
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
RAFT polymer cross-coupling with boronic acids†
Hartwig
Golf
a,
Riley
O'Shea
a,
Carl
Braybrook
b,
Oliver
Hutt
b,
David W.
Lupton
 *a and
Joel F.
Hooper
*a
*a and
Joel F.
Hooper
*a
aSchool of Chemistry, Monash University, Clayton, Melbourne, VIC 3800, Australia. E-mail: david.lupton@monash.edu; joel.hooper@monash.edu
bCSIRO, Research Way, Melbourne, VIC 3168, Australia
First published on 18th July 2018
Abstract
The ability to modify the thiocarbonylthio end-groups of RAFT polymers is important for applications where an inert or highly functionalised material is required. Here we report a copper promoted cross-coupling reaction between RAFT polymer end-groups and aryl boronic acids. This method gives high conversion to the modified polymers, and is compatible with a wide variety of functional molecules.
Introduction
Reversible addition fragmentation chain transfer (RAFT) polymerisation is a powerful method for the synthesis of narrow polydispersity polymers.1 Applicable to most monomers, RAFT is increasingly being deployed in sophisticated applications, such as in biomedical science.2–7 Central to such applications is the ability to prepare robust narrow dispersity polymer conjugates. For example, in 2006 Hong reported formation of polyacrylamide biotin conjugate 2 using prefunctionalised RAFT agent 1 (eqn (1)).6a,8 While this approach provided a biotin polyacrylamide conjugate it retained the thiocarbonylthio end-group, which is generally considered undesirable as it can lead to instability, discolouration, or unpleasant odours.9 An alternate strategy to the formation of polymer conjugates involves post-polymerisation functionalization (Scheme 1b). This often involves a two-step cleavage of the thiocarbonyl group to leave a free thiol,10 and coupling via thiol ene or other sulfur specific conjugations (eqn (2)).11 This common strategy suffers from its multistep nature while leaving a sulphur-containing tether, which may be prone to oxidation, elimination or exchange reactions.12 A number of functionalisations allow removal of the sulfur-containing end-group.13 In early studies, thermal eliminations were developed to generate alkene terminated polymers,14 while more recently re-initiation and quenching with a hydrogen atom source15 or alkyl radical has been developed (eqn (3)).13b,16 Recently reported desulfurisation to generate bromine terminated polymers has been described by Lunn using a 2-step aminolysis/bromination,17 while Armes used oxidative methods to give hydroxyl end-groups.18 In addition, Lunn and Sumerlin have independently exploited photoactivation to deliver hydrogen terminated polymers.19 Although existing techniques to remove sulfur have strengths, they often require multiple reactions and are only capable of introducing simple functionality.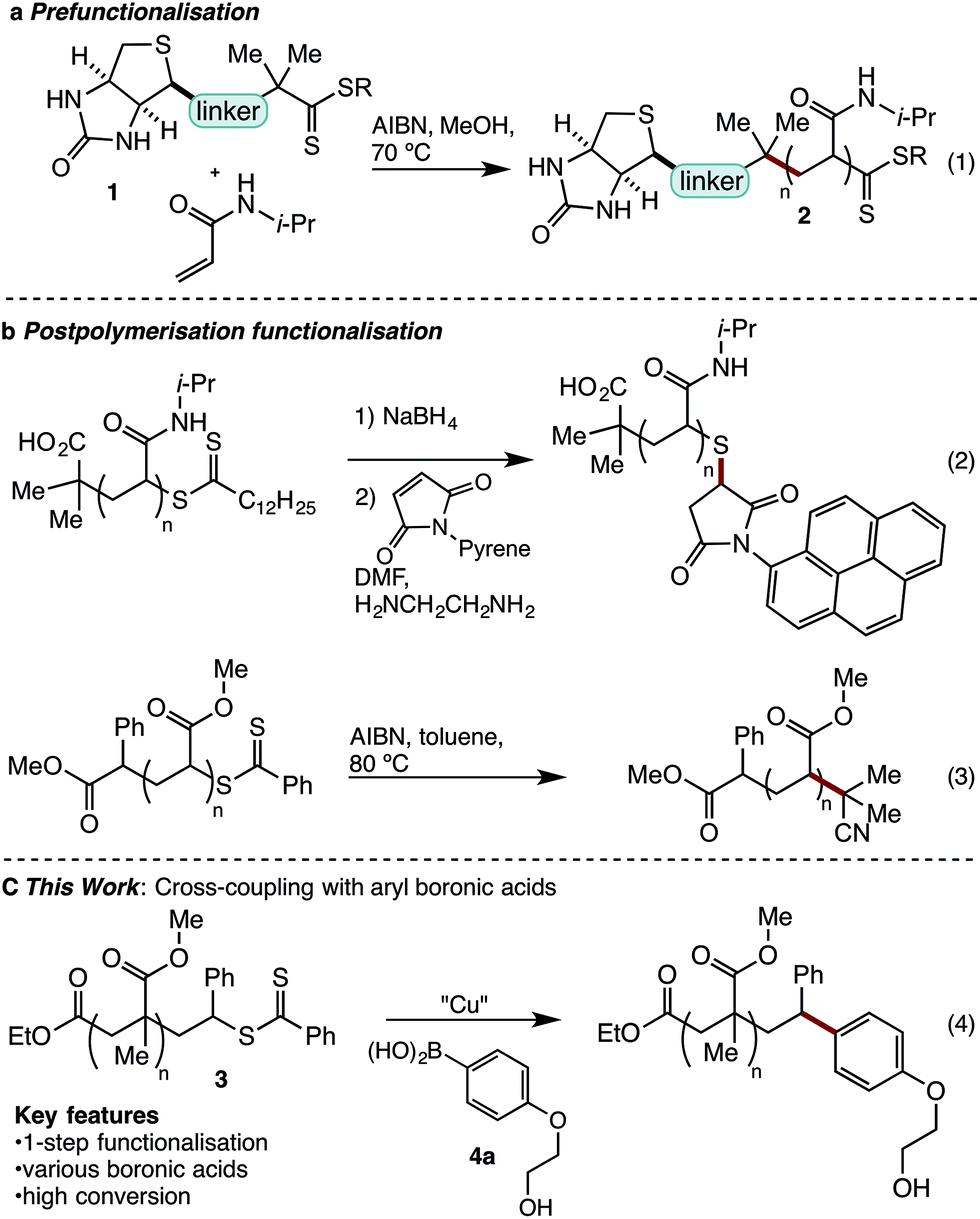 | ||
| Scheme 1 Existing approaches to RAFT end-group removal. | ||
Despite recent advances in the use of transition metals for the activation and functionalisation of C–S bonds,20 we noted with interest that this strategy has not been applied to RAFT endgroups. In principle, such a strategy should allow deletion of the sulfur end-group and introduction of new functionality via a robust C–C bond. Herein, we report studies on this topic that have led to the discovery of a Cu[II] promoted coupling of RAFT polymers 3 with aryl boronic acid 4 (eqn (4)). This method exploits readily available boronic acids, deletes the thiocarbonylthio group, and introduces the conjugate with a stable C–C bond.
