DOI:
10.1039/C8SC02018C
(Edge Article)
Chem. Sci., 2018,
9, 8142-8149
Designing an asymmetrical isomer to promote the LUMO energy level and molecular packing of a non-fullerene acceptor for polymer solar cells with 12.6% efficiency†
Received
4th May 2018
, Accepted 18th August 2018
First published on 20th August 2018
Abstract
Isomers with small structural changes usually exhibit different properties. Rationally designing isomers of some high-performance SMAs can further enhance their function. In this work, an asymmetrical small molecule acceptor (SMA) MeIC1 isomerized from MeIC is reported. Compared with the symmetrical MeIC, the asymmetrical isomer showed almost the same absorption range but an elevated LUMO energy level and simultaneously enhanced π–π stacking and electron mobility by replacing the thieno[3,2-b]thiophene unit with a larger sized dithieno[3,2-b:2′,3′-d]thiophene unit in the ladder-type core of MeIC. As a result, the MeIC1-based PSCs achieved a higher PCE up to 12.58% with a promoted Voc and Jsc and an unchanged FF compared with those of MeIC-based PSCs when blended with PBDB-T. This work reveals that asymmetrical isomerization is effective for PCE promotion.
1. Introduction
In recent years, with the help of non-fullerene small molecule acceptors (NF-SMAs),1–7 especially the 3,9-bis(2-methylene-(3-(1,1-dicyanomethylene)-indanone))-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′]-dithiophene (ITIC) family,8–10 the overall power conversion efficiency (PCE) of PSCs has broken through 12%.11–24 ITIC-like SMAs have shown great promise as alternative acceptors that possess tremendous advantages in terms of low production cost, tunable molecular energy levels, a wide absorption range and high electron mobility.8–39 Moreover, recently published works have revealed that ITIC-like SMAs exhibit universal applicability with different polymeric and small-molecule donors in binary,8–11,14–39 ternary13,40–44 and tandem12,14 PSCs.
In general, ITIC-like SMAs are a perfect combination of electron-withdrawing end groups (EGs), an electron-donating core and outstretched side chains. Numerous endeavors have been devoted to exploring the relationship between molecular structure and performance.8–11,15–39 Interestingly, SMA isomers with very small molecular structure changes may show great differences in device performance. Li and coworkers performed a side-chain isomerization on ITIC by replacing the para-alkylphenyl with meta-alkylphenyl, PSCs based on the isomeric counterpart m-ITIC showed remarkably promoted PCEs due to the enhanced π–π stacking and higher electron mobility of m-ITIC.28 Our group recently reported a novel SMA named ITCPTC with 2-(6-oxo-5,6-dihydro-4H-cyclopenta[c]thiophen-4-ylidene)malononitrile (CPTCN) as an EG,10 while Hou and coworkers reported an EG isomer of ITCPTC (called ITCC).29 Though devices based on ITCPTC and ITCC achieved similar PCEs, their open-circuit voltage (Voc) and short-circuit current (Jsc) differed greatly, attributed to the different electron-withdrawing abilities of the two EG isomers. Chen and coworkers modified ITIC with heptacyclic benzodi(cyclopentadithiophene) as the donor core, resulting in an increased Jsc but a decreased Voc in PSCs compared with those of ITIC-based devices.30 The different device performances of SMA isomers in the side chain, end group and donor core enlightened us to the idea that rationally designing isomers of some high-performance SMAs may further enhance the PCE of PSCs.
After ITCPTC, another high-performance SMA, namely MeIC, was designed and synthesized in our group by substituting a hydrogen atom of CPTCN with a slightly electron-donating methyl (CPTCN-M) group to elevate the lowest unoccupied molecular orbital (LUMO) energy level and enhance the π–π stacking of ITCPTC.18 A MeIC-based PSC realized a high PCE up to 12.54% with a Voc of 0.918 V, a Jsc of 18.41 mA cm−2 and a fill factor (FF) of 74.2% when blended with a famous polymer donor J71.45 Motivated by the high-performance of MeIC, we designed an asymmetrical isomer (MeIC1) of MeIC by replacing two thieno[3,2-b]thiophenes with thiophene and dithieno[3,2-b:2′,3′-d]thiophene in the ladder-type core of MeIC, respectively. Unexpectedly, the asymmetrical structure promotes the LUMO energy level of MeIC1 by 0.05 eV relative to its symmetrical counterpart, and the big dithieno[3,2-b:2′,3′-d]thiophene moiety in MeIC1 helps effective stacking. Finally, the PBDB-T:MeIC1-based PSCs achieved a higher PCE up to 12.58% mainly due to an enhanced Voc and Jsc compared with those of PBDB-T:MeIC-based PSCs. Rationally designing asymmetrical isomers of high-performance SMAs through introducing large packing moieties can synchronously enhance the molecular LUMO energy level and π–π stacking and thus achieve larger Voc and Jsc and thus higher PCEs in non-fullerene PSCs.
2. Results and discussion
2.1. Synthesis and characterization
The synthesis route for MeIC1 is displayed in Scheme S1.† An asymmetrical intermediate 3 was synthesized through two successive Negishi coupling reactions by utilizing thiophen-2-ylzinc(II) chloride and dithieno[3,2-b:2′,3′-d]thiophen-2-ylzinc(II) chloride as organozinc reagents. Then, a double nucleophilic addition reaction was carried out to convert the ester groups of 3 into hydroxyls, which, without further purification, were subjected to an acid-mediated Friedel–Crafts reaction to obtain 4. Then, two formyl groups were introduced into 4 and end-capped with 2-(1-methyl-6-oxo-5,6-dihydro-4H-cyclopenta[c]thiophen-4-ylidene)malononitrile (CPTCN-M) resulting in MeIC1. These two SMAs have been fully characterized and show good solubility in commonly used solvents.
