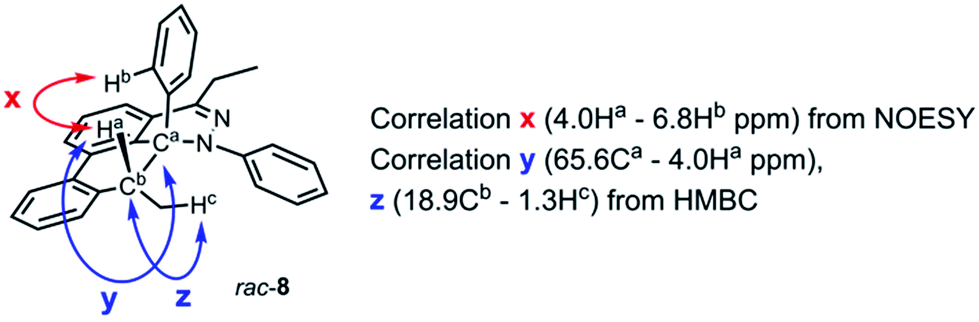DOI:
10.1039/C8SC01796D
(Edge Article)
Chem. Sci., 2018,
9, 7688-7693
The mechanism of the triple aryne–tetrazine reaction cascade: theory and experiment†
Received
19th April 2018
, Accepted 10th August 2018
First published on 23rd August 2018
Abstract
This article describes an experimental and computational investigation on the possible aryne reactivity modes in the course of the reaction of two highly energetic molecules, an aryne and a 1,2,4,5-tetrazine. Beyond the triple aryne–tetrazine (TAT) reaction, it was observed that combinations of several reactivity modes afford several heterocyclic compounds. Density Functional Theory (DFT) calculations of competition between a second Diels–Alder reaction and the nucleophilic addition pathways indicates the latter to be more favorable. Crossover experiments and computational study of the proton transfer step reveal that the reaction proceeds intermolecularly with the assistance of a water molecule, rather than intramolecularly. The resulting enamine intermediate was found to undergo either a stepwise formal [2 + 2] or [4 + 2] cycloaddition, and their energetic profiles were compared against each other. Isolation of an ene-product and a rearranged product shows the potential competition with oxidation/desaturation. These studies show how multiple arynes react with a highly reactive starting material and provide guidance for future applications of aryne-based multicomponent cascade reactions.
Introduction
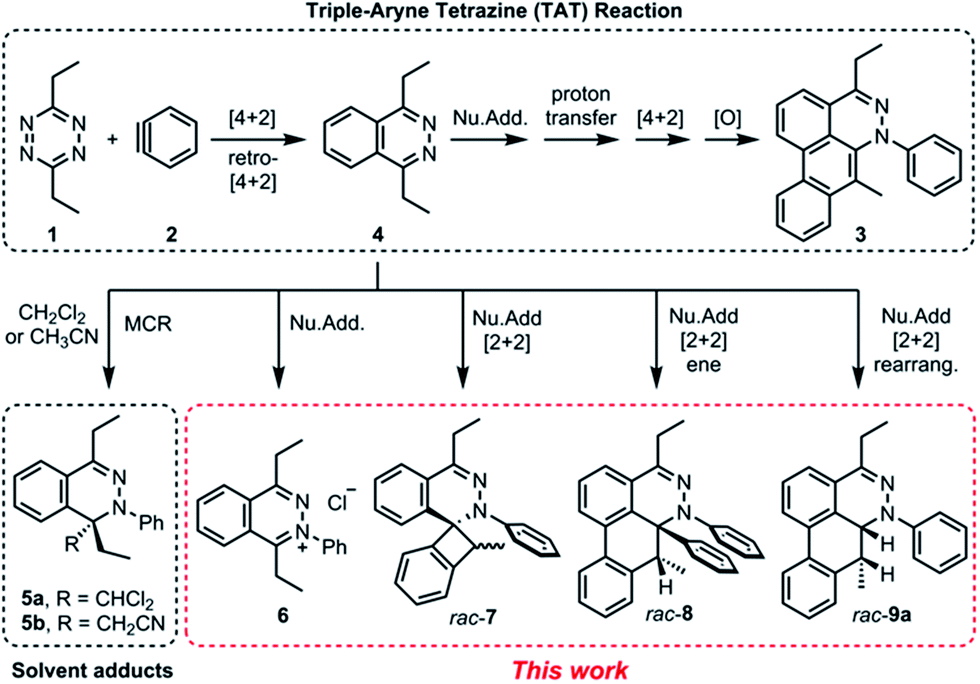
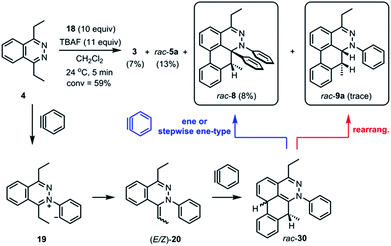
Recently, we reported a new multistep process termed the triple aryne–tetrazine (TAT) reaction.1 The reaction combined diverse reactivity modes between simple 1,2,4,5-tetrazine 1 (henceforth referred to as “tetrazine”) starting materials2 and aryne 2 into a single multistep process, resulting in the addition of three aryne equivalents to a tetrazine core. In the TAT reaction, dibenzocinnoline 3 and the cycloaddition intermediate 4 were isolated. In addition, we reported solvent participation in aryne reactions with the two solvent-trapped intermediates 5a and 5b (Fig. 1).3 Detailed investigation of these side reactions can yield valuable insight into aryne chemistry4 and provide access to new heterocyclic scaffolds.5 Here we report new mechanistic aspects of the reaction between a tetrazines and arynes, elucidating unique divergent pathways to alternative products that arise during the reaction course. The reaction mechanism consists of six consecutive elementary steps. As many as five steps can directly engage the aryne.6 Using a combination of experimental and computational methods, we elucidate the unique competing pathways that are operational throughout the course of the reaction to yield products 6, rac-7, rac-8, and rac-9a.