Results and discussion
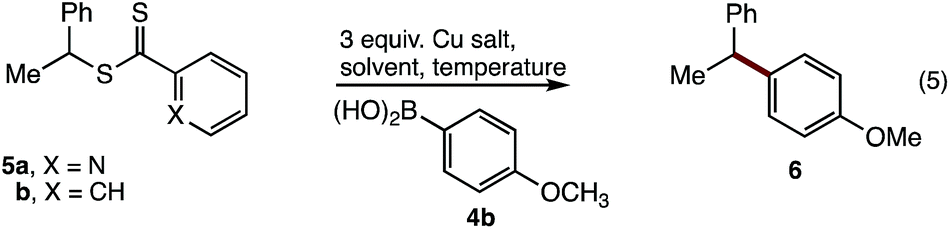
Studies began by examining the cross-coupling of small molecule polystyrene surrogates (i.e.5) with aryl boronic acid (4b) (Table 1). Pleasingly a range of Cu[0] and [II] complexes gave the desired diarylmethane 6, albeit in low yield (Table 1, entries 1–5). Cu(BF4)2·H2O proved the best copper promoter, giving the product in 35% isolated yield (Table 1, entry 5). Solvent screening identified chlorinated solvents, and particularly 1,2-dichloroethane, as suitable for this reaction lifting the yield to 54% (Table 1, entries 6–10). This outcome was improved with the simpler phenyl-thiocarbonylthio substrate (5b) (Table 1, entry 11). Finally, through attempts to dry the Cu(BF4)2·H2O salt over MgSO4 in EtOAc, an amorphous Cu(BF4)2·H2O·EtOAc adduct formed that proved more active than the Cu(BF4)2·H2O salt, leading to the isolation of diarylmethane 6 in 85% yield from the coupling of phenyl-thiocarbonylthio 5b with boronic acid 4b.|
|
|||||
|---|---|---|---|---|---|
| Entry 5 | Cu salt | Solvent | Temp | Yielda | |
| a Isolated yields. | |||||
| 1 | a | Cu(acac) | THF | 66 | 0 |
| 2 | ′′ | CuCl2·H2O | ′′ | ′′ | Trace |
| 3 | ′′ | Cu powder | ′′ | ′′ | 12 |
| 4 | ′′ | Cu(OAc)2·H2O | ′′ | ′′ | 21 |
| 5 | ′′ | Cu(BF4)2·H2O | ′′ | ′′ | 35 |
| 6 | ′′ | ′′ | Dioxane | ′′ | 12 |
| 7 | ′′ | ′′ | DMF | ′′ | 21 |
| 8 | ′′ | ′′ | Toluene | ′′ | 16 |
| 9 | ′′ | ′′ | CH2Cl2 | 40 | 42 |
| 10 | ′′ | ′′ | DCE | 66 | 54 |
| 11a | b | ′′ | DCE | 80 | 67 |
| 12a | b | Cu(BF4)2·H2O·EtOAc | ′′ | 80 | 85 |
With conditions optimised for end-group modification of a polystyrene surrogate, the cross-coupling with a low molecular weight polystyrene bearing a phenyl-thiocarbonylthio end-group (i.e.7) was examined (Fig. 1). Pleasingly, the conditions were well suited to polymer 7 and gave the polymer conjugate 8 with >95% end-group conversion, as judged by 1H NMR spectroscopy (see ESI†). The conditions were equally suited (>95% conversion) to the cross-coupling of polystyrene bearing the recently reported dimethylpyrazole end-group (i.e.9) with aryl boronic acid 4b (eqn (7)).21 The 1H NMR spectra of the resultant conjugate 10 showed clear disappearance of signals assigned to the pyrazole end-group (i.e.Ha and Hb). In addition, a broad signal at ∼3.75 ppm was seen, consistent with the methoxy group from the aryl boronic acid (Fig. 1a).