2.2. Theoretical calculation
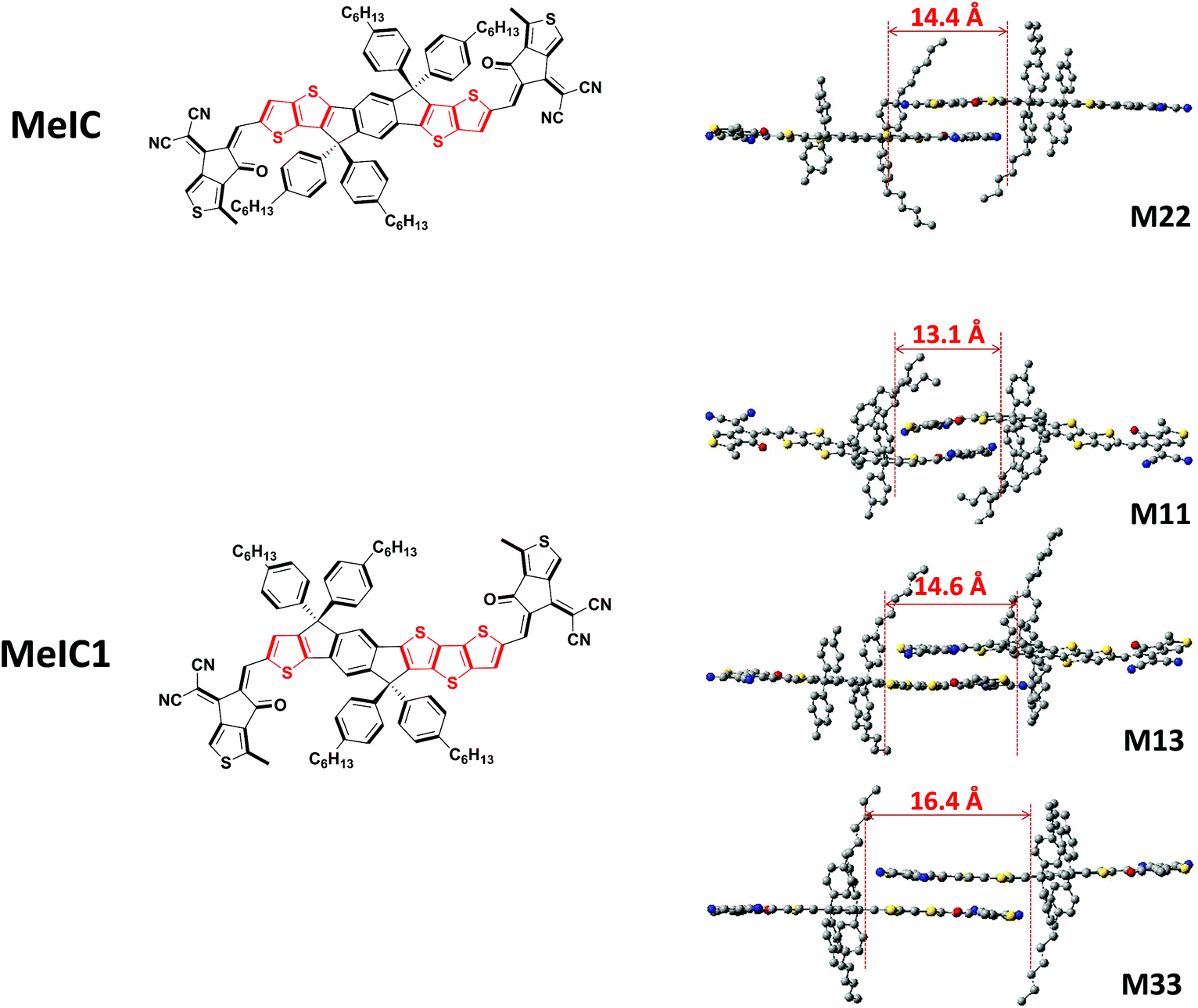
The two isomeric counterparts of MeIC and MeIC1 show small distinctions in the ladder-type core with the number and position of thiophenes being different (Fig. 1). To gain deep insights into the effects on molecule energy levels, optical bandgaps and molecular packing induced by structural differences, theoretical simulations of single and dimer molecules were performed (Fig. S1†). The geometries were optimized at the B3LYP/def2-SVP level with a RIJCOSX46 approximation using the ORCA 4.0 program.47 Grimme’s D3 dispersion correction with Becke–Johnson damping48 and Grimme’s geometrical counterpoise correction (gCP)49 were used during the optimization. The wave functions were obtained using the Gaussian09 program at the B3LYP/6-31G(d,p) level. The transfer integral and electron density overlap integral were evaluated by the J-from-g03 and Multiwfn50 programs, respectively. As calculated, the predicated LUMO/HOMO energy levels of MeIC and MeIC1 were −3.547 eV/−5.538 eV and −3.537 eV/−5.542 eV, respectively, and the corresponding bandgaps were 1.991 eV and 2.005 eV, respectively. It can be seen that asymmetrical MeIC1 has a higher LUMO energy level and a slightly enlarged optical bandgap compared to the symmetric MeIC. Packing geometry configurations of the two dimer molecules are displayed in Fig. 1. As shown, asymmetrical MeIC1 exhibits three possible packing forms: M11 (thiophene stacking on thiophene), M13 (thiophene stacking on dithieno[3,2-b:2′,3′-d]thiophene) and M33 (dithieno[3,2-b:2′,3′-d]thiophene stacking on dithieno[3,2-b:2′,3′-d]thiophene), while symmetrical MeIC shows one form M22 (thieno[3,2-b]thiophene stacking on thieno[3,2-b]thiophene). The overlap lengths are positively correlated with the size of the two stacked units, and these were 13.1 Å, 14.6 Å and 16.4 Å for M11, M13 and M33, and 14.4 Å for M22. Obviously, the overlap length of M13 and average overlap length of M11 and M33 were both longer than that of M22. The intermolecular bonding energies were calculated to be −2.36 eV, −1.99 eV, −2.05 eV and −1.75 eV for M11, M13, M33 and M22, respectively. The larger bonding energy of MeIC1 suggests that it has a stronger tendency for intermolecular stacking during the course of self-assembly. Combining the overlap lengths and the interaction energies, we can speculate that the π–π stacking pattern of M11 and M33 is superior to that of M13 in MeIC1, but each π–π stacking in MeIC1 is better than that of M22 in MeIC. This indicates that the asymmetrical isomerization of MeIC by incorporating the larger dithieno[3,2-b:2′,3′-d]thiophene unit could enhance the π–π stacking in theory. Moreover, the electron transfer integral values of M11, M13, M33 and M22 were 352 cm−1, 466 cm−1, 538 cm−1 and 440 cm−1, respectively, which reveals that MeIC1 has a higher probability for charge transfer from one molecule to another. As the electron transfer integral is very sensitive to the relative position of the two π fragments, we also calculated the electron density overlap integral and obtained the same trends (Table S1†). The aforementioned results reveal that the asymmetrical structure can elevate the LUMO energy level of MeIC1, and the large dithieno[3,2-b:2′,3′-d]thiophene moiety in MeIC1 facilitates intermolecular stacking and charge transfer compared to MeIC.
