
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of cyclic chiral α-amino boronates by copper-catalyzed asymmetric dearomative borylation of indoles†
Lili
Chen
a,
Jun-Jian
Shen
a,
Qian
Gao
a and
Senmiao
Xu
 *ab
*ab
aState Key Laboratory for Oxo Synthesis and Selective Oxidation, Centre for Excellence in Molecular Synthesis, Suzhou Research Institute, Lanzhou Institute of Chemical Physics, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Lanzhou 730000, China. E-mail: senmiaoxu@licp.cas.cn
bKey Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University, Hangzhou 311121, China
First published on 12th June 2018
Abstract
A copper(I)-catalyzed dearomative borylation of N-alkoxycarbonyl protected indole-3-carboxylates has been developed. The boron addition in this reaction occurred regioselectively at the 2-position of indoles followed by diastereoselective protonation, affording the corresponding stable cyclic chiral α-amino boronates (2-borylindolines) in moderate to good yields with excellent diastereo- and enantioselectivities. The product 2c could be used as a versatile precursor to undergo subsequent stereoselective transformations, delivering highly functionalized 2,3,3-trisubstituted chiral indolines.
The importance of chiral α-amino boronic acid derivatives has been demonstrated in pharmaceutically useful protease inhibitors such as bortezomib,1 delanzomib,2 and ixazomib.3 In addition, their use in transition-metal-catalyzed stereospecific C–C bond forming reactions has also gained growing attention.4 Therefore, significant efforts have been devoted to the development of efficient methods to synthesize chiral α-amino boronate esters.5 Most methods rely on a diastereoselective synthesis involving a stoichiometric amount of chiral auxiliaries.6 The recently emerged transition-metal-catalyzed asymmetric borylations by Fernández, Morken, Lin, Liao, Miura, Tang, Parra and Tortosa, and our group also provide efficient methods to access a number of acyclic chiral α-amino boronate esters.7 In contrast, the direct catalytic asymmetric borylation towards cyclic chiral α-amino boronate esters remains elusive,8 although some of these molecules have shown promising bioactivities such as dipeptidyl peptidase-4 (DPP-4) inhibitors, e.g., talabostat and dutogliptin.9
Dearomatization reactions have emerged as powerful approaches to convert readily available planar aromatic compounds into a plethora of three dimensional, highly functionalized cyclic products.10 Among them, dearomative borylation involving N-heteroarenes has gained increasing attention recently as it can provide saturated or partially saturated borylated N-heterocycles that are important building blocks for the synthesis of natural and bioactive compounds. Pioneered by Hill and Suginome,11 many systems including transition-metal catalysis and organocatalysis have been developed to achieve high chemo- and regioselectivity in this area.12 The successes of most aforementioned reactions are probably due to the formation of stable N–B bonds.11b In stark contrast, only a few examples of asymmetric transformations have been documented. In 2015, the Ito group reported a copper-catalyzed asymmetric protoboration of 2-substituted indoles, delivering 3-borylindolines with high regio-, diastereo-, and enantioselectivity (Fig. 1a).13 Subsequently, they developed one-pot sequential dearomative reduction/asymmetric borylation of pyridines and quinolines.14 The reaction produced C3 borylated chiral piperidine derivatives with high diastereo- and enantioselectivity (Fig. 1a). Tang and coworkers recently reported chiral diboron templated dearomative reductive coupling of isoquinolines involving a diastereoselective concerted [3,3]-sigma rearrangement along with the formation of two N–B bonds (Fig. 1b).15 It is quite surprising that the direct asymmetric boryl addition to the carbon adjacent to the nitrogen of N-heteroarenes remains elusive although numerous examples have been shown with carbon nucleophiles.16 The lack of research probably arises from the instability of the product.14a However, the asymmetric nucleophilic addition of a boryl group at N-adjacent carbon could offer a straightforward method that leads to cyclic chiral α-amino boronate esters. Particularly, asymmetric dearomative borylation of the 2-position of 3-substituted indoles could also furnish potentially useful chiral 2,3-disubstituted indolines that may serve as key building blocks in drug discovery and natural product synthesis. In this communication, we disclose a copper(I)-catalyzed asymmetric dearomative borylation of N-alkoxycarbonyl protected indole-3-carboxylates by way of a borylcopper(I) species (Fig. 1c).17 The boron addition takes place regioselectively at the 2-position followed by diastereoselective protonation, affording a series of indoline-based cyclic chiral α-amino boronate esters (2-borylindolines) with high diastereo- and enantioselectivity. Stereospecific transformations of the C–B bond of chiral 2-borylindoline have also been demonstrated.