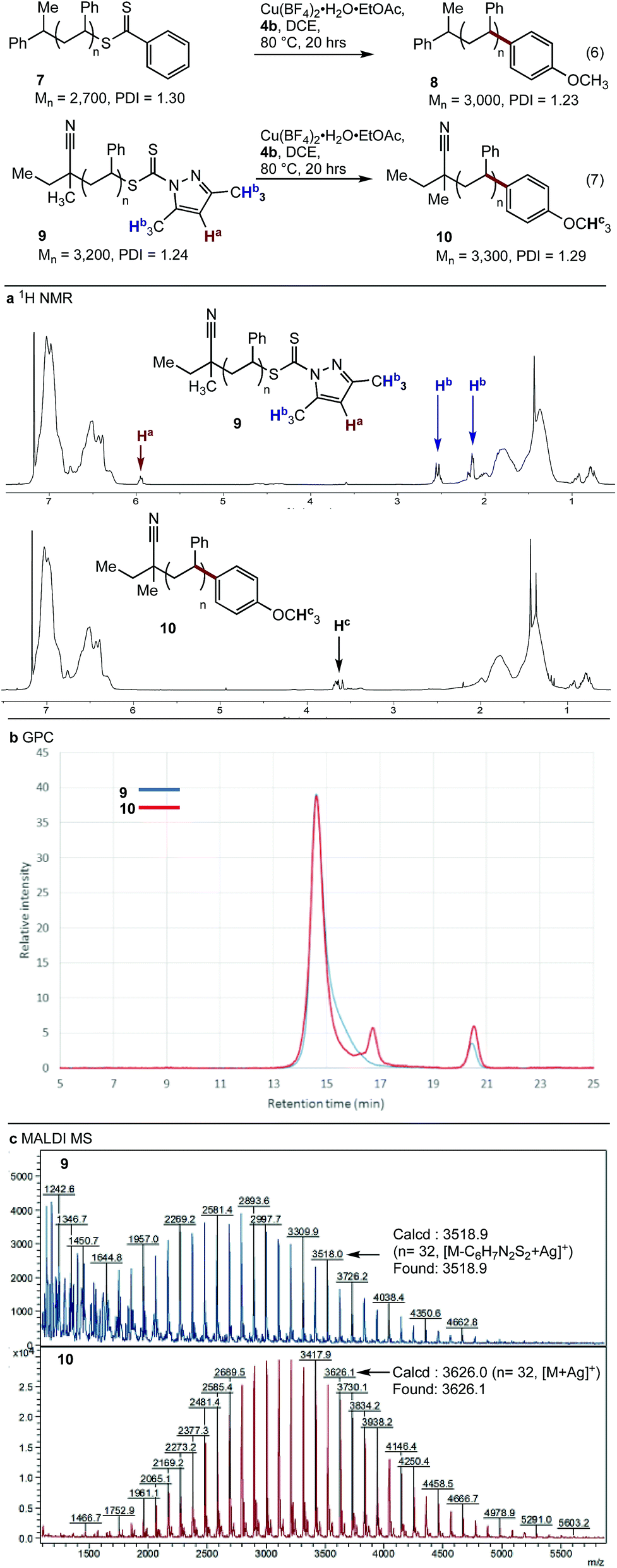 | ||
| Fig. 1 Cross-coupling of polystyrenes 7 and 9 with boronic acid 4b. | ||
As expected, analysis of polymers 9 and 10 by gel permeation chromatography (GPC) showed very little change in the molecular weight or dispersity (Fig. 1b). MALDI-MS analysis of polymers 9 and 10 clearly showed incorporation of the aryl end-group (Fig. 1c). Finally, analysis of polymer 10 by ICP-MS after only simple purification by precipitation showed a residual copper content of <10 ppm.
We next tested our coupling conditions on higher molecular weight polystyrenes 11 and 13 (Fig. 2). Coupling under our standard conditions, with 3 equivalents of Cu relative to the endgroup, resulted in incomplete conversion. However, when the equivalents of Cu and 4b were increased so as to maintain the same concentration as in the previous experiments (∼50 mg mL−1 Cu(BF4)2·H2O·EtOAc, 16 equiv for 11, 36 equiv. for 13), clear incorporation of the new methoxyphenyl end-group could be observed. While quantitative end-group analysis by 1H NMR spectroscopy is difficult at these higher molecular weights, these results clearly show that our coupling conditions are viable with these substrates, as long as the concentration of reagents is maintained at the optimised level.
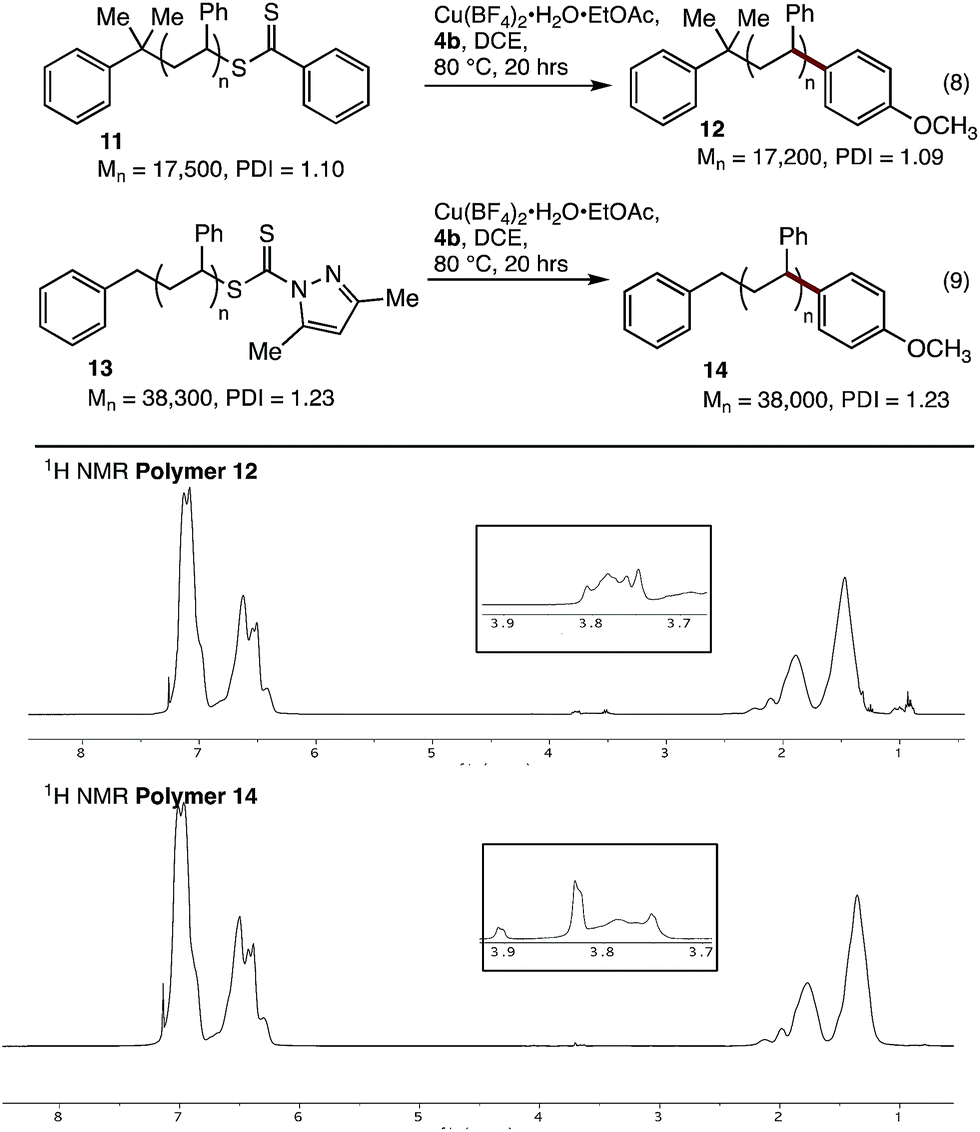 | ||
| Fig. 2 Cross-coupling of polystyrenes 11 and 13 with boronic acid 4b. | ||