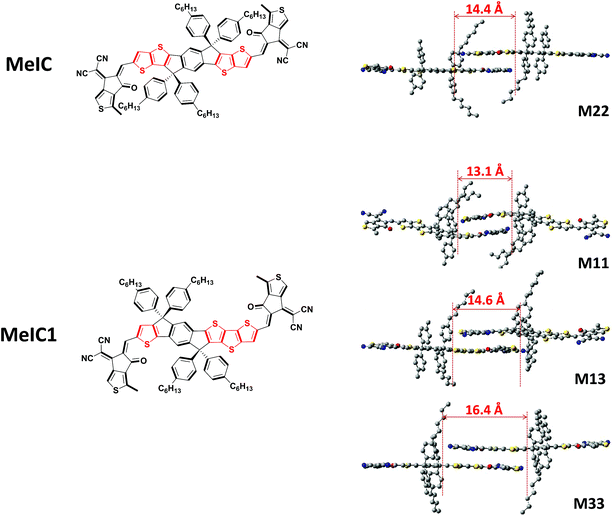 |
| | Fig. 1 The structures of the two isomers (left) and the geometry configurations of dimer molecules (right). | |
2.3. Optical and electrochemical properties
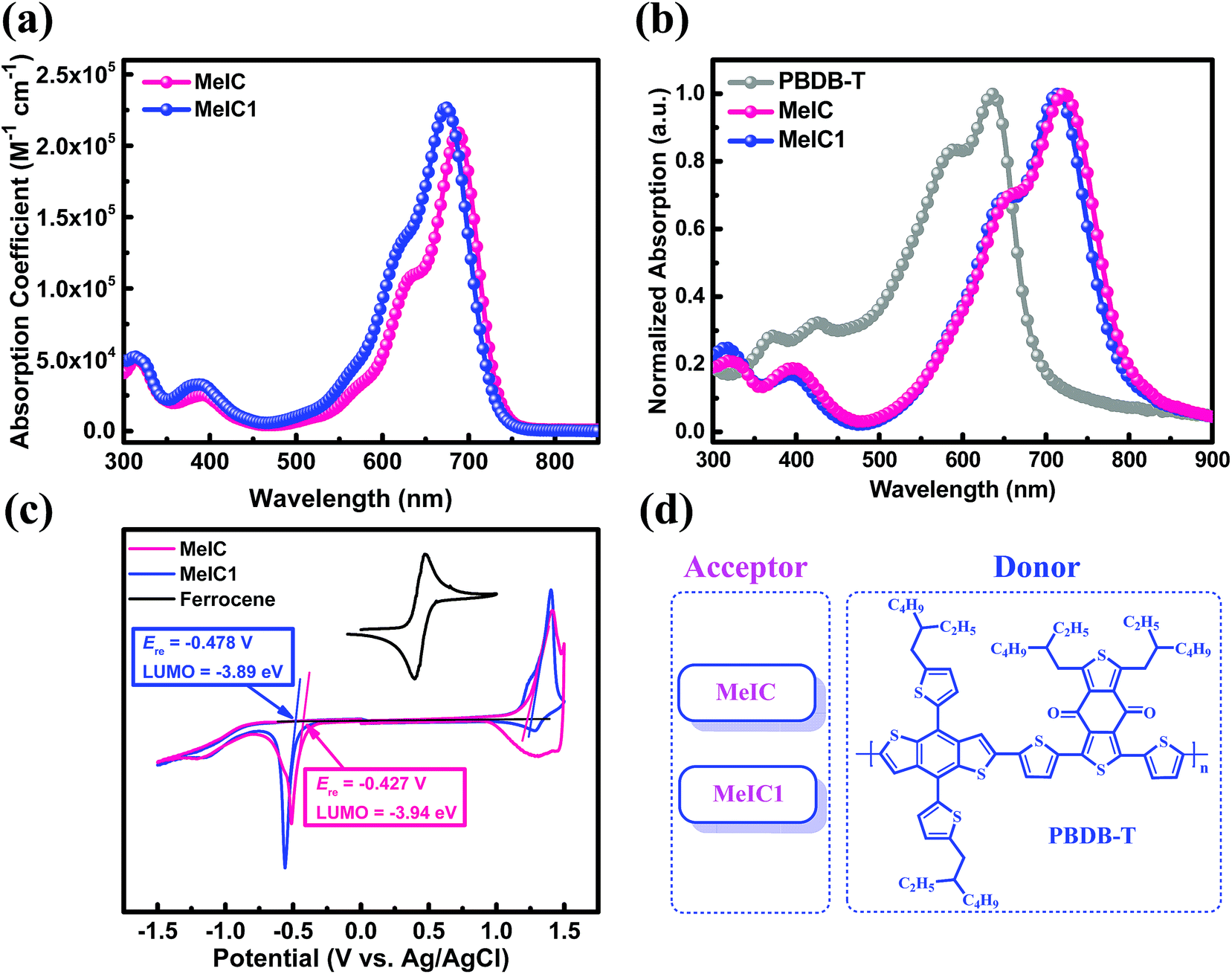
Fig. 2a and b show the absorption spectra of MeIC and MeIC1. In dilute chloroform solution, asymmetrical MeIC1 exhibited a slightly blue-shifted absorption spectrum by 8 nm but a stronger molar absorption coefficient (2.27 × 105 M−1 cm−1) compared with MeIC (2.09 × 105 M−1 cm−1). From solution to films, strong electronic vibration shoulder peaks were formed. In neat films, MeIC1 and MeIC showed almost the same absorption spectra, and the optical bandgaps of MeIC and MeIC1 were 1.53 eV and 1.54 eV, respectively (Table 1). Cyclic voltammetry (CV) measurements were conducted using a Ag/AgCl electrode as a reference and Fc/Fc+ (0.435 V) as a standard to test the HOMO and LUMO differences between the two isomers. As revealed in the CV plots (Fig. 2c), the reduction functional wave onset of MeIC1 obviously falls behind that of MeIC, thus MeIC1 possesses a higher LUMO energy level (−3.89 eV) than MeIC (−3.94 eV) by approximately 0.05 eV, which is consistent with the density functional theory (DFT) calculation results and results in a higher Voc when applied in PSCs.
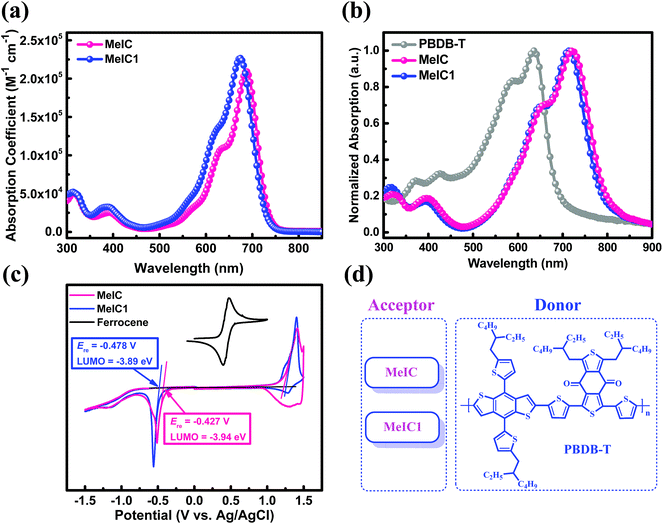 |
| | Fig. 2 (a) Molar absorption coefficient spectra in chloroform solution. (b) Normalized UV-vis absorption spectra of neat films. (c) CV curves and LUMO energy levels. (d) Active materials investigated in the studied PSCs. | |
Table 1 Basic properties of MeIC and MeIC1
| Acceptor |
λ
max
(nm) |
ε
max
(M−1 cm−1) |
λ
onset
(nm) |
λ
max
(nm) |
λ
onset
(nm) |
E
optg
(eV) |
HOMOd (eV) |
LUMOd (eV) |
E
CVg
(eV) |
μ
e
(cm2 V−1 s−1) |
|
In chloroform solution.