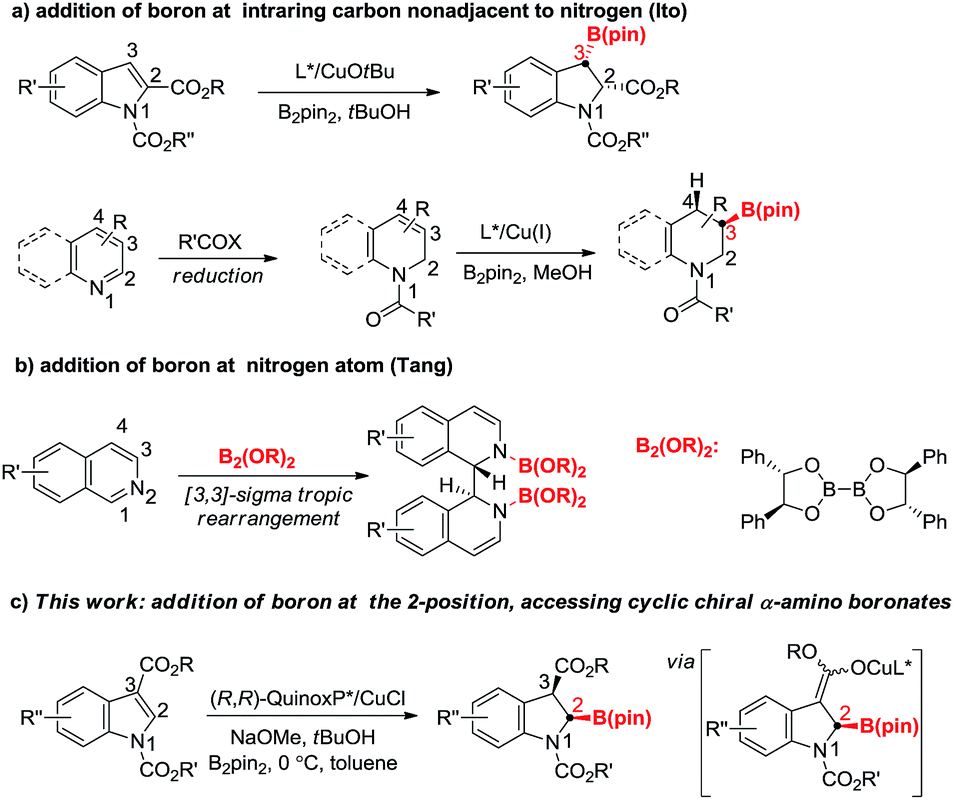 | ||
| Fig. 1 Recent advances in asymmetric dearomative borylation of N-heteroarenes. | ||
To test our hypothesis, we began our reaction with an indole substrate with different combinations of substituents at 1 and 3 positions. The initial results showed that in the presence of dppe/CuCl (10 mol%), NaOtBu (10 mol%) and tBuOH (2.0 equiv.), the reaction of N-alkoxycarbonyl methyl indole-3-carboxylates with bis(pinacolato)diboron (B2pin2) in THF at room temperature for 18 hours gave a significant amount of isolable cis-2-borylindoline whereas the other diastereomer was not stable towards purification.18 Particularly, the N-Boc methyl indole-3-carboxylate 1a gave the cis-isomer preferentially. With 1a in hand, we then turned our attention to the asymmetric version of this reaction. The reaction of 1a with B2pin2 in the presence of 10 mol% of the axially chiral ligand (S)-BINAP (L1) or bulky (R)-DTBM-SEGPHOS (L2) only gave a trace amount of the product (Table 1, entries 1 and 2). Fortunately, when the electron-rich ligand (R,R)-DuPhos (L3) was used, an appreciable amount of cis-product 2a was obtained with an excellent ee value (92%) albeit with almost no dia-stereoselectivity (45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :55) (Table 1, entry 3). Encouraged by this, several electron-rich bidentate phosphines were investigated. For example, the use of (R,R)-Me-BPE resulted in a product with elevated diastereoselectivity (80:20) but decreased enantioselectivity (80%) compared to L3 (Table 1, entry 4). Gratifyingly, when the bulky electron-rich ligand (R,R)-QuinoxP* (L5) was used, the reaction proceeded smoothly, affording cis-2-borylindoline 2a in 90% yield with good stereoselectivity (86% ee, 92:8 d.r.; Table 1, entry 5). The size of R in the ester moiety also played an important role in controlling the stereoselectivity. For example, when R was ethyl (2b), an enhanced stereoselectivity was observed (91% ee, >98:2 d.r.; Table 1, entry 6). With the use of a substrate with R = iPr, the corresponding 2-borylindoline 2c could be obtained with 94% ee and good diastereoselectivity (94:6 d.r.; Table 1, entry 7). Further increasing the size of R such as tBu (2d) led to a diminished ee value (81%) and yield (72%) whereas good diastereoselectivity (97:3 d.r.; Table 1, entry 8) was maintained. Although the other applied alcohols such as MeOH, EtOH or iPrOH gave products with excellent enantiomeric excesses (96–97%), only moderate d.r. values (50:50–70:30) were achieved. When the reaction of 2c was carried out at 0 °C, the product was obtained with a slightly enhanced stereoselectivity (95% ee, 95:5 d.r.; Table 1, entry 12).
