo is anticipated to have an important effect on its redox reactivity. Moreover, it has been suggested that iron binding at mica surfaces leads to the stabilization of uranyl(V) intermediates but the effect of iron binding on the redox properties of isolated uranyl(V) complexes has not been investigated.At first, we explored the chemical oxidation of uranyl(V) by Fe3+. The reaction of 3 with 1 eq. FeCl3 leads to the oxidation of the uranium center (Fig. S21†) and to the formation of the uranyl(VI)–Fe(II) complex 2 [(UO2(trensal))2Fe] as identified by X-ray diffraction crystallography and 1H NMR spectroscopy. The oxidation of uranyl(V) complex to uranyl(VI) by Fe(III) is explained in terms of the respective redox potential (Fe(III)/Fe(II) = 0.0 V; UO22+/UO2+ = −1.6 vs. V (Fc/Fc+)).
In order to probe the possibility of obtaining a uranyl(V)–Fe(III) complex we explored the reactivity of 3 and 6 with increasingly electron-rich FeLCln complexes (L = tpa and tdmba); tpa = (tris(pyridin-2-ylmethyl)amine) and H3tdmba = (tris-(2-hydroxy-3,5-dimethylbenzyl)amine). The reaction of 6 with [Fe(tpa)Cl3] led to the oxidation of uranyl(V) to uranyl(VI) with concomitant formation of [Fe(tpa)Cl2] (as shown by X-ray diffraction studies and 1H NMR spectroscopy). The reaction of 6 with the neutral Fe(III) complex [Fe(tdmba)] did not result in any change observable in the 1H NMR spectrum of 6 (Fig. S23†) indicating that the Fe(III) cation in [Fe(tdmba)] does not form CCIs with the uranyl(V) oxo group but does not oxidize the uranyl(V) either. In contrast, when complex 3 is reacted with [Fe(tdmba)], 1H NMR spectroscopy indicated that a redox reaction occurs yielding uranyl(VI) and Fe(II) species (Fig. S24†). These results are in agreement with the reported influence of chelating agents on the reoxidation by Fe(III) of biogenic products of uranyl(VI) reduction.17
These results suggest that the presence of UO2+⋯Fe2+ CCI stabilizes the uranyl(V) oxidation state with respect to the oxidation. In order to further probe the effect of iron binding on the redox properties of uranyl(V) species we performed comparative cyclic voltammetry studies of complexes 1, 2, 5, 6 and 8 (Fig. 6 and SCV1–SCV4†). The voltammogram of 1 in pyridine (Fig SCV1†) shows an irreversible redox event at −1.75 V, but when the voltammogram of 1 is measured in the presence of the cryptand a reversible redox event assigned to the U(VI)/U(V) couple is observed at E1/2 = −1.69 V vs. Fc/Fc+ (Fig. 7, green curve). The voltammogram of the protonated uranyl(V) complex 2 also shows the presence of a reversible redox event at E1/2 = −1.66 V vs. Fc/Fc+ assigned to the U(VI)/U(V) couple. These values compare well with the values previously measured in pyridine for other uranyl(V) complexes of tetradentate (E1/2 = −1.61 V or −1.67 V vs. Fc/Fc+)15a,13e and pentadentate Schiff bases (E1/2 = −1.58 V vs. Fc/Fc+).18 A second irreversible redox event is observed at E1/2 = −2.47 V vs. Fc/Fc+ for complex 2, but not for complex 1. This event is consistent with the reduction of the metal centre (values of redox potential ranging from −2.02 to −2.88 V vs. Fc/Fc+ were previously assigned to the U(V)/U(IV) couple12a). The possibility that this event could be related to the reduction of the Schiff base ligand is unlikely since this feature is absent from the voltammograms of the H3trensal, K3trensal ligands and of the complex 1. Moreover, the shift of the U(V)/U(IV) couple to a more positive potential in complex 2 could be explained by the presence of a proton on the complex.