In the neat film.
Calculated from the empirical formula: Eoptg = 1240/λonset.
Cyclic voltammetry (CV) method by measuring the neat film in acetonitrile.
E
CVg = ELUMO − EHOMO.
Measured by the space charge limited current (SCLC) method.
|
| MeIC |
687 |
2.09 × 105 |
739 |
722 |
807 |
1.53 |
−5.57 |
−3.94 |
1.63 |
2.03 × 10−3 |
| MeIC1 |
679 |
2.27 × 105 |
732 |
714 |
804 |
1.54 |
−5.59 |
−3.89 |
1.70 |
2.38 × 10−3 |
2.4. Photovoltaic performance
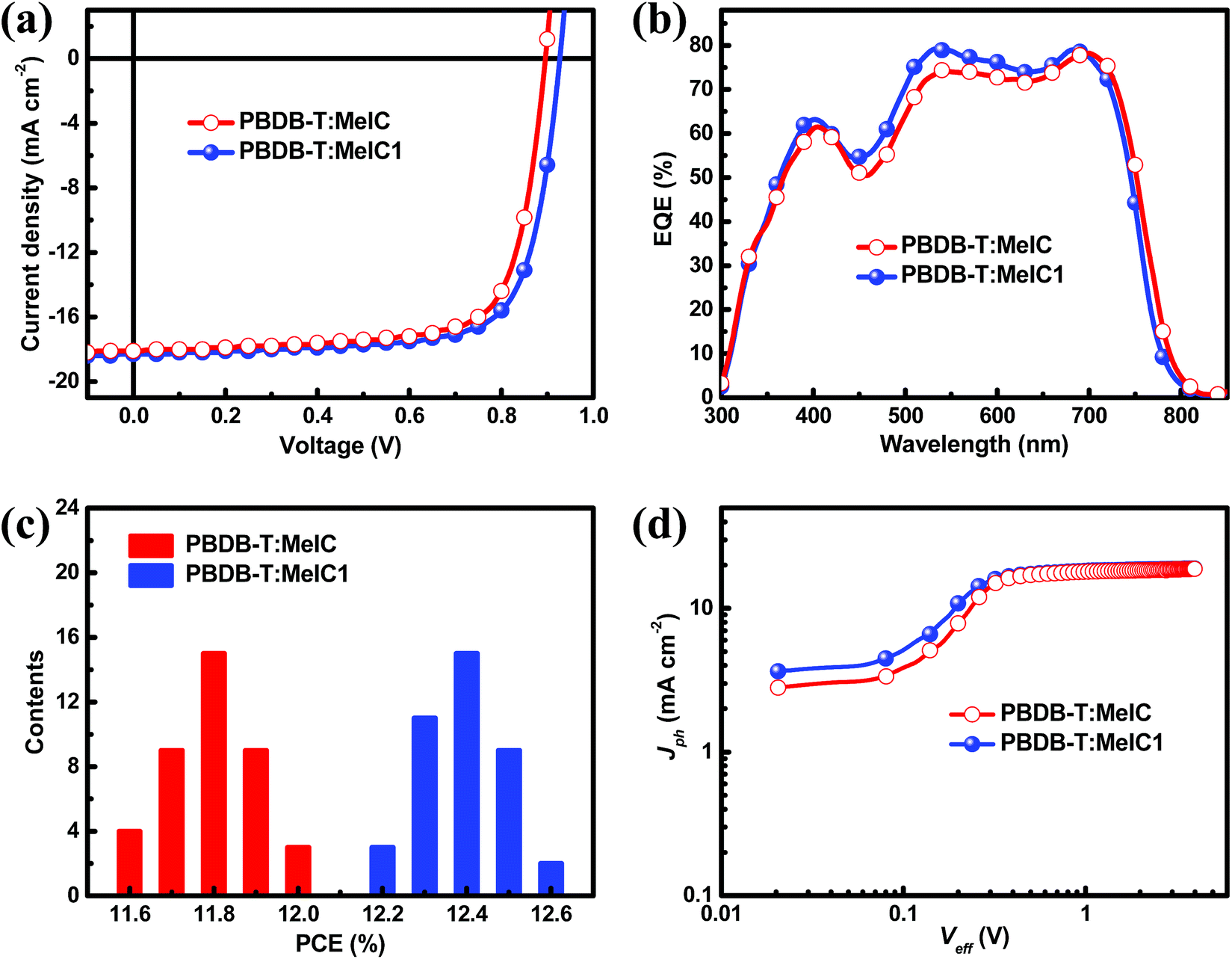
A series of PSC devices with a structure of indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS)/PBDB-T:acceptor/PDIN/Al were fabricated to investigate the photovoltaic performance of the two isomers (Fig. S2†). A wide-bandgap polymer PBDB-T was selected as a donor due to its complementary absorption spectrum and matched energy levels with MeIC and MeIC1. The optimal fabrication conditions were as follows: chlorobenzene (CB) was utilized as the processing solvent and 0.5% (v/v) 1,8-diiodooctane (DIO) was added; the weight ratio and blend solution concentration of PBDB-T:acceptor were 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 20 mg ml−1, respectively; the optimal annealing temperature was 120 °C (Fig. S3 and Table S2†). The characteristic current density–voltage (J–V) curves of MeIC- and MeIC1-based PSCs were measured under illumination of AM 1.5 G at 100 mW cm−2 (Fig. 3a) and the key photovoltaic parameters are summarized in Table 2. As shown, the PBDB-T:MeIC1-based PSCs achieved PCEs up to 12.58% produced from a Voc of 0.927 V, a Jsc of 18.32 mA cm−2 and a FF of 74.1%, while the PCE for PBDB-T:MeIC-based PSCs was 12.03% along with a Voc of 0.896 V, a Jsc of 18.07 mA cm−2 and a FF of 74.3%. The higher PCE for asymmetrical isomer-based PSCs is mainly attributed to their higher Voc and Jsc and almost unchanged FF compared to those of MeIC-based PSCs. The asymmetrical structure of MeIC1 results in LUMO energy level promotion which is 0.05 eV higher than that of symmetrical MeIC. Thus, the Voc of MeIC1-based PSCs is 0.03 V higher than that of MeIC-based PSCs. Furthermore, the dithieno[3,2-b:2′,3′-d]thiophene moiety in the asymmetrical core enhances the intermolecular π–π stacking and charge transfer of MeIC1 (this is discussed below), thus the FF of MeIC1-based PSCs can stay at a very high level. Though the absorption range becomes narrower, the MeIC1-based PSCs still achieve a larger Jsc than MeIC-based PSCs, which can be observed from the higher external quantum efficiency (EQE) spectrum of MeIC1-based PSCs in the range of 400 nm to 700 nm (Fig. 3b). The two isomer based PSCs show great repeatability and the PCE distribution histogram from 40 devices of different batches is displayed in Fig. 3c. The average PCEs and mean square errors for PBDB-T:MeIC- and PBDB-T:MeIC1-based PSCs were 11.80% ± 0.11% and 12.38% ± 0.10%, respectively. The detailed photovoltaic parameters of the typical 40 samples are shown in Fig. S4† and their average values and mean square errors are shown in Table 2.