:55) (Table 1, entry 3). Encouraged by this, several electron-rich bidentate phosphines were investigated. For example, the use of (R,R)-Me-BPE resulted in a product with elevated diastereoselectivity (80:20) but decreased enantioselectivity (80%) compared to L3 (Table 1, entry 4). Gratifyingly, when the bulky electron-rich ligand (R,R)-QuinoxP* (L5) was used, the reaction proceeded smoothly, affording cis-2-borylindoline 2a in 90% yield with good stereoselectivity (86% ee, 92:8 d.r.; Table 1, entry 5). The size of R in the ester moiety also played an important role in controlling the stereoselectivity. For example, when R was ethyl (2b), an enhanced stereoselectivity was observed (91% ee, >98:2 d.r.; Table 1, entry 6). With the use of a substrate with R = iPr, the corresponding 2-borylindoline 2c could be obtained with 94% ee and good diastereoselectivity (94:6 d.r.; Table 1, entry 7). Further increasing the size of R such as tBu (2d) led to a diminished ee value (81%) and yield (72%) whereas good diastereoselectivity (97:3 d.r.; Table 1, entry 8) was maintained. Although the other applied alcohols such as MeOH, EtOH or iPrOH gave products with excellent enantiomeric excesses (96–97%), only moderate d.r. values (50:50–70:30) were achieved. When the reaction of 2c was carried out at 0 °C, the product was obtained with a slightly enhanced stereoselectivity (95% ee, 95:5 d.r.; Table 1, entry 12).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | 1: R | Yieldb (%) | d.r.c | eed (%) |
| a Unless otherwise noted, all the reactions were carried out with 1 (0.2 mmol), L (0.02 mol), CuCl (0.02 mmol), NaOMe (0.02 mmol), alcohol (0.4 mmol), and B2pin2 (0.3 mmol) in toluene (1 mL) at 25 °C for 16 h. b The yield of isolated cis-product 2. c The diastereoselective ratio (cis/trans) was determined by 1H NMR of crude reaction mixtures. d The enantiomeric excess was determined by HPLC on a chiral IE column. e MeOH was used instead of tBuOH. f EtOH was used instead of tBuOH. g iPrOH was used instead of tBuOH. h The reaction was carried out at 0 °C for 18 h. | |||||
| 1 | L1 | 1a: Me | Trace | n.d. | n.d. |
| 2 | L2 | 1a: Me | Trace | n.d. | n.d. |
| 3 | L3 | 1a: Me | 44 | 45:55 |
92 |
| 4 | L4 | 1a: Me | 60 | 80:20 |
80 |
| 5 | L5 | 1a: Me | 90 | 92:8 |
86 |
| 6 | L5 | 1b: Et | 86 | >98:2 |
91 |
| 7 | L5 | 1c: iPr | 93 | 94:6 |
94 |
| 8 | L5 | 1d: tBu | 72 | 97:3 |
81 |
| 9e | L5 | 1c: iPr | 46 | 50:50 |
96 |
| 10f | L5 | 1c: iPr | 55 | 63:37 |
97 |
| 11g | L5 | 1c: iPr | 56 | 70:30 |
97 |
| 12h | L5 | 1c: iPr | 85 | 95:5 |
95 |
|
|||||
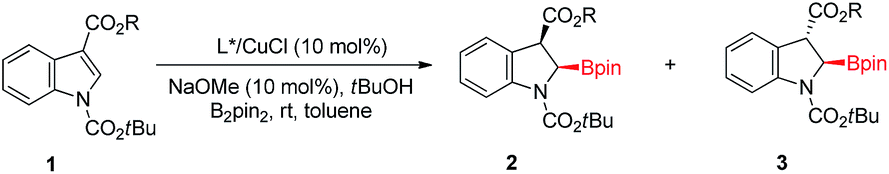