Having established cross-coupling with polystyrene, the applicability of these conditions to the coupling of model polyacrylate (15) and polymethacrylate (16) small molecules with aryl boronic acid 4b was examined. Unfortunately, these reactions resulted in decomposition of the starting materials (Scheme 2), indicating that benzylic activation of the reactive site is required for effective coupling.
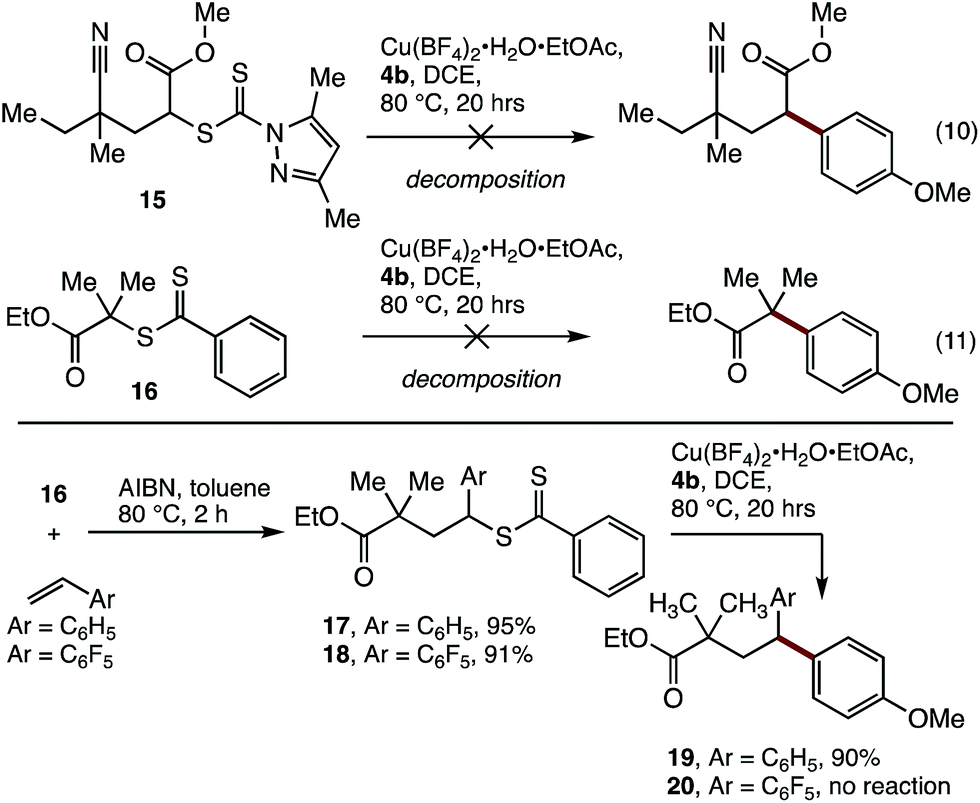 | ||
| Scheme 2 SUMI/coupling strategy for the modification of methacrylates. | ||
The requirement for benzylic activation at the reactive site is consistent with previous Cu-promoted activation of Csp3–S bonds,22 where benzylic activation is thought to stabilise a proposed carbocation intermediate. In addition, a radical-based mechanism is also possible, where copper coordination to the dithiocarbonyl group is followed by C–S bond clevage to produce a benzylic radical and concomitant single-electron oxidation of Cu. It has been previously shown that Cu[0]23 or Fe[0]24 can be used to initiate RAFT polymerisations, suggesting that Csp3 radicals can be generated under these conditions.
To address this limitation, we examined the incorporation of styrene units to the end of a polymethacrylate to modify its reactivity. The synthesis of a block co-polymer was considered, although this would complicate the characterisation of the polymer by MALDI-MS, and would limit the utility of the material produced. As an alternative, a single unit monomer insertion (SUMI)25 strategy was examined, where a single styrene unit may be inserted into the end of the polymer. This strategy would provide the required reactivity for functionalization and deliver a well-defined material for analysis, while having minimal impact on the bulk properties of the polymethacrylate.