:1 and 20 mg ml−1, respectively; the optimal annealing temperature was 120 °C (Fig. S3 and Table S2†). The characteristic current density–voltage (J–V) curves of MeIC- and MeIC1-based PSCs were measured under illumination of AM 1.5 G at 100 mW cm−2 (Fig. 3a) and the key photovoltaic parameters are summarized in Table 2. As shown, the PBDB-T:MeIC1-based PSCs achieved PCEs up to 12.58% produced from a Voc of 0.927 V, a Jsc of 18.32 mA cm−2 and a FF of 74.1%, while the PCE for PBDB-T:MeIC-based PSCs was 12.03% along with a Voc of 0.896 V, a Jsc of 18.07 mA cm−2 and a FF of 74.3%. The higher PCE for asymmetrical isomer-based PSCs is mainly attributed to their higher Voc and Jsc and almost unchanged FF compared to those of MeIC-based PSCs. The asymmetrical structure of MeIC1 results in LUMO energy level promotion which is 0.05 eV higher than that of symmetrical MeIC. Thus, the Voc of MeIC1-based PSCs is 0.03 V higher than that of MeIC-based PSCs. Furthermore, the dithieno[3,2-b:2′,3′-d]thiophene moiety in the asymmetrical core enhances the intermolecular π–π stacking and charge transfer of MeIC1 (this is discussed below), thus the FF of MeIC1-based PSCs can stay at a very high level. Though the absorption range becomes narrower, the MeIC1-based PSCs still achieve a larger Jsc than MeIC-based PSCs, which can be observed from the higher external quantum efficiency (EQE) spectrum of MeIC1-based PSCs in the range of 400 nm to 700 nm (Fig. 3b). The two isomer based PSCs show great repeatability and the PCE distribution histogram from 40 devices of different batches is displayed in Fig. 3c. The average PCEs and mean square errors for PBDB-T:MeIC- and PBDB-T:MeIC1-based PSCs were 11.80% ± 0.11% and 12.38% ± 0.10%, respectively. The detailed photovoltaic parameters of the typical 40 samples are shown in Fig. S4† and their average values and mean square errors are shown in Table 2.
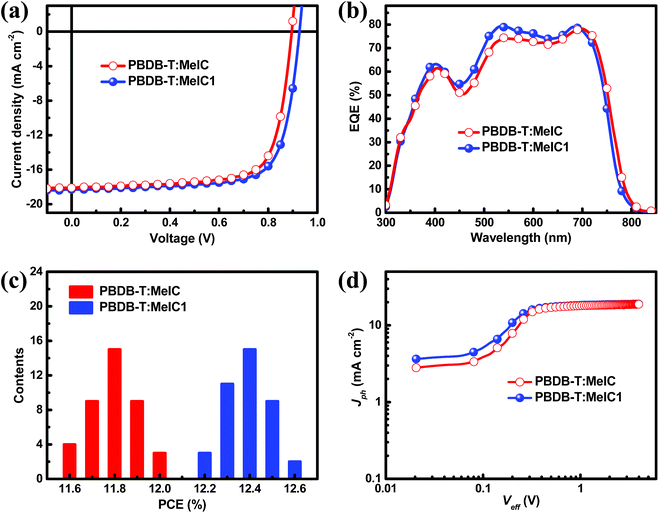 |
| | Fig. 3 (a) Characteristic current–voltage (J–V) curves. (b) Corresponding EQE spectra. (c) PCE distribution histogram (40 devices). (d) Photocurrent density (Jph) versus effective voltage (Veff). | |
Table 2 Key photovoltaic parameters of PSCs based on the optimal devices
| Active layersa |
V
oc
(V) |
J
sc
(mA cm−2) |
FFb (%) |
PCEmaxb (%) |
|
The area of active layers was 3.8 mm2.
The values in brackets are the average values and mean square errors calculated from 40 devices.
|
| PBDB-T:MeIC |
0.896 (0.890 ± 0.003) |
18.07 (18.05 ± 0.08) |
74.3 (73.4 ± 0.6) |
12.03 (11.80 ± 0.11) |
| PBDB-T:MeIC1 |
0.927 (0.922 ± 0.003) |
18.32 (18.29 ± 0.10) |
74.1 (73.4 ± 0.4) |
12.58 (12.38 ± 0.10) |
2.5. Hole and electron mobility
To evaluate the charge transport ability of MeIC and MeIC1 in neat and blend films, space charge limited current (SCLC) measurements were carried out. The electron-only device was a stack of ITO/ZnO/active layer/Al while the hole-only device was a stack of ITO/PEDOT:PSS/active layer/MoO3/Ag. As shown in Fig. S5,† the electron mobilities of MeIC and MeIC1 neat films were determined to be 2.03 × 10−3 cm2 V−1 s−1 and 2.38 × 10−3 cm2 V−1 s−1, respectively. The electron/hole mobilities (μe/μh) of PBDB-T:MeIC and PBDB-T:MeIC1 blend films were 3.63 × 10−4 cm2 V−1 s−1/7.61 × 10−4 cm2 V−1 s−1 and 5.10 × 10−4 cm2 V−1 s−1/10.80 × 10−4 cm2 V−1 s−1 with μe/μh as 0.47 and 0.48, respectively. The two isomers both showed high electron mobility up to 10−3 cm2 V−1 s−1 orders of magnitude in neat films, which is comparable with that of fullerene derivatives. It can be seen that the asymmetrical SMA exhibited slightly higher electron mobility than its symmetrical isomer. This suggests that the asymmetrical MeIC1 was able to form better π–π stacking during the self-assembly process, which is in agreement with the aforementioned theoretical calculations. When the polymer donor PBDB-T was incorporated, the courses of crystallization of the two isomers were restrained to different degrees, and the electron mobilities of donor and acceptor hybrids were dramatically decreased compared to those of acceptor neat films. The PBDB-T:MeIC1 blend film showed approximately 1.5 times the electron mobility relative to that of PBDB-T:MeIC and a much higher hole mobility, indicative of a more ordered blend film of PBDB-T:MeIC1. We speculated that the big dithieno[3,2-b:2′,3′-d]thiophene unit in MeIC1 induced a faster crystallization process than that of the thieno[3,2-b]thiophene unit in MeIC. Thus, better phase domain sizes, nanofibers and interpenetrating network structures can be achieved in MeIC1-based blend films, which can be clearly observed in the atomic force microscope (AFM) and transmission electron microscope (TEM) images (Fig. 4) which we are discussed in detail below.