With the optimized reaction conditions (Table 1, entry 12) in hand,19 we then explored the substrate scope of this reaction as illustrated in Fig. 2. Generally, the N-protecting group affected enantioselectivity significantly, with less influence on diastereoselectivity. For example, the smaller groups MeOCO and Cbz provided inferior results (78% and 68% ee, respectively; Fig. 2e and f) compared to the substrate with bulkier Boc (86% ee; Fig. 2a). The size of the ester at the 3-position of indole also played a pivotal role in chiral induction. The reaction of the substrate with R = iPr (1c) afforded corresponding cis-2-borylindoline (2c) with a superior ee value (95%) compared to those with R = Me (2a, 86% ee), Et (2b, 93% ee) and tBu (2d, 91% ee). In most cases, the reaction of N-Boc isopropyl indole-3-carboxylate 1 resulted in good yields (81–93%) with uniformly excellent stereoselectivities (92–96% ee, ≥94:6 d.r.). The use of an electron-withdrawing group such as F or cyano at the 5-position in 1 afforded a decreased yield (67% and 50%, respectively) and stereoselectivity (2k: 93% ee, 85:15 d.r.; 2m: 85% ee, 71:29 d.r.). Interestingly, when 7-bromo indole 1t was employed, the reaction gave trans-product 3t predominantly (cis/trans = 16:84) with 78% ee. The destruction of coplanarity of Boc and indole caused by steric repulsion between bromo and Boc may give rise to reversed diastereoselectivity. The proton might approach the copper O-bound enolate intermediate from the opposite side of Boc's tBu group that would be in the trans position of the boryl group, thereby leading to trans-2-borylindoline 3t as the major product. 2-Methylindole (1u) failed to yield any product (2u or 3u). The reaction of 3-cyano indole 1v could also give a cis-product in good yield with reasonable stereoselectivity (2v: 92:8 d.r., 73% ee). However, when the EWG was formyl or acetyl, only a labile trans-product was observed (3w and 3x). The absolute configuration of 2s was determined to be 2R, 3R by X-ray analysis.20 The configurations of the other products were provisionally assigned as the same by analogy. Because the proton at the 3 position of product 2 is relatively acidic, we tested the stability of its stereochemistry. The results of control experiments clearly show that no isomerization was observed when 2c was subjected to reaction conditions at 40 °C for 18 hours or in its CDCl3 solution at room temperature for 24 hours (see the ESI† for more information).
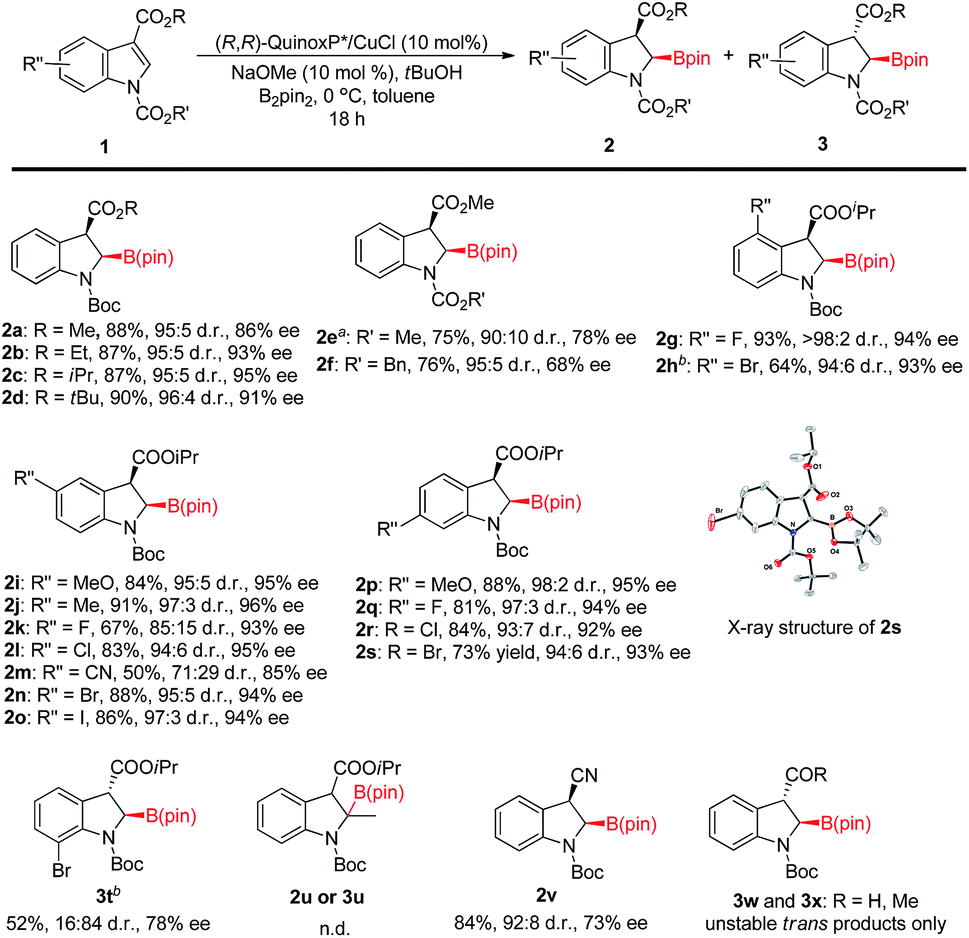 | ||
| Fig. 2 Substrate scope of reaction. Unless otherwise noted, all the reactions were carried out with 1 (0.2 mmol), (R,R)-QuinoxP* (0.02 mol), CuCl (0.02 mmol), NaOMe (0.02 mmol), tBuOH (0.4 mmol), and B2pin2 (0.3 mmol) in toluene (1 mL) at 0 °C for 18 h. The d.r. values (cis/trans) were determined by 1H NMR of crude reaction mixtures. The enantiomeric excesses were determined by chiral HPLC. aThe d.r. value was determined by GC of crude reaction mixtures. bThe reaction time was 48 hours. | ||
