
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic Barbier reaction – visible-light induced allylation and benzylation of aldehydes and ketones†
Anna Lucia
Berger
,
Karsten
Donabauer
and
Burkhard
König
 *
*
Institut für Organische Chemie, Universität Regensburg, Universitätsstrasse 31, 93053 Regensburg, Germany. E-mail: Burkhard.Koenig@chemie.uni-regensburg.de; Fax: +49-941-943-1717; Tel: +49-941-943-4575
First published on 2nd August 2018
Abstract
We report a photocatalytic version of the Barbier type reaction using readily available allyl or benzyl bromides and aromatic aldehydes or ketones as starting materials to generate allylic or benzylic alcohols. The reaction proceeds at room temperature under visible light irradiation with the organic dye 3,7-di(4-biphenyl)1-naphthalene-10-phenoxazine as a photocatalyst and DIPEA as sacrificial electron donor. The proposed cross-coupling mechanism of a ketyl- and an allyl or benzyl radical is supported by spectroscopic investigations and cyclic voltammetry measurements.
Introduction
Although first reported over a century ago, Barbier-type reactions are still important tools for carbon–carbon bond formations in organic synthesis today.1 In the classical Barbier reaction a metal, e.g. zinc2 or magnesium3 is able to insert in the carbon–halide bond of a reactive organic halide to form a nucleophilic organometallic intermediate 4 which can undergo a reaction with various electrophiles, like aldehydes or ketones to form the corresponding secondary or tertiary alcohols as products (Scheme 1a). One of the main application of Barbier reactions is the synthesis of allylic or benzylic alcohols from an aldehyde or ketone and allyl or benzyl bromide using a metal as reductant.4 Over the years, Barbier-type reactions have been developed further, and today they are known for many different substrates5 and with various metals e.g. tin,6 indium,7 praseodymium8 or manganese.9 While these methods offer a wide variety of reaction conditions, they all are overall two-electron processes which is why they require the use of a stoichiometric amount of metal as a reductant. Using a photoredox catalyst to access an organic electron source instead of a metal would represent an interesting and more environmentally benign alternative. However, photoredox catalyzed two-electron processes are scarce, as photocatalytic reactions usually proceed via radical intermediates that are generated by a photoinduced single electron transfer (SET).10 To generate carbanion synthons with similar reactivity as the nucleophilic organometallic intermediate in classical Barbier-type reactions, two consecutive SETs would be required to generate a radical first followed by another reduction to the corresponding carbanion.11 Due to the high reactivity of most radicals and their low concentration in photocatalytic reactions this process is rather unlikely. Another