 |
| | Fig. 4 AFM height sensor images of (a) PBDB-T:MeIC and (b) PBDB-T:MeIC1. AFM phase images of (c) PBDB-T:MeIC and (d) PBDB-T:MeIC1. TEM images of (e) PBDB-T:MeIC and (f) PBDB-T:MeIC1. | |
2.6. Exciton dissociation and charge collection
The exciton dissociation and charge extraction of the two isomer based optimal active layers were investigated by measuring the photocurrent density (Jph) versus effective voltage (Veff). Jph is defined as Jph = JL − JD, where JL and JD are the current densities under AM 1.5 G light illumination and in the dark, respectively. Veff is defined as Veff = V0 − Vbias, where V0 is the voltage at which Jph = 0 and Vbias is the applied external voltage bias. It is assumed that all the photoinduced excitons can be separated into free charges and collected in corresponding electrodes when Veff is high enough. As displayed in Fig. 3d, the Jph of PBDB-T:MeIC- and PBDB-T:MeIC1-based devices approached saturation (Jsat) at a relatively low voltage of about 0.5 V, indicating an efficient course of exciton separation, charge transport and collection in PSCs, which accords with their high FFs. The Jsat of MeIC- and MeIC1-based PSCs extracted from Jph–Veff plots was 18.81 mA cm−2 and 18.91 mA cm−2, respectively (Table S3†). The probability of exciton dissociation calculated from values of Jph/Jsat under short circuit conditions was 96.06% and 96.88% for MeIC- and MeIC1-based devices, respectively. Similarly, the probability of charge collection of MeIC- and MeIC1-based devices calculated from values of Jph/Jsat at maximal power output conditions was 84.79% and 84.45%, respectively. The results indicated that the asymmetrical MeIC1 was the same or even superior to the symmetrical MeIC in aspects of exciton dissociation and charge collection.
2.7. Morphology study
To show the distinctions in superficial morphology of the blend films based on MeIC and MeIC1, AFM and TEM measurements were carried out. AFM height sensor images, AFM phase images and TEM images are displayed in Fig. 4. As shown in Fig. 4a and b, the MeIC- and MeIC1-based optimal blend films exhibited quite smooth and uniform surfaces with root-mean-square (RMS) roughness values of 2.18 nm and 2.37 nm, respectively. AFM images (Fig. 4a–d) and TEM images (Fig. 4e and f) revealed that MeIC- and MeIC1-based blend films possess excellent phase separation. It is generally acknowledged that good phase separation is beneficial to exciton dissociation, thus the active layers fabricated with PBDB-T:MeIC and PBDB-T:MeIC1 exhibited high exciton separation probability, Jsc and EQE values. Moreover, the nanofiber structures in PBDB-T:MeIC- and PBDB-T:MeIC1-based blend films can be clearly observed from the flocculent features in the AFM images (Fig. 4a–d) and the capillary structures in the TEM images (Fig. 4e and f), while the continuous interpenetrating network structures were also well-developed, which is in favour of efficient charge transport for achieving a large Jsc and FF in PBDB-T:MeIC and PBDB-T:MeIC1 fabricated PSCs. It can be clearly observed that MeIC1-based blend films showed thicker nanofibers compared with MeIC. This was very likely due to the faster crystallization speed of MeIC1 which resulted from bigger dithieno[3,2-b:2′,3′-d]thiophene units leading to the more ordered condensed-state and larger phase domain sizes in MeIC1-based active layers, which are beneficial to obtain higher phase purity to achieve a larger Jsc and FF in PSCs. This phenomenon also supports a smaller electron mobility loss from neat film to blend film and a much higher hole mobility when compared to that of MeIC.
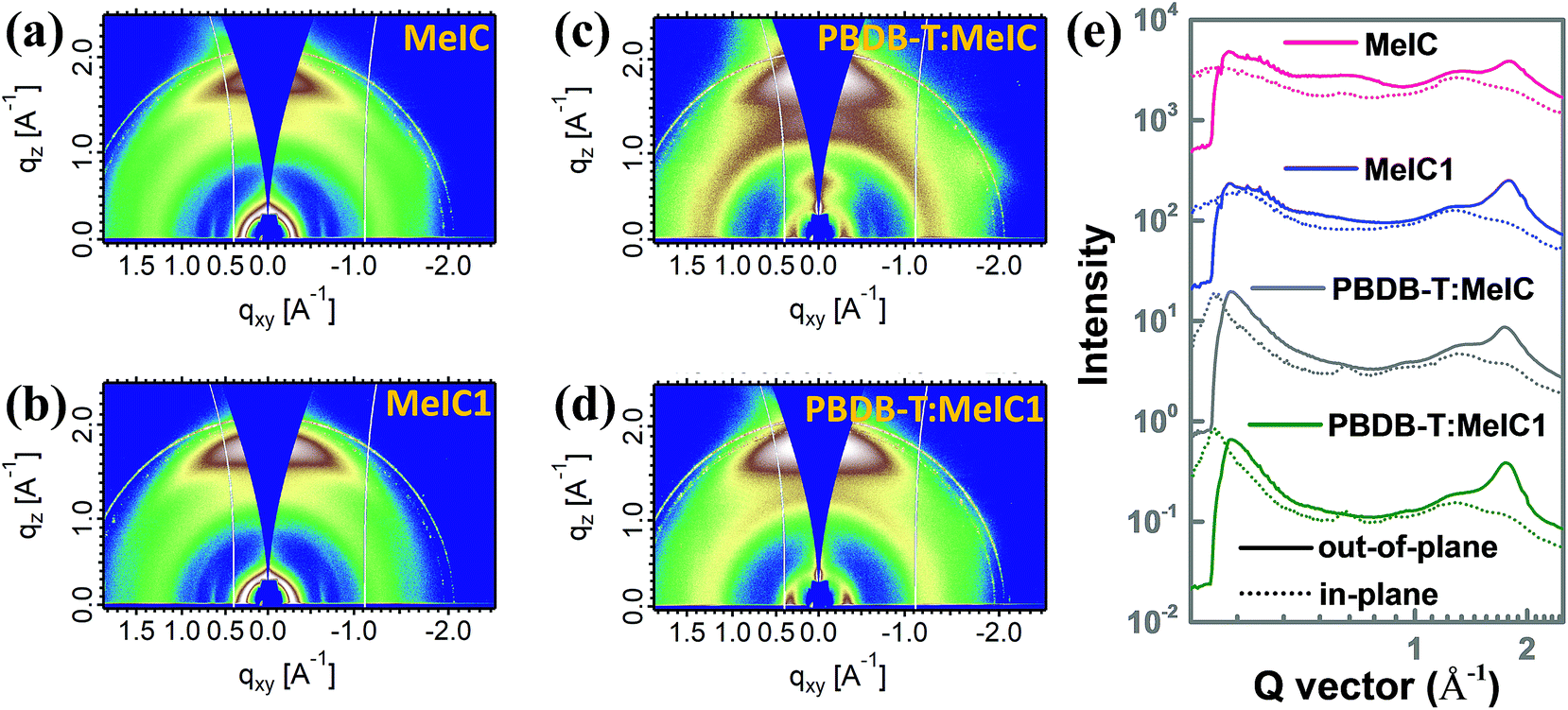
Grazing incidence wide-angle X-ray scattering (GIWAXS) measurements were employed to survey the intermolecular π–π stacking of the two isomer based neat and blend films, and the corresponding out-of-plane (solid line) and in-plane (dotted line) cutline profiles are shown in Fig. 5e. As shown, MeIC1 showed a well-defined face-on orientation with a significantly stronger (010) diffraction peak compared to MeIC in the out-of-plane direction. The d-spacing and coherence length (CL) calculated from the cutline profiles were 3.55 Å and 23.7 Å for MeIC-based neat films and 3.49 Å and 28.2 Å for MeIC1-based neat films, which indicates the enhanced π–π stacking effect between MeIC1 molecules and is consistent with the theoretical simulation results and SCLC measurements. The strong crystallization properties of MeIC1 also facilitate the effective π–π stacking of PBDB-T:MeIC1-based blend films. It can be clearly observed that the (010) diffraction intensity coming from PBDB-T:MeIC1-blend films was significantly stronger than that of PBDB-T:MeIC-based blend films, which well accounts for the higher charge mobility and Jsc of PBDB-T:MeIC1-based PSCs.