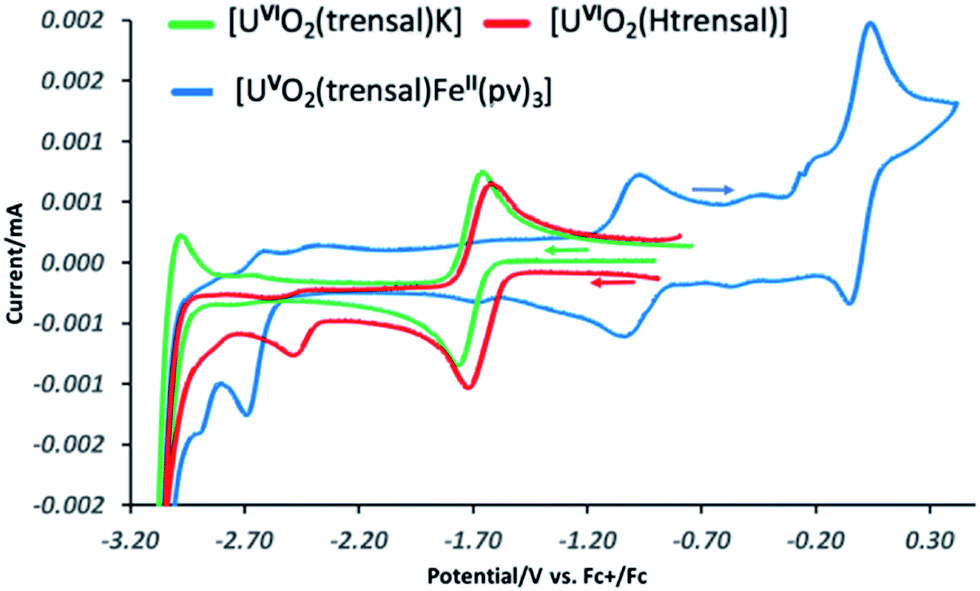 | ||
| Fig. 7 Room temperature cyclic voltammograms of 4 mM pyridine solutions of [UVIO2(trensal)K] 1 in the presence of 1 eq. of cryptand (green), of [UVIO2(Htrensal)] 2 (red) and of [UVO2(trensal)FeII(py)3] 6 (blue) recorded in 0.1 M [Bu4N][PF6] at 100 mV s−1 scan rate, Cp2Fe/Cp2Fe+ (Fc/Fc+) corrected. | ||
Similar redox events are observed in the voltammogram of complex 6 in addition to the quasi-reversible wave at E1/2 = 0.0 V vs. Fc/Fc+, assigned to the Fe(III)/Fe(II) couple. However, the U(VI)/U(V) reduction process is found at −1.03 V vs. Fc/Fc+ in the voltammogram of 6 and the second reduction event occurs at E1/2 = −2.7 V vs. Fc/Fc+ demonstrating that the range of stability of the uranyl(V) species is significantly extended compared to complex 2 as a result of Fe(II) binding. Both reduction and oxidation of the uranyl(V) cation are more difficult in the presence of Fe(II). No additional redox stabilisation was observed upon addition of two or more equivalents of Fe(II) to complex 6 as indicated by the voltammogram of complex 8 (Fig. SCV4†). This is probably due to the labile binding of the second Fe(II) cation to the uranyl(V) oxo group in pyridine.
Moreover, in the voltammogram of complex 5 (Fig. SCV2†) the redox event assigned to the U(VI)/U(V) couple is found at E1/2 = −1.66 V vs. Fc/Fc+ as in complexes 1 and 2 in spite of the presence of a Fe(II) ion bound through the Schiff base acting as a bridging ligand. These results indicate that cation–cation interaction between the uranyl(V) oxygen and the Fe2+ is essential for the stabilization of U(V) while the presence of a Fe(II) bound through the ligand has no significant effect on the redox properties of uranyl(V).