strategy to enable photocatalytic two-electron processes would be a reductive radical–radical cross coupling where one electron is transferred to each starting material, generating two radical intermediates that can recombine to give the desired product (Scheme 1b).12 Photocatalytic reductions of aromatic aldehydes to the corresponding alcohols have been known since a report by Pac et al. in 1983 (ref. 13) and in 1990 the formation of diols as homocoupling products of ketyl radical anions has been observed.14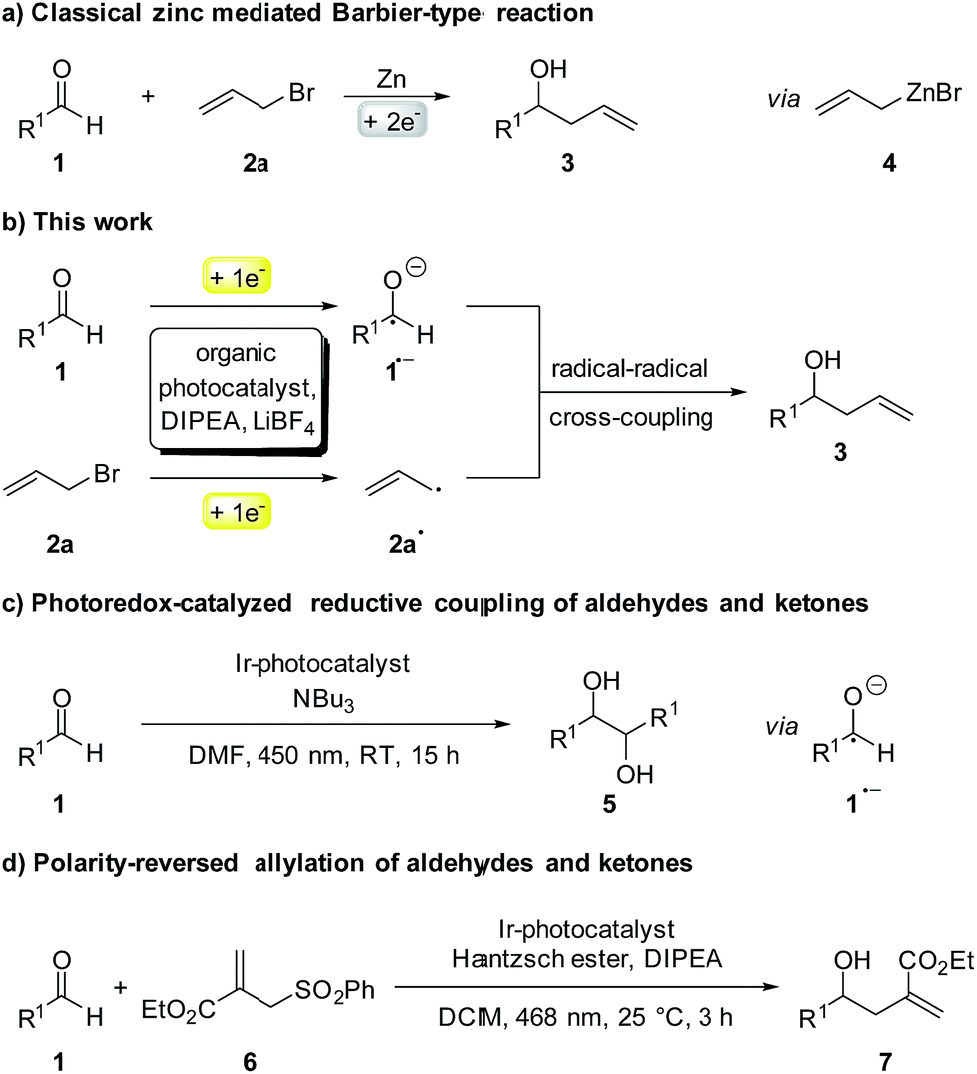 | ||
| Scheme 1 (a) Classical and (b) photocatalytic version of the Barbier-type reaction; (c and d) other photocatalytic reactions with ketyl radicals. | ||
After having been used only rarely in photoredox catalysis for many years, there has been an increasing number of reports about photocatalytic reductions of aldehydes and ketones recently. Ketyl radicals have often been used for radical–radical coupling reactions,12,15e.g. in the work of Rueping and co-workers about a photoredox-catalyzed reductive dimerization of aldehydes and ketones (Scheme 1c)15b or in the reductive arylation of carbonyl derivatives by Xia et al. in 2017.12 Apart from radical–radical coupling reactions, it is also possible to use ketyl radicals for cyclization reactions16 or to trap them intermolecularly with alkenes.17 In the work of Chen and co-workers, hydroxymethyl radicals derived from the photocatalytic reduction of aldehydes or ketones are added to allyl sulfones (Scheme 1d) to form the corresponding homoallylic alcohols as products.17a While this is an elegant method for the photocatalytic allylation of aldehydes and ketones, it is only possible for allyl sulfones with electron withdrawing CO2Et-groups. A photochemical method for the allylation and benzylation of ketones and 1,2-diketones using organotrifluoroborate has been reported in 2009 by Nishigaichi et. al.18 We developed a method for the direct photocatalytic synthesis of allylic and benzylic alcohols from ketones or aldehydes and allyl or benzyl bromides with an organic photocatalyst via a reductive radical–radical cross coupling.
Results and discussion
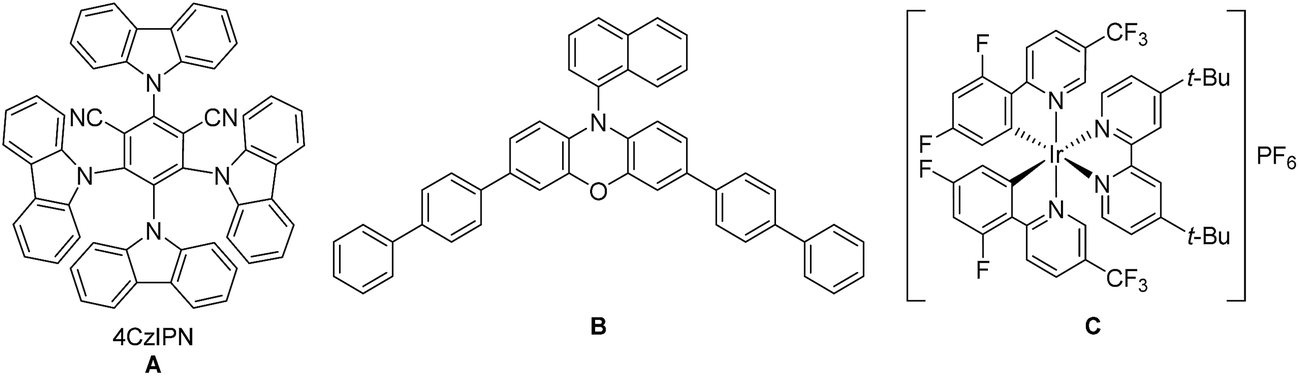
For the optimization of the reaction conditions, we used the readily available substrates benzaldehyde (1a) and allyl bromide (2a) as starting materials. Initial experiments have shown that we could obtain 22% of the desired product 3a when the reaction was performed in dry DMF with 4CzIPN (A) as a photocatalyst and DIPEA as sacrificial electron donor (Table 1, entry 1).|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | PC (mol%, hν, [nm]) | Solvent | DIPEA (eq.) | Additive (eq.) | t [h] | Yield 3ab [%] | Yield 5ac [%] | Yield 8ad [%] |