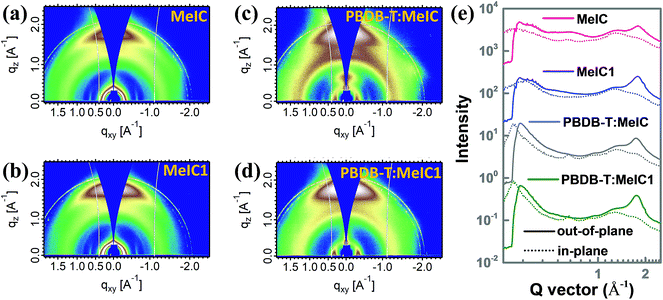 |
| | Fig. 5 (a–d) 2D GIWAXS patterns of MeIC- and MeIC1-based neat and blend films. (e) Corresponding out-of-plane (solid line) and in-plane (dotted line) GIWAXS cutline profiles. | |
3. Conclusion
In summary, an asymmetrical SMA isomer MeIC1 was designed and synthesized to further boost the performance of MeIC by replacing two thieno[3,2-b]thiophene units in the ladder-type core of MeIC with thiophene and dithieno[3,2-b:2′,3′-d]thiophene units, respectively. The asymmetrical ladder-type core enabled MeIC1 to show a higher LUMO energy level by 0.05 eV compared to that of MeIC without sacrificing absorption. Moreover, theoretical simulations and GIWAXS results reveal that the bulk mass dithieno[3,2-b:2′,3′-d]thiophene unit in MeIC1 facilitates more effective π–π stacking than the thieno[3,2-b]thiophene unit in MeIC, which is beneficial for higher charge mobility in MeIC1-based neat and blend films. Using MeIC1 as acceptors in fabricated PSCs resulted in PCEs up to 12.58% with a promoted Voc and Jsc and an unchanged FF relative to MeIC-based PSCs (12.03%). The higher Voc originates from the higher LUMO energy level of MeIC1, and the larger Jsc and unchanged FF are mainly attributed to the higher charge mobility, more perfect morphology and more ordered condensed state of MeIC1-based blend films. These results showed us that rationally designing asymmetrical isomers by introducing large packing units is an effective approach for PCE promotion.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21572171 and 61675017).
Notes and references
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131 CrossRef.
- J. Hou, O. Inganas, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119 CrossRef PubMed.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef.
- J. Zhang, Y. Li, J. Huang, H. Hu, G. Zhang, T. Ma, P. C. Y. Chow, H. Ade, D. Pan and H. Yan, J. Am. Chem. Soc., 2017, 139, 16092 CrossRef PubMed.
- Q. Wu, D. Zhao, A. M. Schneider, W. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7248 CrossRef PubMed.
- D. Meng, H. Fu, C. Xiao, X. Meng, T. Winands, W. Ma, W. Wei, B. Fan, L. Hou, N. L. Doltsinis, Y. Li, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2016, 138, 10184 CrossRef PubMed.
- H. Li, Y.-J. Hwang, B. A. E. Courtright, F. N. Eberle, S. Subramaniyan and S. A. Jenekhe, Adv. Mater., 2015, 27, 3266 CrossRef PubMed.
- Y. Lin, J. Wang, Z.-G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27, 1170 CrossRef PubMed.
- S. Li, L. Ye, W. Zhao, S. Zhang, S. Mukherjee, H. Ade and J. Hou, Adv. Mater., 2016, 28, 9423 CrossRef PubMed.
- D. Xie, T. Liu, W. Gao, C. Zhong, L. Huo, Z. Luo, K. Wu, W. Xiong, F. Liu, Y. Sun and C. Yang, Sol. RRL, 2017, 1, 1700044 CrossRef.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148 CrossRef PubMed.
- Y. Cui, H. Yao, B. Gao, Y. Qin, S. Zhang, B. Yang, C. He, B. Xu and J. Hou, J. Am. Chem. Soc., 2017, 139, 7302 CrossRef PubMed.
- Z. Xiao, X. Jia and L. Ding, Sci. Bull., 2017, 62, 1562 CrossRef.
- X. Xu, T. Yu, Z. Bi, W. Ma, Y. Li and Q. Peng, Adv. Mater., 2017, 29, 1703973 Search PubMed.
- Z. Fei, F. D. Eisner, X. Jiao, M. Azzouzi, J. A. Röhr, Y. Han, M. Shahid, A. S. R. Chesman, C. D. Easton, C. R. MeNeill, T. D. Anthopoulos, J. Nelson and M. Heeney, Adv. Mater., 2018, 30, 1705209 CrossRef PubMed.
- C. Sun, F. Pan, H. Bin, J. Zhang, L. Xue, B. Qiu, Z. Wei, Z.-G. Zhang and Y. Li, Nat. Commun., 2018, 9, 743 CrossRef PubMed.