To demonstrate the practicality of our method, a gram-scale reaction and synthetic applications of 2c were performed as illustrated in Fig. 3. Firstly, the current method could be amendable to the gram-scale with reduced catalyst loading (2.5 mol%) and elevated temperature. The reaction of 1c (1.21 grams, 4.0 mmol) at room temperature for 18 hours gave corresponding 2-borylindoline 2c (1.64 grams, 3.8 mmol) in 95% yield with excellent stereoselectivity (97:3 d.r. and 96% ee). The acidity of the C3 proton allows further functionalization at this position. The deprotonation of 2c with LDA at −78 °C in THF followed by the addition of electrophiles afforded 2,3,3-trisubstituted 2-borylindolines 4 in good yields with good stereoselectivities.21 The C–B bond in 4a could be transformed to a C–O bond in the presence of NaBO3. After benzoylation, the corresponding indolin-2-yl benzoate 5 was obtained in 60% overall yield (2 steps) with 95% ee. The C–B bond in 4a could also undergo stereospecific C–C bond forming reactions. For example, the reaction of 4a with vinylMgBr followed by the sequential addition of methanolic solution of I2 and NaOMe could provide 2-vinylindoline 6 in 98% yield with 95% ee.22 In addition, the arylation of 4a with furyl-2-lithium followed by the addition of NBS was able to produce 2-(2-furyl)-indoline 7 in 40% yield with 95% ee.23
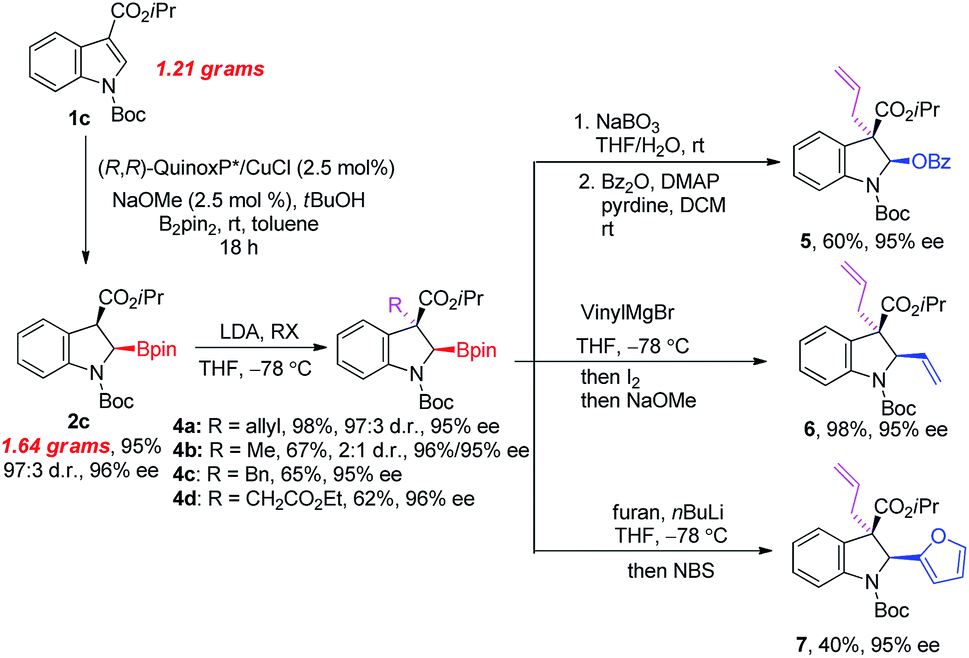 | ||
| Fig. 3 Gram-scale synthesis and transformations of 2-borylindoline 2c. | ||
The plausible reaction mechanism for the current copper(I)-catalyzed dearomative borylation of 3-substituted indoles is depicted in Fig. 4. Because the borylation only worked for indole with an EWG at its 3-position, the reaction should proceed in a similar way to the copper-catalyzed conjugate boration of α,β-unsaturated carbonyl compounds.24 The reaction of LCu-OtBu (A) with B2pin2 would generate active species borylcopper(I) B. The coordination of complex B to the C2–C3 π bond of indole 1c followed by the subsequent syn-addition of the Cu–B bond to the C2–C3 π bond would give C-bound enolate D. The protolytic cleavage of the copper–carbon bond of D by tBuOH would result in trans-product 3c, which is not consistent with the experimental outcome. To release large steric congestion between the Bpin group and LCu, D would isomerize into O-bound enolate E.25 To avoid the steric repulsion between the Bpin group and bulky tBuOH, the protonation of E would take place from the opposite side of Bpin to liberate cis-product 2c and A for the next catalytic cycle.