| a The reactions were performed using 1 eq. (0.2 mmol) 1a and 2 eq. (0.4 mmol) 2a in 2 mL degassed solvent under nitrogen. b Yields were determined by GC analysis with 1-naphthol as an internal standard. c Yields were determined by crude NMR with 1,3,5-trimethoxybenzene as an internal standard. d Yields were determined by GC analysis with 1-naphthol as an internal standard. | ||||||||
| 1 | A (5, 455) | DMF (dry) | 6 | — | 18 | 22 | 15 | 31 |
| 2 | B (5, 455) | DMF (dry) | 6 | — | 18 | 38 | 8 | 17 |
| 3 | B (5, 400) | DMA | 6 | — | 18 | 54 | 14 | 23 |
| 4 | C (2, 455) | DMA | 6 | — | 18 | 21 | 19 | 43 |
| 5 | B (5, 400) | DMA | 6 | LiBF4 (1.5) | 18 | 64 | 13 | 23 |
| 6 | B (5, 400) | DMA | 6 | LiBF4 (1.5) | 2 | 59 | 12 | 28 |
| 7 | B (5, 400) | DMA | — | LiBF4 (1.5) | 18 | 0 | 0 | 0 |
| 8 | B (5, dark) | DMA | 6 | LiBF4 (1.5) | 18 | 0 | 0 | 0 |
| 9 | — (400) | DMA | 6 | LiBF4 (1.5) | 15 | 46 | 3 | 15 |
| 10 | — (400) | DMA | 6 | LiBF4 (1.5) | 4 | 3 | 2 | 6 |
| 11 | — (455) | DMA | 6 | LiBF4 (1.5) | 15 | 0 | 0 | 0 |
|
||||||||
By using 3,7-di(4-biphenyl) 1-naphthalene-10-phenoxazine (B) as a photocatalyst19 the yield could be increased to 38% (Table 1 – entry 2) and by changing the irradiation wavelength from 455 to 400 nm and the solvent from DMF to DMA a yield of 54% could be obtained (Table 1 – entry 3). With the iridium-based photocatalyst C only 21% of 3a was formed (Table 1 – entry 4). By adding 1.5 equivalents of LiBF4 to the reaction mixture the formation of the diol homocoupling product of 1a could be suppressed, which further increased the yield to 64% (Table 1 – entry 5).15a Reducing the reaction time from 18 to 2 hours only slightly decreased the yield (Table 1 – entry 6).
While light and DIPEA are necessary for product formation (Table 1 – entries 7 and 8), the reaction also works in moderate yields without photocatalyst at 400 nm (46%, Table 1 – entry 9). However, the presence of the photocatalyst significantly accelerates the reaction as we already obtain complete conversion after 3 hours with 5 mol% of B, while only traces of 3a were formed after the same time without photocatalyst (Table 1 – entry 10). When the reaction was performed at 455 nm without B no product formation could be observed (Table 1 – entry 11). As shown in Table 1, varying amounts of the homocoupling products 5a and 8a are formed under all tested reaction conditions. Due to the use of an excess of allyl bromide (2a), the generation of 8a has little influence on the yield of the reaction. In contrast, the formation of the diol homocoupling product 5a decreases the yield of the desired product significantly, as two equivalents of the stoichiometric reagent 1a are required to form one equivalent of 5a.
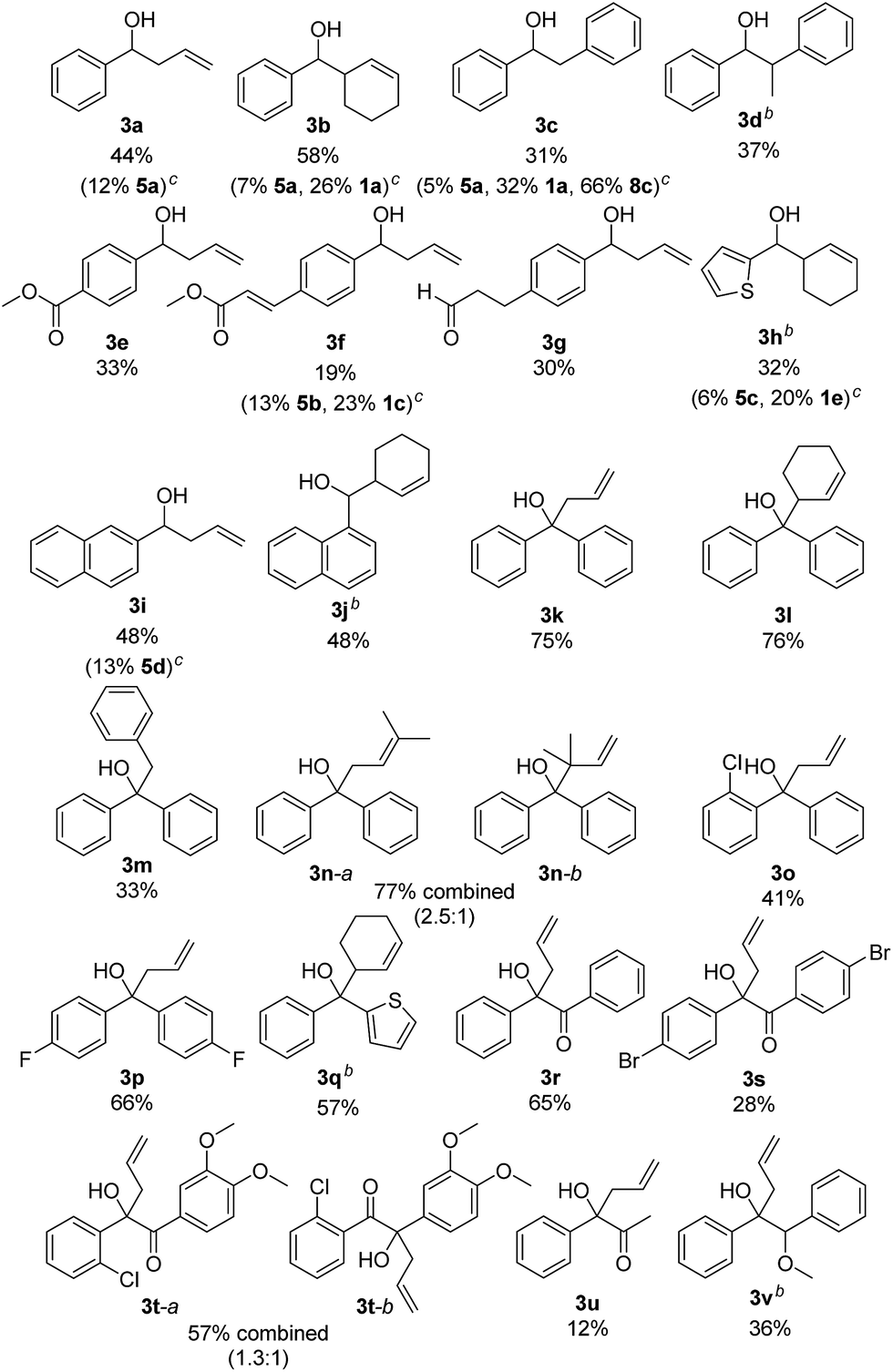