Thus, treatment of the model substrate 16 with AIBN and two equivalents of styrene gave the SUMI surrogate 17 in excellent yield (Scheme 2). The incorporation of pentafluorostyrene was also successful (18), which was included to probe the effect of increased cation/radical stabilisation at the benzylic position. While 18 proved to be unreactive under the cross-coupling conditions, the styrene insertion product 17 underwent efficient coupling with boronic acid 4b to deliver 19 in 90% yield.26
Having demonstrated the viability of a SUMI strategy to modify the endgroup reactivity of a methacrylate surrogate, this approach was examined with polymethylmethacrylate 21. Single monomer insertion of styrene gave 22, which underwent smooth cross-coupling with boronic acid 4b, to give the endgroup modified polymethacrylate 23. While the aryl methoxy peak was obscured in the 1H NMR spectra of 23, characteristic aryl peaks could be observed at 6.7 ppm (Hd), consistent with >95% incorporation of the end group. This was further confirmed by MALDI-MS analysis (Fig. 3).
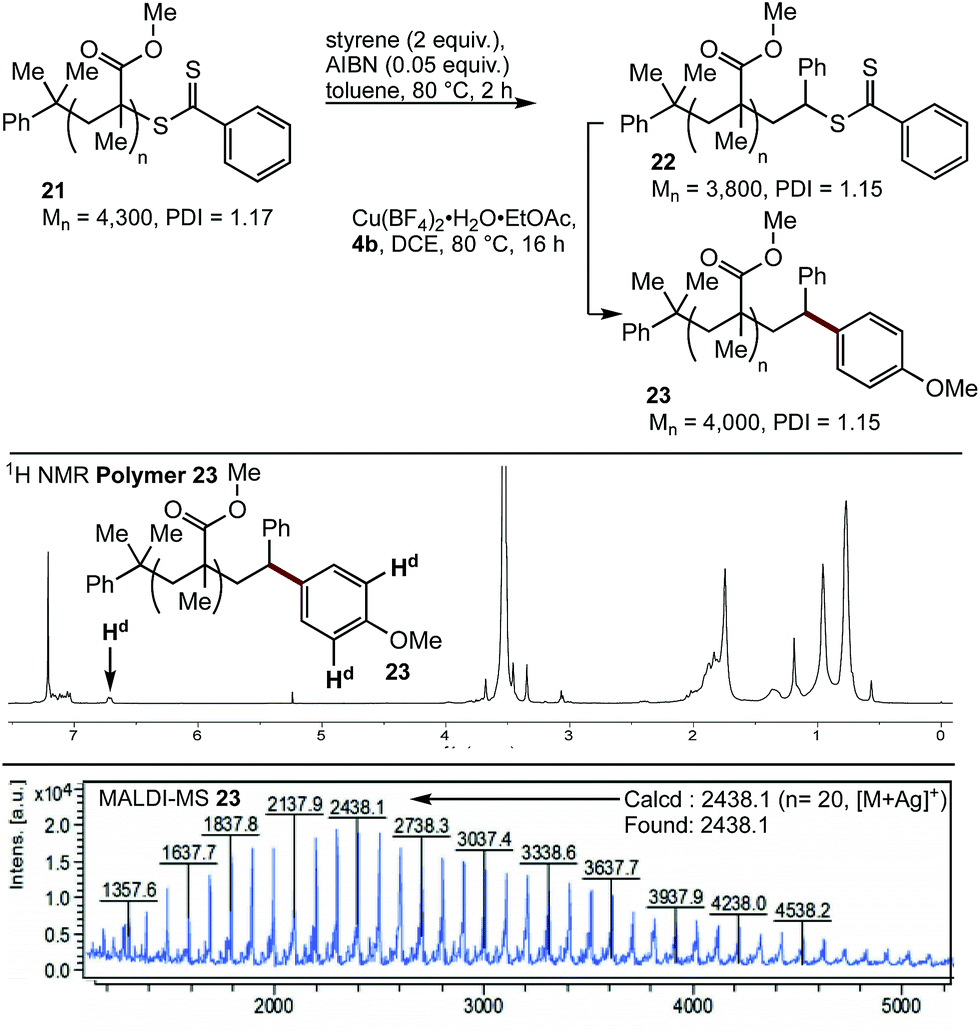 | ||
| Fig. 3 SUMI/coupling strategy for modification of polymer 22, NMR and MALDI-MS of 23. | ||
The styrene SUMI strategy was also applied to a larger polymethacrylate (Mn = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600), to generate polymer 24. Coupling of this material with the ethylene glycol substituted aryl boronic acid 4a gave the modified polymer 25 (Fig. 4). The 1H NMR spectra clearly shows the endgroups from both the ethyl ester (Hd) and the ethylene glycol groups (He and Hf, Fig. 4), while 1H DOESY NMR analysis shows that He and Hf have the same diffusion coefficient (8.4 × 10−11 m2 s−1) as the bulk polymer, indicating successful incorporation of the aryl boronic acid into the polymer chain.
600), to generate polymer 24. Coupling of this material with the ethylene glycol substituted aryl boronic acid 4a gave the modified polymer 25 (Fig. 4). The 1H NMR spectra clearly shows the endgroups from both the ethyl ester (Hd) and the ethylene glycol groups (He and Hf, Fig. 4), while 1H DOESY NMR analysis shows that He and Hf have the same diffusion coefficient (8.4 × 10−11 m2 s−1) as the bulk polymer, indicating successful incorporation of the aryl boronic acid into the polymer chain.