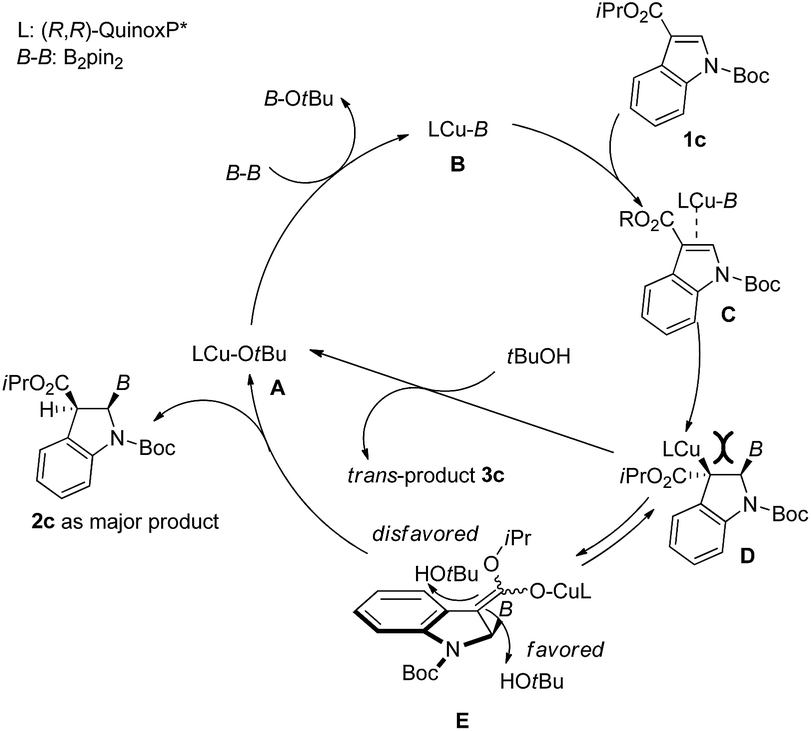 | ||
| Fig. 4 Plausible reaction pathway of the current dearomative borylation. | ||
Conclusions
In conclusion, we have developed a copper-catalyzed asymmetric dearomative borylation of N-alkoxycarbonyl protected indole-3-carboxylates under mild reaction conditions, providing a straightforward method to achieve cyclic chiral α-amino boronate esters with high diastereo- and enantioselectivity. The obtained products could undergo subsequent stereoselective transformations, affording highly functionalized 2,3,3-trisubstituted chiral indolines. This method provides not only a route to cyclic chiral α-amino boronate esters but also a series of versatile chiral precursors for chiral indoline synthesis. The further application of chiral 2-borylindolines and the development of other dearomative process are currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the 1000-Youth Talents Plan, a Start-up Grant from the Lanzhou Institute of Chemical Physics, National Natural Science Foundation of China (21573262) and Natural Science Foundation of Jiangsu Province (BK20161259, BK20170422) for generous financial support.Notes and references
- (a) J. Adams and M. Kauffman, Cancer Invest., 2004, 22, 304 CrossRef PubMed; (b) L. R. Dick and P. E. Fleming, Drug Discovery Today, 2010, 15, 243 CrossRef PubMed.
- (a) E. Gallerani, M. Zucchetti, D. Brunelli, E. Marangon, C. Noberasco, D. Hess, A. Delmonte, G. Martinelli, S. Böhm, C. Driessen, F. De Braud, S. Marsoni, R. Cereda, F. Sala, M. D'Incalci and C. Sessa, Eur. J. Cancer, 2013, 49, 290 CrossRef PubMed; (b) R. C. Roemmele and M. A. Christie, Org. Process Res. Dev., 2013, 17, 422 CrossRef.