The scope of the reaction was investigated using the optimized reaction conditions (Table 2). Apart from allyl bromide (2a) the reaction also worked in moderate to good yields with 3-bromo cyclohexene (2b), benzyl bromide (2c) (1-bromoethyl)benzene (2d). When 3,3-dimethylallyl bromide (2e) was used, a mixture of product 3n-a and 3n-b was obtained, with 3n-a being the main product. Using alkyl or phenyl bromides did not lead to any product formation, probably because the radicals formed upon reduction and debromination are too unstable to undergo the coupling reaction. Aromatic aldehydes containing ester groups (3e, 3f), or aliphatic aldehydes (3g) were also tolerated in the reaction with moderate yields. Notably, the reaction selectively takes place at the carbonyl group in benzylic position, while other carbonyl groups in the molecule remain unchanged. Apart from benzaldehydes which gave moderate yields (3a–3g) the reaction also works well with 1- and 2-naphthaldehyde (3i, 3j) and with the heterocyclic 2-thiophenecarboxaldehyde (3h). Good yields were obtained when benzophenone was used (3k–3n) and 1,2-diketones (3r–3u) are also viable substrates. Using a non-symmetric diketone with an electron rich and an electron poor arene gave a mixture of product 3t-a and 3t-b with only a slight preference of the less electron rich position (3t-a). Product 3u shows an important advantage over the classical Barbier reaction, as the reaction selectively takes place at the sterically more hindered ketone next to the aromatic system. Halogen substituted substrates (3o, 3p, 3s, 3t) and substrates containing a methoxy group (3v) were also tolerated. Alkyl aldehydes and ketones did not yield any product as they have significantly lower reduction potentials and can therefore not be reduced by B (Ered1/2(benzophenone 1e) = −1.83 V vs. SCE,20 compared to Ered1/2(cyclohexanone 2m) = −2.79 V vs. SCE15a). Additionally, an aromatic system in α-position to the carbonyl group seems to be required, probably due to the enhanced stability of the ketyl radical.
|
|
|---|
a The reactions were performed using 1 eq. (0.2 mmol) 1 and 2 eq. (0.4 mmol) 2 in 2 mL degassed DMA under nitrogen, all yields are of the isolated products.
b A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the syn- and anti-product was obtained.
c Yields of the side products were determined by crude NMR with 1,3,5-trimethoxybenzene as an internal standard. :1 mixture of the syn- and anti-product was obtained.
c Yields of the side products were determined by crude NMR with 1,3,5-trimethoxybenzene as an internal standard.
|
|
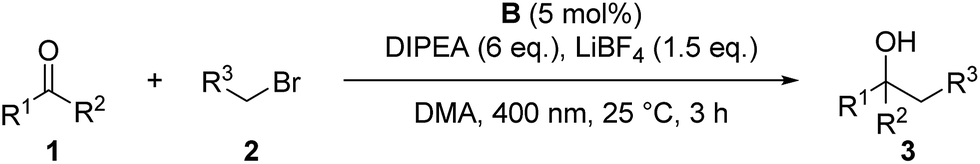
As moderate yields are obtained in many cases, the side products of the reaction were determined for selected examples (3a–3c, 3f, 3h, 3i). The diol homocoupling products 5 were observed in all examples. In some cases, remaining starting material was observed (3b, 3c, 3f, 3h) which indicates an incomplete reaction. While the homocoupling of 2 did not have any influence on the yield in most cases, it seems to have significant effect when benzyl bromide (2c) was used. This can be seen in the case of 3c, where 66% of the homocoupling product 8a was formed.