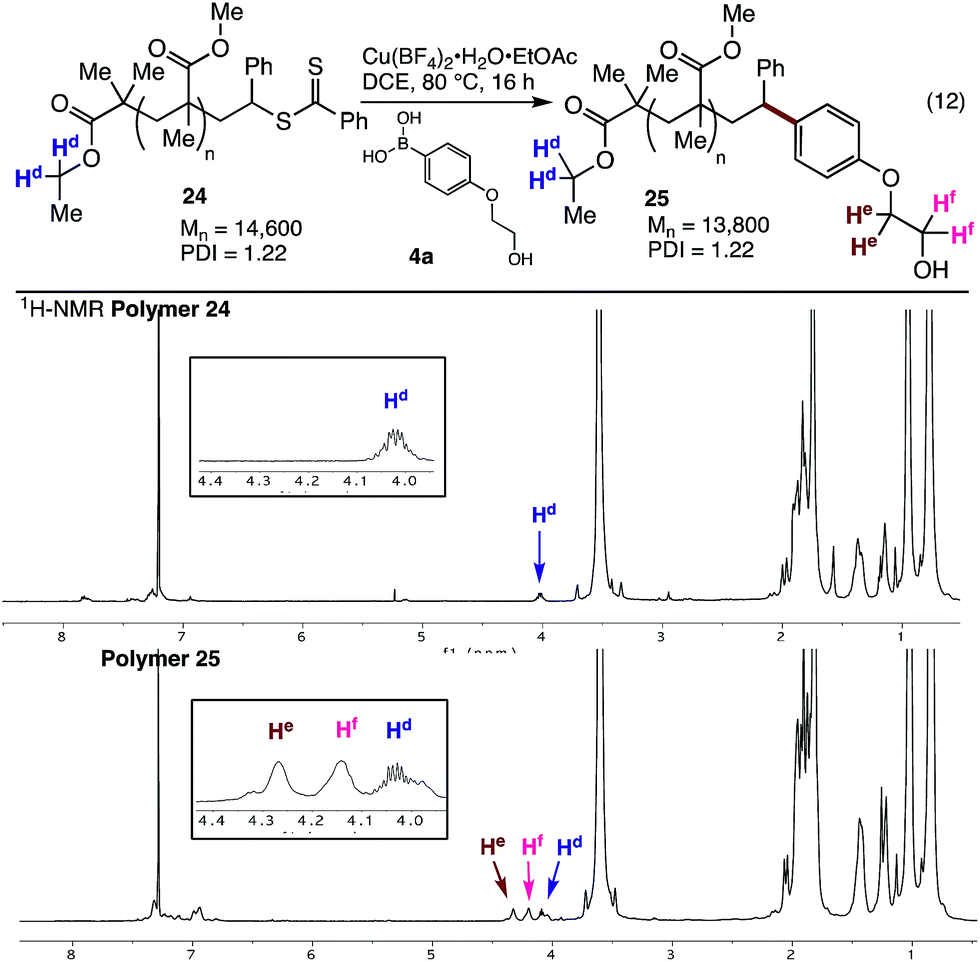 | ||
| Fig. 4 Cross-coupling with 24 and 1H NMR spectra of polymers 24 and 25. | ||
As a demonstration of the utility of this method, we coupled two highly functional molecules to methacrylate polymers (Fig. 5). Boronic acid 4a was covalently linked to a BODIPY dye, before coupling with polymer 22 to give the functionalised material 26. UV-vis analysis of the polymer shows a strong absorbance at 490–510 nm, which overlaps well with the absorbance of the BODIPY dye (Fig. 5). A similar strategy was used to synthesise polymer 27, with a biotin functional group linked to a water soluble polymethacrylate bearing a triethylene glycol methyl ether (TEG-OMe) group. Analysis of the 1H NMR spectra of this polymer shows clear signals at 4.13 and 4.40 ppm, consistent with the methylene groups of the linker, along with signals at ∼6.8 ppm which we assign to the newly introduced aryl-oxy group (see ESI†).
 | ||
| Fig. 5 Polymer functionalisation with complex functional groups. | ||
The copper-promoted cross-coupling of RAFT polymers with aryl boronic acids is a powerful method to access functionalised polymers with controlled molecular weight and low polydispersity. This methodology has been demonstrated with a variety of polymers, including polystyrenes, and several polymethacrylates. We believe that this method will aide in the discovery of new polymers for functional materials and biological applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Australian Research Council through the Discovery programs (DWL) and a DECRA fellowship (JFH) is acknowledged. JFH would like to acknowledge valuable advice from Dr Graham Moad (CSIRO), and Professors San Thang and Tanja Junkers (Monash University).References
- (a) J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef; (b) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef; (c) G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef; (d) W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef; (e) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef; (f) G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef; (g) G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef; (h) Y. Zhang, R. A. Mulvenna, S. Qu, B. W. Boudouris and W. A. Phillip, ACS Macro Lett., 2017, 6, 726–732 CrossRef; (i) A. Kerr, M. Hartlieb, J. Sanchis, T. Smith and S. Perrier, Chem. Commun., 2017, 53, 11901–11904 RSC; (j) J. Jonikaite-Svegzdiene, A. Kudresova, S. Paukstis, M. Skapas and R. Makuska, Polym. Chem., 2017, 8, 5621–5632; (k) C. Boyer, M. H. Stenzel and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 551–595; (l) S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14–22 RSC; (m) M. Baum and W. J. Brittain, Macromolecules, 2002, 35, 610–615 CrossRef.
- (a) J. Hu, R. Qiao, M. R. Whittaker, J. F. Quinn and T. P. Davis, Aust. J. Chem., 2017, 70, 1161–1170 CrossRef; (b) B. D. Fairbanks, P. A. Gunatillake and L. Meagher, Adv. Drug Delivery Rev., 2015, 91, 141–152 CrossRef PubMed; (c) S. E. York, A. W. York and C. L. McCormick, ACS Symp. Ser., 2010, 1053, 49–63 CrossRef; (d) C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402–5436 CrossRef PubMed.