Conclusions
In conclusion the tripodal heptadentate Schiff base trensal3− ligand allowed the synthesis and characterization of uranyl(V) complexes presenting UO2+⋯K+ or UO2+⋯Fe2+ cation–cation interactions. The reported uranyl(V) complexes show similar stability in pyridine solution, but the presence of Fe2+ bound to the uranyl(V) oxygen leads to increased stability with respect to proton induced disproportionation. A stable Fe2+–UO2+–U4+ intermediate (7b) containing both UO2+⋯Fe2+ and UO2+⋯U4+ cation–cation interactions formed upon addition of 2 eq. of PyHCl to the iron bound uranyl(V) complex (6). In contrast, the addition of 2 eq. of PyHCl to the potassium bound uranyl(V) complexes (3 and 4) resulted in the immediate formation of U(IV) and UO22+ complexes. The UO2+⋯Fe2+ (6) complex reacts with an additional Fe2+ cation leading to the formation of a pentanuclear Fe2+–UO2+–Fe2+–UO2+–Fe2+ complex (8) but the additional Fe2+–UO2+ cation–cation interactions do not lead to increased stability. Redox reactivity and cyclic voltammetry studies also show an increased range of stability of the uranyl(V) species in the presence of Fe2+ with respect both to oxidation and reduction reactions, while the presence of a proton in the uranyl(VI) complex (2) results in a smaller stability range for the uranyl(V) species. Cyclic voltammetry studies also show that the presence of a Fe2+ cation bound only through one trensal3− arm in the trinuclear complex [{UO2(trensal)}2Fe], 5 does not lead to increased redox stability of the uranyl(V) demonstrating the important role of UO2+⋯Fe2+ cation–cation interactions in increasing the stability of uranyl(V). These results provide an important insight into the role that iron binding may play in stabilizing uranyl(V) species in the environmental mineral-mediated reduction of uranium(VI).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr F. Fadaei Tirani for her contribution to the X-ray single crystal structure data collection and analyses. We thank Dr Euro Solari for elemental analysis, Dr A. Sienkiewicz and L. Barluzzi for EPR data collection and L. Chatelain for preliminary experiments. We thank Prof. R. Bernier-Latmani for useful discussions. This work was supported by the Ecole Polytechnique Fédérale de Lausanne (EPFL) and by the Swiss National Science Foundation grant (number CR23I2_16645).Notes and references
- (a) P. L. Arnold, J. B. Love and D. Patel, Coord. Chem. Rev., 2009, 253, 1973–1978 CrossRef; (b) L. S. Natrajan, A. N. Swinburne, M. B. Andrews, S. Randall and S. L. Heath, Coord. Chem. Rev., 2014, 266, 171–193 CrossRef; (c) H. Steele and R. J. Taylor, Inorg. Chem., 2007, 46, 6311–6318 CrossRef PubMed; (d) J. C. Renshaw, L. J. C. Butchins, F. R. Livens, I. May, J. M. Charnock and J. R. Lloyd, Environ. Sci. Technol., 2005, 39, 5657–5660 CrossRef PubMed; (e) E. S. Ilton, A. Haiduc, C. L. Cahill and A. R. Felmy, Inorg. Chem., 2005, 44, 2986–2988 CrossRef PubMed.
- (a) K. Yuan, E. S. Ilton, M. R. Antonio, Z. R. Li, P. J. Cook and U. Becker, Environ. Sci. Technol., 2015, 49, 6206–6213 CrossRef PubMed; (b) E. S. Ilton, J.-F. Boily, E. C. Buck, F. N. Skomurski, K. M. Rosso, C. L. Cahill, J. R. Bargar and A. R. Felmy, Environ. Sci. Technol., 2010, 44, 170–176 CrossRef PubMed; (c) F. N. Skomurski, E. S. Ilton, M. H. Engelhard, B. W. Arey and K. M. Rosso, Geochim. Cosmochim. Acta, 2011, 75, 7277–7290 CrossRef.
- I. Pidchenko, K. O. Kvashnina, T. Yokosawa, N. Finck, S. Bahl, D. Schild, R. Polly, E. Bohnert, A. Rossberg, J. Gottlicher, K. Dardenne, J. Rothe, T. Schafer, H. Geckeis and T. Vitova, Environ. Sci. Technol., 2017, 51, 2217–2225 CrossRef PubMed.
- (a) E. S. Ilton, J. S. L. Pacheco, J. R. Bargar, Z. Shi, J. Liu, L. Kovarik, M. H. Engelhard and A. R. Felmy, Environ. Sci. Technol., 2012, 46, 9428–9436 CrossRef PubMed; (b) H. E. Roberts, K. Morris, G. T. W. Law, J. F. W. Mosselmans, P. Bots, K. Kvashnina and S. Shaw, Environ. Sci. Technol. Lett., 2017, 4, 421–426 CrossRef.