Control reactions have shown that under the reaction conditions the homocoupling products of benzaldehyde (5a) and allyl or benzyl bromide (8) could be observed (Scheme 2a). This confirms that the ketyl- (1a˙−) as well as the allyl- (2a˙) or benzyl radical (2c˙) are present in the reaction mixture and lead to product formation via a radical–radical cross-coupling reaction. Notably, the homocoupling products of 2a and 2c are formed also without photocatalyst just by irradiating a mixture of the bromide 2 and DIPEA with 400 nm while the photocatalyst is required for the formation of the diol 5a from benzaldehyde. However, DIPEA and 400 nm light are both crucial for the formation of allyl radicals, as irradiating only 2a at 400 nm as well as stirring a mixture of 2a and DIPEA in the dark or at 455 nm did not lead to the formation of homocoupling product 8a. It was also possible to trap the allyl radical, which was formed upon irradiation with 1,1-diphenylethylene (9) yielding product 10 (Scheme 2b).
 | ||
| Scheme 2 Control reactions for radical–radical cross coupling. | ||
Stern–Volmer fluorescence quenching experiments of photocatalyst B show that the excited state of B is quenched efficiently by benzaldehyde 1a, but not by allyl bromide (2a) or DIPEA (Fig. 1). These results are in accord with the prior observations, as they show that radical 1a˙− is generated by a SET from B to 1a while the allyl radical (2a˙) is formed without photocatalyst. According to cyclic voltammetry, benzaldehyde has a reduction potential of −2.0 V vs. SCE in DMF and should therefore not be in the range of photocatalyst B (E0* = −1.80 V vs. SCE).19b,19c However, it is known that the potential of aldehydes and ketones can be lowered by activating the carbonyl group