- (a) S. Bandyopadhyay, X. Xia, A. Maiseiyeu, G. Mihai, S. Rajagopalan and D. Bong, Macromolecules, 2012, 45, 6766–6773 CrossRef PubMed; (b) J.-R. Chen, H.-T. Lin, G.-W. Hwang, Y.-J. Chan and P.-W. Li, Nanotechnology, 2009, 20, 255706–255709 CrossRef PubMed.
- (a) C. C. Williams, S. H. Thang, T. Hantke, U. Vogel, P. H. Seeberger, J. Tsanaktsidis and B. Lepenies, ChemMedChem, 2012, 7, 281–291 CrossRef PubMed; (b) Y. Xiao, R. Xiao, J. Tang, Q. Zhu, X. Li, Y. Xiong and X. Wu, Talanta, 2017, 162, 415–422 CrossRef PubMed; (c) L. Qiu, J.-W. Li, C.-Y. Hong and C.-Y. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 40887–40897 CrossRef PubMed; (d) B. Louage, M. J. van Steenbergen, L. Nuhn, M. D. P. Risseeuw, I. Karalic, J. Winne, S. Van Calenbergh, W. E. Hennink and B. G. De Geest, ACS Macro Lett., 2017, 6, 272–276 CrossRef; (e) Y. Bao and J. Nicolas, Polym. Chem., 2017, 8, 5174–5184 RSC; (f) B. Louage, L. Nuhn, M. D. P. Risseeuw, N. Vanparijs, R. De Coen, I. Karalic, S. Van Calenbergh and B. G. De Geest, Angew. Chem., Int. Ed., 2016, 55, 11791–11796 CrossRef PubMed; (g) A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60, 1018–1036 CrossRef PubMed; (h) M. H. Stenzel, Chem. Commun., 2008, 3486–3503 RSC.
- (a) C. Chen, F. Kong, X. Wei and S. H. Thang, Chem. Commun., 2017, 53, 10776–10779 RSC; (b) N. Isahak, G. Gody, L. R. Malins, N. J. Mitchell, R. J. Payne and S. Perrier, Chem. Commun., 2016, 52, 12952–12955 RSC; (c) R. Chapman, K. A. Jolliffe and S. Perrier, Polym. Chem., 2011, 2, 1956–1963; (d) J. Hentschel, K. Bleek, O. Ernst, J.-F. Lutz and H. G. Boerner, Macromolecules, 2008, 41, 1073–1075 CrossRef; (e) M. G. J. ten Cate, H. Rettig, K. Bernhardt and H. G. Boerner, Macromolecules, 2005, 38, 10643–10649 CrossRef.
- (a) C.-Y. Hong and C.-Y. Pan, Macromolecules, 2006, 39, 3517–3524 CrossRef; (b) M. Bathfield, F. D'Agosto, R. Spitz, M.-T. Charreyre and T. Delair, J. Am. Chem. Soc., 2006, 128, 2546–2547 CrossRef PubMed.
- (a) M. M. Herth, M. Barz, D. Moderegger, M. Allmeroth, M. Jahn, O. Thews, R. Zentel and F. Rosch, Biomacromolecules, 2009, 10, 1697–1703 CrossRef PubMed; (b) D. Pissuwan, C. Boyer, K. Gunasekaran, T. P. Davis and V. Bulmus, Biomacromolecules, 2010, 11, 412–420 CrossRef PubMed.
- (a) S. Martens, F. Driessen, S. Wallyn, O. Turunc, F. E. Du Prez and P. Espeel, ACS Macro Lett., 2016, 5, 942–945 CrossRef; (b) M. Chen, K. P. Ghiggino, A. W. H. Mau, E. Rizzardo, W. H. F. Sasse, S. H. Thang and G. J. Wilson, Macromolecules, 2004, 37, 5479–5481 CrossRef; (c) D. S. W. Benoit, S. Srinivasan, A. D. Shubin and P. S. Stayton, Biomacromolecules, 2011, 12, 2708–2714 CrossRef PubMed.
- J. Xu, J. He, D. Fan, W. Tang and Y. Yang, Macromolecules, 2006, 39, 3753–3759 CrossRef.
- (a) J. Mazzolini, I. Mokthari, R. Briquel, O. Boyron, F. Delolme, V. Monteil, D. Bertin, D. Gigmes, F. D'Agosto and C. Boisson, Macromolecules, 2010, 43, 7495–7503 CrossRef; (b) P. J. Roth, D. Kessler, R. Zentel and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3118–3130 CrossRef; (c) P. J. Roth and P. Theato, Polym. Prepr., 2010, 51, 746–747 Search PubMed; (d) W. Shen, Q. Qiu, Y. Wang, M. Miao, B. Li, T. Zhang, A. Cao and Z. An, Macromol. Rapid Commun., 2010, 31, 1444–1448 CrossRef PubMed.
- (a) A. B. Lowe, RSC Polym. Chem. Ser., 2013, 6, 28–58 Search PubMed; (b) A. B. Lowe, Polym. Chem., 2014, 5, 4820–4870 RSC; (c) H. W. Ooi, K. S. Jack, A. K. Whittaker and H. Peng, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4626–4636 Search PubMed; (d) C. W. Scales, A. J. Convertine and C. L. McCormick, Biomacromolecules, 2006, 7, 1389–1392 CrossRef PubMed; (e) J. M. Spruell, B. A. Levy, A. Sutherland, W. R. Dichtel, J. Y. Cheng, J. F. Stoddart and A. Nelson, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 346–356 CrossRef; (f) M. M. Stamenovic, P. Espeel, W. Van Camp and F. E. Du Prez, Macromolecules, 2011, 44, 5619–5630 CrossRef; (g) K. J. Sykes, S. Harrisson and D. J. Keddie, Macromol. Chem. Phys., 2016, 217, 2310–2320 CrossRef; (h) N. Toda, S. Asano and C. F. Barbas, III, Angew. Chem., Int. Ed., 2013, 52, 12592–12596 CrossRef PubMed; (i) Y. Wu, Y. Zhou, J. Zhu, W. Zhang, X. Pan, Z. Zhang and X. Zhu, Polym. Chem., 2014, 5, 5546–5550 RSC; (j) Q. Zhang, L. Voorhaar, R. Hoogenboom and G. De Geest Bruno, Macromol. Rapid Commun., 2015, 36, 1177–1183 CrossRef PubMed.