- L. R. Morss, N. M. Edelstein and J. Fuger, The Chemistry of the Actinide and Transactinide Elements, Springer, Dordrecht, 2006 Search PubMed.
- N. N. Krot and M. S. Grigoriev, Russ. Chem. Rev., 2004, 73, 89–100 CrossRef.
- (a) M. Sundararajan, A. J. Campbell and I. H. Hillier, J. Phys. Chem. A, 2008, 112, 4451–4457 CrossRef PubMed; (b) V. Mougel, B. Biswas, J. Pecaut and M. Mazzanti, Chem. Commun., 2010, 46, 8648–8650 RSC.
- (a) M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137–1198 CrossRef PubMed; (b) V. Mougel, P. Horeglad, G. Nocton, J. Pecaut and M. Mazzanti, Chem.–Eur. J., 2010, 16, 14365–14377 CrossRef PubMed; (c) L. Chatelain, V. Mougel, J. Pecaut and M. Mazzanti, Chem. Sci., 2012, 3, 1075–1079 RSC; (d) V. Mougel, P. Horeglad, G. Nocton, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2009, 48, 8477–8480 CrossRef PubMed; (e) G. Nocton, P. Horeglad, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2008, 130, 16633–16645 CrossRef PubMed; (f) F. Burdet, J. Pecaut and M. Mazzanti, J. Am. Chem. Soc., 2006, 128, 16512–16513 CrossRef PubMed.
- V. Mougel, J. Pecaut and M. Mazzanti, Chem. Commun., 2012, 48, 868–870 RSC.
- (a) P. L. Arnold, N. A. Potter, N. Magnani, C. Apostolidis, J. C. Griveau, E. Colineau, A. Morgenstern, R. Caciuffo and J. B. Love, Inorg. Chem., 2010, 49, 5341–5343 CrossRef PubMed; (b) J. L. Brown, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 7248–7249 CrossRef PubMed; (c) D. D. Schnaars, G. Wu and T. W. Hayton, Inorg. Chem., 2011, 50, 4695–4697 CrossRef PubMed; (d) T. W. Hayton and G. Wu, Inorg. Chem., 2009, 48, 3065–3072 CrossRef PubMed.
- D. D. Schnaars, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2009, 131, 17532–17533 CrossRef PubMed.
- (a) J. R. Pankhurst, N. L. Bell, M. Zegke, L. N. Platts, C. A. Lamfsus, L. Maron, L. S. Natrajan, S. Sproules, P. L. Arnold and J. B. Love, Chem. Sci., 2017, 8, 108–116 RSC; (b) N. L. Bell, B. Shaw, P. L. Arnold and J. B. Love, J. Am. Chem. Soc., 2018, 140, 3378–3384 CrossRef PubMed; (c) J. J. Kiernicki, M. Zeller and S. C. Bart, Angew. Chem., Int. Ed. Engl., 2017, 56, 1097–1100 CrossRef PubMed.