with Lewis acids15d or with the oxidized form of the tertiary amine (DIPEA˙+).15b Indeed, CV-measurements show, that the signal for the reduction of 1a is clearly shifted to lower potentials upon addition of DIPEA and LiBF4 (Fig. 2). This effect could only be observed when both additives were present in the reaction mixture, which explains the role of LiBF4 in the reaction.
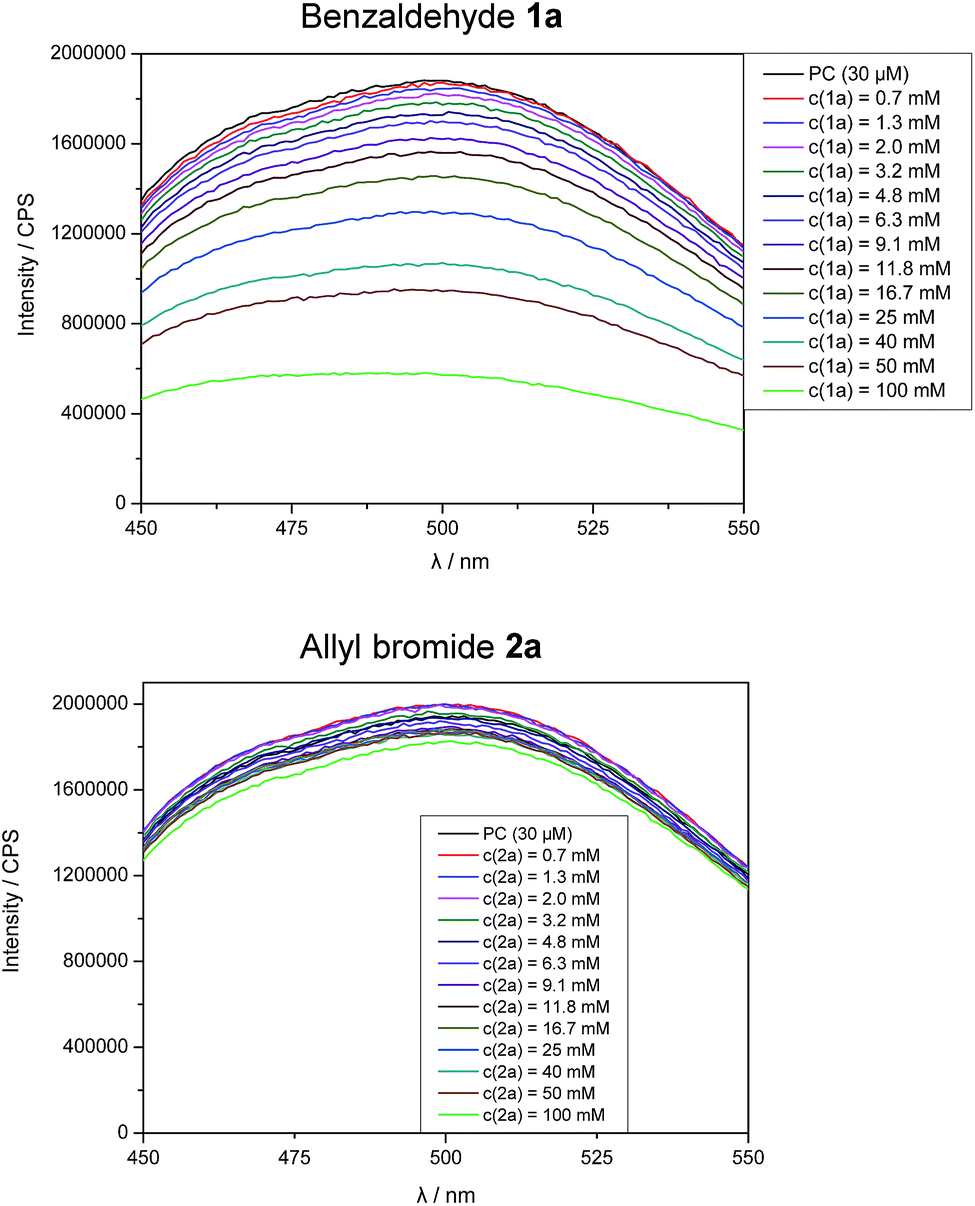 | ||
| Fig. 1 Fluorescence quenching experiments of photocatalyst B upon addition of benzaldehyde (1a) and allyl bromide (2a). | ||
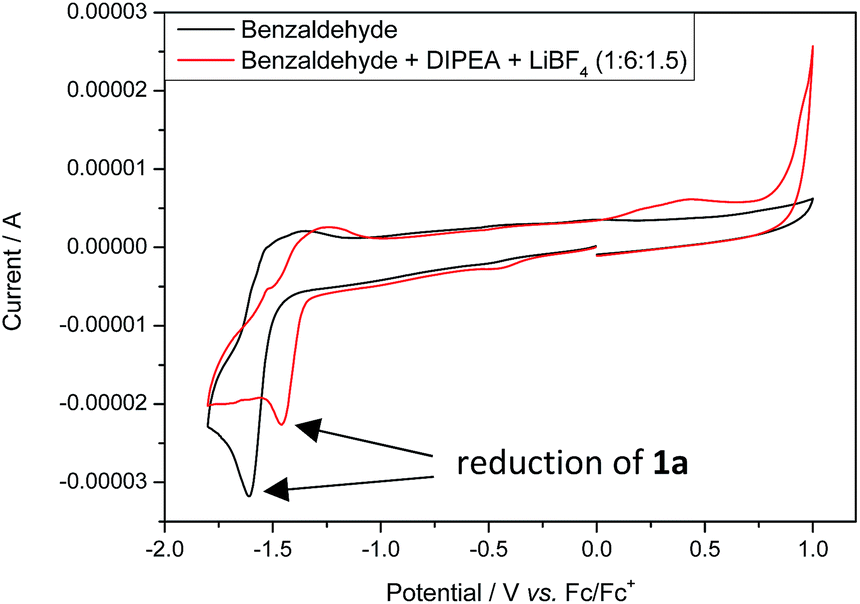 | ||
| Fig. 2 Cyclic voltammograms of benzaldehyde (1a, black) and a mixture of 1a (1 eq.), DIPEA (6 eq.) and LiBF4 (1.5 eq.) (red); the peak that corresponds to the reduction of 1a is shifted to lower potentials upon addition of DIPEA and LiBF4. | ||
Although the mixture of allyl bromide and DIPEA has no detectable absorbance at 400 nm, there seems to be a weak interaction between 2a and DIPEA leading to the absorption of small amounts of light and initiating an electron transfer from the amine to 2a. After a few minutes of irradiation, the absorption spectrum of the reaction mixture changes and an absorbance band with λmax,abs = 413 nm arises, therefore enabling the efficient absorbance of 400 nm light and speeding up the reaction (Fig. 3).
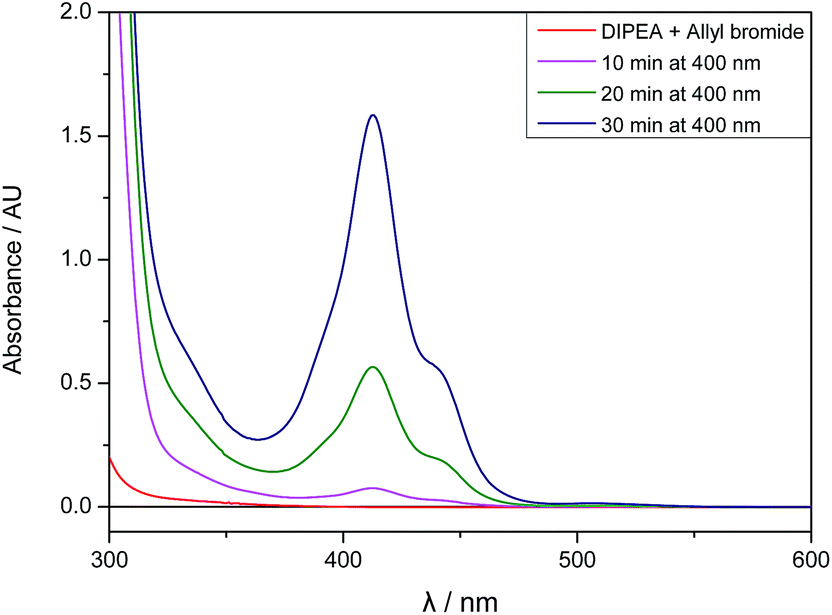 | ||
| Fig. 3 UV/Vis absorption spectra of allyl bromide (2a, 1 eq.) and DIPEA (3 eq.) in DMA before irradiation and after 10, 20 and 30 minutes of 400 nm irradiation. | ||
To gain further insight, the quantum yield of the reaction was measured. While the determined value of ϕ = 7.6% is rather high for photocatalytic reactions, it is in accordance with the fast reaction times.