- (a) S. P. S. Koo, M. M. Stamenovic, R. A. Prasath, A. J. Inglis, F. E. Du Prez, C. Barner-Kowollik, W. Van Camp and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1699–1713 CrossRef; (b) M. H. Stenzel, ACS Macro Lett., 2013, 2, 14–18 CrossRef.
- (a) G. Moad, E. Rizzardo and S. H. Thang, Polym. Int., 2011, 60, 9–25 CrossRef; (b) S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef; (c) H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- (a) C. H. Hornung, A. Postma, S. Saubern and J. Chiefari, Polymer, 2014, 55, 1427–1435 CrossRef; (b) X. Zhang, J. Jiang and Y. Zhang, J. Appl. Polym. Sci., 2016, 133, 43992 Search PubMed; (c) Y. Liu, J. He, J. Xu, D. Fan, W. Tang and Y. Yang, Macromolecules, 2005, 38, 10332–10335 CrossRef.
- Y. K. Chong, G. Moad, E. Rizzardo and S. Thang, Macromolecules, 2007, 40, 4446–4455 CrossRef.
- (a) M. Chen, G. Moad and E. Rizzardo, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6704–6714 CrossRef; (b) B. J. Kim, S. Given-Beck, J. Bang, C. J. Hawker and E. J. Kramer, Macromolecules, 2007, 40, 1796–1798 CrossRef.
- I.-H. Lee, E. H. Discekici, S. L. Shankel, A. Anastasaki, J. Read de Alaniz, C. J. Hawker and D. J. Lunn, Polym. Chem., 2017, 8, 7188–7194 RSC.
- C. P. Jesson, C. M. Pearce, H. Simon, A. Werner, V. J. Cunningham, J. R. Lovett, M. J. Smallridge, N. J. Warren and S. P. Armes, Macromolecules, 2017, 50, 182–191 CrossRef.
- (a) R. N. Carmean, C. A. Figg, G. M. Scheutz, T. Kubo and B. S. Sumerlin, ACS Macro Lett., 2017, 6, 185–189 CrossRef; (b) E. H. Discekici, S. L. Shankel, A. Anastasaki, B. Oschmann, I.-H. Lee, J. Niu, A. J. McGrath, P. G. Clark, D. S. Laitar, J. Read de Alaniz, C. J. Hawker and D. J. Lunn, Chem. Commun., 2017, 53, 1888–1891 RSC.
- (a) J. F. Hooper, A. B. Chaplin, C. Gonzalez-Rodriguez, A. L. Thompson, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 2906–2909 CrossRef PubMed; (b) J. F. Hooper, R. D. Young, I. Pernik, A. S. Weller and M. C. Willis, Chem. Sci., 2013, 4, 1568–1572 RSC; (c) J. F. Hooper, R. D. Young, A. S. Weller and M. C. Willis, Chem.–Eur. J., 2013, 19, 3125–3130 CrossRef PubMed; (d) L. Wang, W. He and Z. Yu, Chem. Soc. Rev., 2013, 42, 599–621 RSC; (e) L. S. Liebeskind and J. Srogl, J. Am. Chem. Soc., 2000, 122, 11260–11261 CrossRef.
- J. Gardiner, I. Martinez-Botella, J. Tsanaktsidis and G. Moad, Polym. Chem., 2016, 7, 481–492 RSC.
- G. Picazo, A. Arvalo, S. Bernes and J. J. Garcia, Organometallics, 2003, 22, 4734–4738 CrossRef.
- Q. Zhang, Z. Zhang, W. Wang, J. Zhu, Z. Cheng, N. Zhou, W. Zhang and X. Zhu, J. Polym. Sci. Part A, 2012, 50, 1424–1433 CrossRef.
- Z. Zhang, W. Wang, Z. Cheng, J. Zhu, N. Zhou, Y. Yang, Y. Tu and X. Zhu, Macromolecules, 2010, 43, 7979–7984 CrossRef.
- (a) J. Vandenbergh, G. Reekmans, P. Adriaensens and T. Junkers, Chem. Sci., 2015, 6, 5753–5761 RSC; (b) J. Xu, S. Shanmugam, C. Fu, K.-F. Aguey-Zinsou and C. Boyer, J. Am. Chem. Soc., 2016, 138, 3094–3106 CrossRef PubMed; (c) J. Xu, C. Fu, S. Shanmugam, C. J. Hawker, G. Moad and C. Boyer, Angew. Chem., Int. Ed., 2017, 56, 8376–8383 CrossRef PubMed.
- The effect of monomer-type and RAFT agent on end-group reactivity is well established. For example, the range of temperatures required to thermalise RAFT end-group across polybutylacrylate (PBA), Polystyrene (PS), Polymethylmethacrylate (PMMA). See: (a) B. Chong, G. Moad, E. Rizzardo, M. Skidmore and S. H. Thang, Aust. J. Chem., 2006, 59, 755–762 CrossRef; (b) A. Postma, T. P. Davis, R. A. Evans, G. Li, G. Moad and M. S. O'Shea, Macromolecules, 2006, 39, 5293–5306 CrossRef; (c) A. Postma, T. P. Davis, G. Li, G. Moad and M. S. O'Shea, Macromolecules, 2006, 39, 5307–5318 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra of all new compounds, GPC traces, MALDI-MS. See DOI: 10.1039/c8sc01862f |
| This journal is © The Royal Society of Chemistry 2018 |