- M. Gentile, M. Offidani, E. Vigna, L. Corvatta, A. G. Recchia, L. Morabito, F. Morabito and S. Gentili, Expert Opin. Invest. Drugs, 2015, 24, 1287 CrossRef PubMed.
- (a) T. Ohmura, T. Awano and M. Suginome, J. Am. Chem. Soc., 2010, 132, 13191 CrossRef PubMed; (b) T. Awano, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2011, 133, 20738 CrossRef PubMed; (c) A. W. Buesking and J. A. Ellman, Chem. Sci., 2014, 5, 1983 RSC.
- P. Andres, G. Ballano, M. I. Calaza and C. Cativiela, Chem. Soc. Rev., 2016, 45, 2291 RSC.
- For a review, see: (a) D. S. Matteson, Chem. Rev., 1989, 89, 1535 CrossRef. For selected examples, see: (b) D. S. Matteson, K. M. Sadhu and G. E. Lienhard, J. Am. Chem. Soc., 1981, 103, 5241 CrossRef; (c) D. S. Matteson, D. Maliakal and L. J. Fabry-Asztalos, Organomet. Chem., 2008, 693, 2258 CrossRef; (d) Z. He, A. Zajdlik, J. D. St. Denis, N. Assem and A. K. Yudin, J. Am. Chem. Soc., 2012, 134, 9926 CrossRef PubMed; (e) A. Zajdlik, Z. Wang, J. L. Hickey, A. Aman, A. D. Schimmer and A. K. Yudin, Angew. Chem., Int. Ed., 2013, 52, 8411 CrossRef PubMed; (f) M. A. Beenen, C. An and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 6910 CrossRef PubMed; (g) A. W. Buesking, V. Bacauanu, I. Cai and J. A. Ellman, J. Org. Chem., 2014, 79, 3671 CrossRef PubMed; (h) J.-b. Xie, J. Luo, T. R. Winn, D. B. Cordes and G. Li, Beilstein J. Org. Chem., 2014, 10, 746 CrossRef PubMed. For substrate control, see: (i) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Science, 2017, 356, 1045 Search PubMed.
- (a) C. Sole, H. Gulyas and E. Fernández, Chem. Commun., 2012, 48, 3769 RSC; (b) K. Hong and J. P. Morken, J. Am. Chem. Soc., 2013, 135, 9252 CrossRef PubMed; (c) S.-S. Zhang, Y.-S. Zhao, P. Tian and G.-Q. Lin, Synlett, 2013, 24, 437 CrossRef; (d) D. Wang, P. Cao, B. Wang, T. Jia, Y. Lou, M. Wang and J. Liao, Org. Lett., 2015, 17, 2420 CrossRef PubMed; (e) D. Nishikawa, K. Hirano and M. Miura, J. Am. Chem. Soc., 2015, 137, 15620 CrossRef PubMed; (f) N. Hu, G. Zhao, Y. Zhang, X. Liu, G. Li and W. Tang, J. Am. Chem. Soc., 2015, 137, 6746 CrossRef PubMed; (g) A. López, T. B. Clark, A. Parra and M. Tortosa, Org. Lett., 2017, 19, 6272 CrossRef PubMed; (h) L. Chen, X. Zou, H. Zhao and S. Xu, Org. Lett., 2017, 19, 3676 CrossRef PubMed.
- Catalytic synthesis of cyclic chiral α-amino boronates via 1,2-rearrangement of indol-2-yl boronates: (a) S. Panda and J. M. Ready, J. Am. Chem. Soc., 2017, 139, 6038 CrossRef PubMed; (b) S. Das, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 4053 CrossRef PubMed.
- (a) S. J. Baker, C. Z. Ding, T. Akama, Y.-K. Zhang, V. Hernandez and Y. Xia, Future Med. Chem., 2009, 1, 1275 CrossRef PubMed; (b) S. E. Poplawski, J. H. Lai, Y. Li, Y. Z. Jin, Y. Liu., W. Wu, Y. Wu, Y. Zhou, J. L. Sudmeier, D. G. Sanford and W. W. Bachovchin, J. Med. Chem., 2013, 56, 3467 CrossRef PubMed.
- (a) C.-X. Zhuo, C. Zheng and S.-L. You, Acc. Chem. Res., 2014, 47, 2558 CrossRef PubMed; (b) C.-X. Zhuo, W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2012, 51, 12662 CrossRef PubMed; (c) Q. Ding, X. Zhou and R. Fan, Org. Biomol. Chem., 2014, 12, 4807 RSC; (d) S. P. Roche, J.-J. Youte Tendoung and B. Tréguier, Tetrahedron, 2015, 71, 3549 CrossRef.