Based on these mechanistic investigations and recent literature reports,15,21 we propose the reaction mechanism depicted in Scheme 3. Photocatalyst B is excited upon irradiation with 400 nm light and benzaldehyde (1a) can be reduced to the ketyl radical 1a˙− by a SET from the excited photocatalyst B*. DIPEA acts as a sacrificial electron donor to regenerate the photocatalyst from its oxidized form B˙+ to the ground state B. Irradiation of allyl bromide and DIPEA initiates an electron transfer, which after the cleavage of Br−, leads to the formation of the allyl radical 2a˙. The more persistent ketyl radical 1a˙−21 and the transient allyl radical 2a˙22 recombine in a radical–radical cross-coupling, which is in accordance with the persistent radical effect,21,23 and after protonation, the desired product 3a is formed.
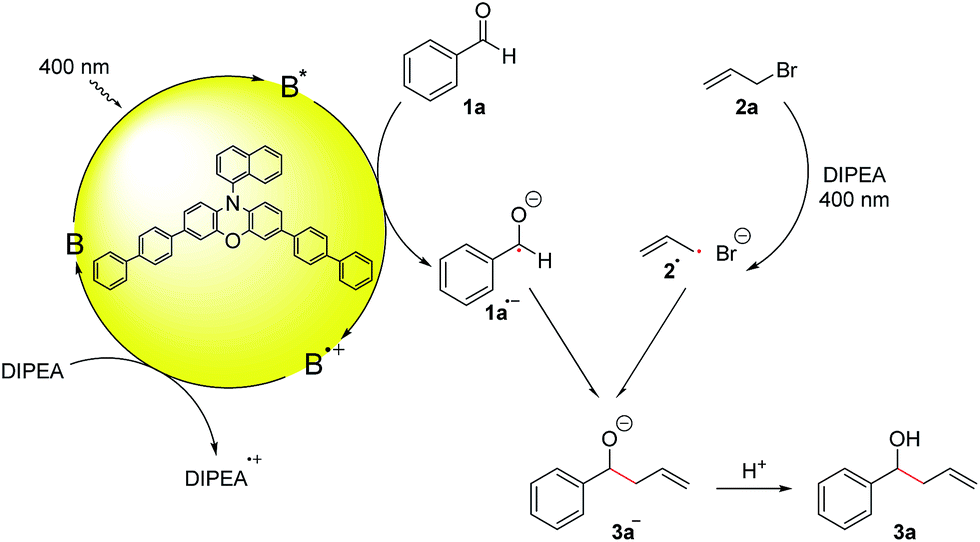 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusion
In summary, we have developed a photocatalytic version of the Barbier-type reaction, which generates allylic or benzylic alcohols from aldehydes or ketones and allyl- or benzyl bromides under mild conditions via a radical–radical cross-coupling. Instead of using stoichiometric amounts of zerovalent metal as a reductant to generate an organometallic carbanion synthon, we use an organic photocatalyst, a tertiary amine and visible light to reduce both substrates to the corresponding radicals. The cross-coupling of these radicals leads to the desired product and enables a photocatalytic two electron process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the German Science Foundation (DFG, GRK 1626). This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. 741623). We thank Dr Rudolf Vasold for GS-MS measurements and Regina Hoheisel for cyclic voltammetry measurements.Notes and references
- P. Barbier, C. R. Acad. Sci., 1899, 128, 110–111 Search PubMed.
- (a) T. A. Killinger, N. A. Boughton, T. A. Runge and J. Wolinsky, J. Organomet. Chem., 1977, 124, 131–134 CrossRef; (b) C. Petrier and J. L. Luche, J. Org. Chem., 1985, 50, 910–912 CrossRef; (c) F. Zhou and C. J. Li, Nat. Commun., 2014, 5, 4254 CrossRef PubMed; (d) L. Keinicke, P. Fristrup, P. O. Norrby and R. Madsen, J. Am. Chem. Soc., 2005, 127, 15756–15761 CrossRef PubMed.
- (a) C.-J. Li and W.-C. Zhang, J. Am. Chem. Soc., 1998, 120, 9102–9103 CrossRef; (b) S. Li, J.-X. Wang, X. Wen and X. Ma, Tetrahedron, 2011, 67, 849–855 CrossRef.
- Y. Yamamoto and N. Asao, Chem. Rev., 1993, 93, 2207–2293 CrossRef.
- (a) A. Rizzo and D. Trauner, Org. Lett., 2018, 20, 1841–1844 CrossRef PubMed; (b) B. M. Trost and A. B. Pinkerton, Tetrahedron Lett., 2000, 41, 9627–9631 CrossRef.
- J. Nokami, J. Otera, T. Sudo and R. Okawara, Organometallics, 1983, 2, 191–193 CrossRef.
- (a) K. Frimpong, J. Wzorek, C. Lawlor, K. Spencer and T. Mitzel, J. Org. Chem., 2009, 74, 5861–5870 CrossRef PubMed; (b) T.-H. Chan and M.-C. Lee, J. Org. Chem., 1995, 60, 4228–4232 CrossRef.