- (a) L. Natrajan, F. Burdet, J. Pecaut and M. Mazzanti, J. Am. Chem. Soc., 2006, 128, 7152–7153 CrossRef PubMed; (b) J. C. Berthet, G. Siffredi, P. Thuery and M. Ephritikhine, J. Chem. Soc., Dalton Trans., 2009, 3478–3494 RSC; (c) K. Takao, S. Tsushima, S. Takao, A. C. Scheinost, G. Bernhard, Y. Ikeda and C. Hennig, Inorg. Chem., 2009, 48, 9602–9604 CrossRef PubMed; (d) P. Horeglad, G. Nocton, Y. Filinchuk, J. Pecaut and M. Mazzanti, Chem. Commun., 2009, 1843–1845 RSC; (e) G. Nocton, P. Horeglad, V. Vetere, J. Pecaut, L. Dubois, P. Maldivi, N. M. Edelstein and M. Mazzanti, J. Am. Chem. Soc., 2010, 132, 495–508 CrossRef PubMed; (f) T. W. Hayton and G. Wu, J. Am. Chem. Soc., 2008, 130, 2005–2014 CrossRef PubMed; (g) T. W. Hayton and G. Wu, Inorg. Chem., 2008, 47, 7415–7423 CrossRef PubMed; (h) P. L. Arnold, B. E. Cowie, M. Suvova, M. Zegke, N. Magnani, E. Colineau, J. C. Griveau, R. Caciuffo and J. B. Love, Angew. Chem., Int. Ed. Engl., 2017, 56, 10775–10779 CrossRef PubMed; (i) P. L. Arnold, E. Hollis, G. S. Nichol, J. B. Love, J. C. Griveau, R. Caciuffo, N. Magnani, L. Maron, L. Castro, A. Yahia, S. O. Odoh and G. Schreckenbach, J. Am. Chem. Soc., 2013, 135, 3841–3854 CrossRef PubMed; (j) P. L. Arnold, A. F. Pecharman, E. Hollis, A. Yahia, L. Maron, S. Parsons and J. B. Love, Nat. Chem., 2010, 2, 1056–1061 CrossRef PubMed; (k) P. L. Arnold, A.-F. Pecharman, R. M. Lord, G. M. Jones, E. Hollis, G. S. Nichol, L. Maron, J. Fang, T. Davin and J. B. Love, Inorg. Chem., 2015, 54, 3702–3710 CrossRef PubMed; (l) P. L. Arnold, M. S. Dutkiewicz, M. Zegke, O. Walter, C. Apostolidis, E. Hollis, A. F. Pecharman, N. Magnani, J. C. Griveau, E. Colineau, R. Caciuffo, X. B. Zhang, G. Schreckenbach and J. B. Love, Angew. Chem., Int. Ed. Engl., 2016, 55, 12797–12801 CrossRef PubMed.
- (a) V. Mougel, L. Chatelain, J. Pecaut, R. Caciuffo, E. Colineau, J. C. Griveau and M. Mazzanti, Nat. Chem., 2012, 4, 1011–1017 CrossRef PubMed; (b) L. Chatelain, J. P. S. Walsh, J. Pecaut, F. Tuna and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2014, 53, 13434–13438 CrossRef PubMed; (c) P. L. Arnold, D. Patel, C. Wilson and J. B. Love, Nature, 2008, 451, 315–318 CrossRef PubMed; (d) L. Chatelain, F. Tuna, J. Pecaut and M. Mazzanti, J. Chem. Soc., Dalton Trans., 2017, 46, 5498–5502 RSC; (e) L. Chatelain, F. Tuna, J. Pecaut and M. Mazzanti, Chem. Commun., 2015, 51, 11309–11312 RSC; (f) L. Chatelain, J. Pecaut, F. Tuna and M. Mazzanti, Chem.–Eur. J., 2015, 21, 18038–18042 CrossRef PubMed; (g) V. Mougel, L. Chatelain, J. Hermle, R. Caciuffo, E. Colineau, F. Tuna, N. Magnani, A. de Geyer, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2014, 53, 819–823 CrossRef PubMed.
- (a) C. Camp, L. Chatelain, V. Mougel, J. Pecaut and M. Mazzanti, Inorg. Chem., 2015, 54, 5774–5783 CrossRef PubMed; (b) K. Takao and Y. Ikeda, Inorg. Chem., 2007, 46, 1550–1562 CrossRef PubMed.
- M. Basile, D. K. Unruh, E. Flores, A. Johns and T. Z. Forbes, J. Chem. Soc., Dalton Trans., 2015, 44, 2597–2605 RSC.
- B. D. Stewart, C. Girardot, N. Spycher, R. K. Sani and B. M. Peyton, Environ. Sci. Technol., 2013, 47, 364–371 CrossRef PubMed.
- K. Takao, M. Kato, S. Takao, A. Nagasawa, G. Bernhard, C. Hennig and Y. Ikeda, Inorg. Chem., 2010, 49, 2349–2359 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, NMR, ESI/MS and EPR spectra, cyclic voltammograms and detailed X-ray crystallographic data in CIF format. CCDC 1842376–1842382. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02099j |
| This journal is © The Royal Society of Chemistry 2018 |