- (a) M. Arrowsmith, M. S. Hill, T. Hadlington, G. Kociok-Köhn and C. Weetman, Organometallics, 2011, 30, 5556 CrossRef; (b) K. Oshima, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2011, 133, 7324 CrossRef PubMed.
- For transition-metal-catalyzed dearomative borylation, see: (a) K. Oshima, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2012, 134, 3699 CrossRef PubMed; (b) A. S. Dudnik, V. L. Weidner, A. Motta, M. Delferro and T. J. Marks, Nat. Chem., 2014, 6, 1100 CrossRef PubMed; (c) A. Kaithal, B. Chatterjee and C. Gunanathan, Org. Lett., 2016, 18, 3402 CrossRef PubMed; (d) F. Zhang, H. Song, X. Zhuang, C.-H. Tung and W. Wang, J. Am. Chem. Soc., 2017, 139, 17775 CrossRef PubMed. For dearomative borylation catalyzed by orgnocatalysts, see: (e) X. Fan, J. Zheng, Z. H. Li and H. Wang, J. Am. Chem. Soc., 2015, 137, 4916 CrossRef PubMed; (f) E. N. Keyzer, S. S. Kang, S. Hanf and D. S. Wright, Chem. Commun., 2017, 53, 9434 RSC; (g) T. Ohmura, Y. Morimasa and M. Suginome, J. Am. Chem. Soc., 2015, 137, 2852 CrossRef PubMed; (h) K. Oshima, T. Ohmura and M. Suginome, Chem. Commun., 2012, 48, 8571 RSC; (i) B. Rao, C. C. Chong and R. Kinjo, J. Am. Chem. Soc., 2018, 140, 652 CrossRef PubMed; (j) A. Jayaraman, L. C. Misal Castro, V. Desrosiers and F.-G. Fontaine, Chem. Sci., 2018, 9, 5057 RSC.
- K. Kubota, K. Hayama, H. Iwamoto and H. Ito, Angew. Chem., Int. Ed., 2015, 54, 8809 CrossRef PubMed.
- (a) K. Kubota, Y. Watanabe, K. Hayama and H. Ito, J. Am. Chem. Soc., 2016, 138, 4338 CrossRef PubMed; (b) K. Kubota, Y. Watanabe and H. Ito, Adv. Synth. Catal., 2016, 358, 2379 CrossRef.
- D. Chen, G. Xu, Q. Zhou, L. W. Chung and W. Tang, J. Am. Chem. Soc., 2017, 139, 9767 CrossRef PubMed.
- J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642 CrossRef PubMed.
- For selected reviews on Cu-catalyzed borylation, see: (a) K. Semba, T. Fujihara, J. Terao and Y. Tsuji, Tetrahedron, 2015, 71, 2183 CrossRef; (b) J. A. Schiffner, K. Müther and M. Oestreich, Angew. Chem., Int. Ed., 2010, 49, 1194 CrossRef PubMed; (c) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091 CrossRef PubMed; (d) J. Cid, H. Gulyas, J. J. Carbo and E. Fernández, Chem. Soc. Rev., 2012, 41, 3558 RSC; (e) L. Dang, Z. Lin and T. B. Marder, Chem. Commun., 2009, 3987 RSC; (f) V. Lillo, A. Bonet and E. Fernández, Dalton Trans., 2009, 2899 RSC.
- For details see Table S1 in ESI.†.
- The reason we chose Table 1, entry 12 as optimal because enantioselectivities of most substrates were not satisfying when the reactions were carried out at room temperature.
- Crystallographic data for 2s could be found in the ESI.† CCDC 1836254 contains the supplementary crystallographic data for this paper.
- The relative configuration of major product was determined by 2D NMR NOSEY spectrum. For details see ESI.†.
- R. P. Sonawane, V. Jheengut, C. Rabalakos, R. Larouche-Gauthier, H. K. Scott and V. K. Aggarwal, Angew. Chem., Int. Ed., 2011, 50, 3760 CrossRef PubMed.
- A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584 CrossRef PubMed.
- L. Dang, Z. Lin and T. B. Marder, Organometallics, 2008, 27, 4443 CrossRef.
- Although conversion of C-bound enolate to O-bound enolate is disfavored in borylation of methyacrylate according to the calculations (ref. 24), the large steric congestion between substituents at 2- and 3-positions of D would probably force this conversion to occur in the current reaction.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1836254. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01815d |
| This journal is © The Royal Society of Chemistry 2018 |