- S. Wu, Y. Li and S. Zhang, J. Org. Chem., 2016, 81, 8070–8076 CrossRef PubMed.
- C.-J. Li, Y. Meng, X.-H. Yi, J. Ma and T.-H. Chan, J. Org. Chem., 1997, 62, 8632–8633 CrossRef.
- (a) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef PubMed; (b) J. Xuan and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef PubMed; (c) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef PubMed; (d) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef PubMed; (e) J. P. Goddard, C. Ollivier and L. Fensterbank, Acc. Chem. Res., 2016, 49, 1924–1936 CrossRef PubMed.
- (a) K. Hironaka, S. Fukuzumi and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1984, 1705 RSC; (b) Y. Zhang, R. Qian, X. Zheng, Y. Zeng, J. Sun, Y. Chen, A. Ding and H. Guo, Chem. Commun., 2015, 51, 54–57 RSC.
- M. Chen, X. Zhao, C. Yang and W. Xia, Org. Lett., 2017, 19, 3807–3810 CrossRef PubMed.
- (a) O. Ishitani, C. Pac and H. Sakurai, J. Org. Chem., 1983, 48, 2941–2942 CrossRef; (b) O. Ishitani, S. Yanagida, S. Takamuku and C. Pac, J. Org. Chem., 1987, 52, 2790–2796 CrossRef; (c) T. Ghosh, T. Slanina and B. König, Chem. Sci., 2015, 6, 2027–2034 RSC.
- T. Shibata, A. Kabumoto, T. Shiragami, O. Ishitani, C. Pac and S. Yanagida, J. Phys. Chem., 1990, 94, 2068–2076 CrossRef.
- (a) F. R. Petronijevic, M. Nappi and D. W. MacMillan, J. Am. Chem. Soc., 2013, 135, 18323–18326 CrossRef PubMed; (b) M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828–8832 CrossRef PubMed; (c) E. Fava, A. Millet, M. Nakajima, S. Loescher and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 6776–6779 CrossRef PubMed; (d) W. Ding, L. Q. Lu, J. Liu, D. Liu, H. T. Song and W. J. Xiao, J. Org. Chem., 2016, 81, 7237–7243 CrossRef PubMed; (e) C. Wang, J. Qin, X. Shen, R. Riedel, K. Harms and E. Meggers, Angew. Chem., Int. Ed., 2016, 55, 685–688 CrossRef PubMed.
- (a) G. Pandey, S. Hajra, M. K. Ghorai and K. R. Kumar, J. Org. Chem., 1997, 62, 5966–5973 CrossRef; (b) J. Du and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 14604–14605 CrossRef PubMed; (c) M. A. Ischay, M. E. Anzovino, J. Du and T. P. Yoon, J. Am. Chem. Soc., 2008, 130, 12886–12887 CrossRef PubMed; (d) K. T. Tarantino, P. Liu and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 10022–10025 CrossRef PubMed; (e) L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 17735–17738 CrossRef PubMed; (f) E. Fava, M. Nakajima, A. L. Nguyen and M. Rueping, J. Org. Chem., 2016, 81, 6959–6964 CrossRef PubMed; (g) W. Li, Y. Duan, M. Zhang, J. Cheng and C. Zhu, Chem. Commun., 2016, 52, 7596–7599 RSC.
- (a) L. Qi and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 13312–13315 CrossRef PubMed; (b) K. N. Lee, Z. Lei and M. Y. Ngai, J. Am. Chem. Soc., 2017, 139, 5003–5006 CrossRef PubMed.
- Y. Nishigaichi, T. Orimi and A. Takuwa, J. Organomet. Chem., 2009, 694, 3837–3839 CrossRef.
- (a) J. C. Theriot, C. H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef PubMed; (b) R. M. Pearson, C. H. Lim, B. G. McCarthy, C. B. Musgrave and G. M. Miyake, J. Am. Chem. Soc., 2016, 138, 11399–11407 CrossRef PubMed; (c) Y. Du, R. M. Pearson, C. H. Lim, S. M. Sartor, M. D. Ryan, H. Yang, N. H. Damrauer and G. M. Miyake, Chem.–Eur. J., 2017, 23, 10962–10968 CrossRef PubMed; (d) B. G. McCarthy, R. M. Pearson, C. H. Lim, S. M. Sartor, N. H. Damrauer and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 5088–5101 CrossRef PubMed.
- P. J. Wagner, R. J. Truman, A. E. Puchalski and R. Wake, J. Am. Chem. Soc., 1986, 108, 7727–7738 CrossRef PubMed.
- J. L. Jeffrey, F. R. Petronijevic and D. W. MacMillan, J. Am. Chem. Soc., 2015, 137, 8404–8407 CrossRef PubMed.
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1976, 9, 13–19 CrossRef.
- (a) A. Studer, Chem.–Eur. J., 2001, 7, 1159–1164 CrossRef PubMed; (b) J. D. Cuthbertson and D. W. MacMillan, Nature, 2015, 519, 74–77 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02038h |
| This journal is © The Royal Society of Chemistry 2018 |