- J. Zhu, Z. Ke, Q. Zhang, J. Wang, S. Dai, Y. Wu, Y. Xu, Y. Lin, W. Ma, W. You and X. Zhan, Adv. Mater., 2017, 29, 1704713 Search PubMed.
- Z. Luo, H. Bin, T. Liu, Z.-G. Zhang, Y. Yang, C. Zhong, B. Qiu, G. Li, W. Gao, D. Xie, K. Wu, Y. Sun, F. Liu, Y. Li and C. Yang, Adv. Mater., 2018, 30, 1706124 CrossRef PubMed.
- T. Li, S. Dai, Z. Ke, L. Yang, J. Wang, C. Yan, W. Ma and X. Zhan, Adv. Mater., 2018, 30, 1705969 CrossRef PubMed.
- S. Xu, Z. Chun, W. Liu, Z. Zhang, F. Liu, H. Yan and X. Zhu, Adv. Mater., 2017, 29, 1704510 CrossRef PubMed.
- Q. An, W. Gao, F. Zhang, J. Wang, M. Zhang, K. Wu, X. Ma, Z. Hu, C. Jiao and C. Yang, J. Mater. Chem. A, 2018, 6, 2468 RSC.
- Q. An, F. Zhang, W. Gao, Q. Sun, M. Zhang, C. Yang and J. Zhang, Nano Energy, 2018, 45, 177 CrossRef.
- F. Zhao, S. Dai, Y. Wu, Q. Zhang, J. Wang, L. Jiang, Q. Ling, Z. Wei, W. Ma, W. You, C. Wang and X. Zhan, Adv. Mater., 2017, 29, 1700144 CrossRef PubMed.
- S. Li, L. Ye, W. Zhao, X. Liu, J. Zhu, H. Ade and J. Hou, Adv. Mater., 2017, 29, 1704510 CrossRef PubMed.
- S. Dai, F. Zhao, Q. Zhang, T.-K. Lau, T. Li, K. Liu, Q. Ling, C. Wang, X. Lu, W. You and X. Zhan, J. Am. Chem. Soc., 2017, 139, 1336 CrossRef PubMed.
- H. Yao, Y. Cui, R. Yu, B. Gao, H. Zhang and J. Hou, Angew. Chem., Int. Ed., 2017, 56, 3045 CrossRef PubMed.
- W. Gao, Q. An, R. Ming, D. Xie, K. Wu, Z. Luo, Y. Zou, F. Zhang and C. Yang, Adv. Funct. Mater., 2017, 27, 1702194 CrossRef.
- Y. Yang, Z. G. Zhang, H. Bin, S. Chen, L. Gao, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 15011 CrossRef PubMed.
- H. Yao, L. Ye, J. Hou, B. Jang, G. Han, Y. Cui, G. M. Su, C. Wang, B. Gao, R. Yu, H. Zhang, Y. Yi, H. Y. Woo, H. Ade and J. Hou, Adv. Mater., 2017, 29, 1700254 CrossRef PubMed.
- B. Kan, H. Feng, X. Wan, F. Liu, X. Ke, Y. Wang, Y. Wang, H. Zhang, C. Li, J. Hou and Y. Chen, J. Am. Chem. Soc., 2017, 139, 4929 CrossRef PubMed.
- Z.-G. Zhang, Y. Yang, J. Yao, L. Xue, S. Chen, X. Li, W. Morrison, C. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 1350 Search PubMed.
- J. Wang, W. Wang, X. Wang, Y. Wu, Q. Zhang, C. Yan, W. Ma, W. You and X. Zhan, Adv. Mater., 2017, 29, 1702125 CrossRef PubMed.
- B. Kan, J. Zhang, F. Liu, X. Wan, C. Li, X. Ke, Y. Wang, H. Feng, Y. Zhang, G. Long, R. H. Friend, A. A. Bakulin and Y. Chen, Adv. Mater., 2017, 29, 1704904 Search PubMed.
- Y. Li, J.-D. Lin, X. Che, Y. Qu, F. Liu, L.-S. Liao and S. R. Forrest, J. Am. Chem. Soc., 2017, 139, 17114 CrossRef PubMed.
- S. Feng, C. Zhang, Y. Liu, Z. Bi, Z. Zhang, X. Xu, W. Ma and Z. Bo, Adv. Mater., 2017, 29, 1703527 CrossRef PubMed.
- T. J. Aldrich, S. M. Swick, F. S. Melkonyan and T. J. Marks, Chem. Mater., 2017, 29, 10294 CrossRef.
- S. Li, L. Zhan, F. Liu, J. Ren, M. Shi, C.-Z. Li, T. P. Russell and H. Chen, Adv. Mater., 2017, 29, 1705208 Search PubMed.
- W. Gao, M. Zhang, T. Liu, R. Ming, Q. An, K. Wu, D. Xie, Z. Luo, C. Zhong, F. Liu, F. Zhang, H. Yan and C. Yang, Adv. Mater., 2018, 30, 1800052 CrossRef PubMed.
- W. Gao, T. Liu, C. Zhong, G. Zhang, Y. Zhang, R. Ming, L. Zhang, J. Xin, K. Wu, Y. Guo, W. Ma, H. Yan, Y. Liu and C. Yang, ACS Energy Lett., 2018, 3, 1760 CrossRef.
- K. Jiang, G. Zhang, G. Yang, J. Zhang, Z. Li, T. Ma, H. Hu, W. Ma, H. Ade and H. Yan, Adv. Energy Mater., 2017, 29, 1701370 Search PubMed.
- H. Zhang, X. Wang, L. Yang, S. Zhang, Y. Zhang, C. He, W. Ma and J. Hou, Adv. Mater., 2017, 29, 1703777 CrossRef PubMed.
- M. Zhang, W. Gao, F. Zhang, Y. Mi, W. Yang, Q. An, J. Wang, X. Ma, J. Miao, Z. Hu, X. Liu, J. Zhang and C. Yang, Energy Environ. Sci., 2018, 11, 841 RSC.
- T. Liu, X. Xue, L. Huo, X. Sun, Q. An, F. Zhang, T. P. Russell, F. Liu and Y. Sun, Chem. Mater., 2017, 29, 2914 CrossRef.
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Röhr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. M. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant and L. McCulloch, Nat. Mater., 2017, 16, 363 CrossRef PubMed.
- H. Bin, L. Gao, Z.-G. Zhang, Y. Yang, Y. Zhang, C. Zhang, S. Chen, L. Xue, C. Yang, M. Xiao and Y. Li, Nat. Commun., 2016, 7, 13651 CrossRef PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98 CrossRef.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef PubMed.
- H. Kruse and S. Grimme, J. Chem. Phys., 2012, 136, 154101 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials synthesis, device fabrication, SCLC measurement, and 1H and 13C NMR spectra. See DOI: 10.1039/c8sc02018c |
| ‡ These two authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c and
Chuluo
Yang
*c and
Chuluo
Yang