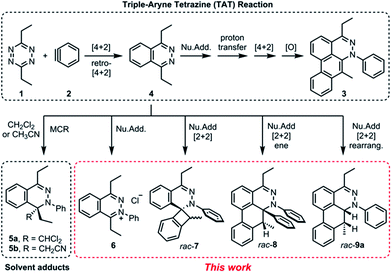 |
| | Fig. 1 Reactivity modes of 1,2,4,5-tetrazine and arynes. [4 + 2], [4 + 2] cycloaddition; Nu.Add, nucleophilic addition; [O], oxidation; MCR, multicomponent reaction; ene, ene-reaction; rearrang., rearrangement. | |
Results and discussion
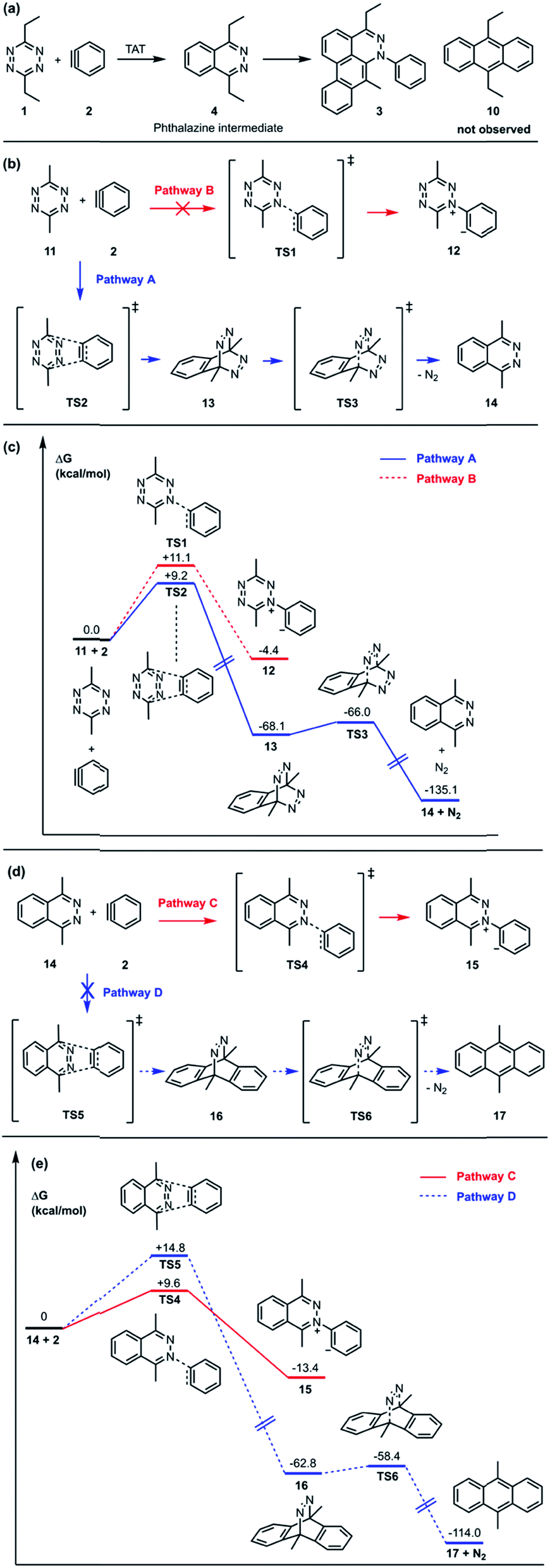
In the TAT reaction, it was observed that 1 undergoes [4 + 2] cycloaddition with 2 and loss of N2 affords the phthalazine intermediate 47 followed by several further mechanistic steps to give dibenzocinnoline 3 (Fig. 2A). In the TAT reaction, anthracene 10 was not observed,8 indicating that the phthalazine intermediate 4 does not undergo a further [4 + 2] cycloaddition. Instead, 4 is observed to undergo a nucleophilic addition to another equivalent of 2. We used Density Functional Theory (DFT) calculations to compare free energy profiles of the Diels–Alder versus the nucleophilic addition reaction pathways for both the tetrazine and benzyne pair and the phthalazine and benzyne pair. To simplify the calculations, the ethyl substituents on tetrazine 1 were modeled with methyl groups (Fig. 2A and B). Two pathways are possible: nucleophilic addition of tetrazine 11 with aryne 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 or a Diels–Alder cycloaddition. The two possible pathways were evaluated using DFT and the free energy profile for the reactions are shown in Fig. 2C. The Diels–Alder transition state TS2 is lower in energy than the nucleophilic addition transition state TS1 by 1.9 kcal mol−1 (Fig. 2B, C and 3). For phthalazine 14 and aryne 2, the nucleophilic addition is more favorable by 5.2 kcal mol−1 (Fig. 2D, E, and 3). These results are in good agreement with the experimental observation that the first Diels–Alder reaction is favored over the nucleophilic addition pathway, but the second Diels–Alder reaction is outcompeted by nucleophilic addition. Both nucleophilic addition pathways are overall somewhat exergonic, whereas both Diels–Alder pathways are strongly exergonic, with very low barriers for dinitrogen extrusion from the Diels–Alder adducts.10,11
9 or a Diels–Alder cycloaddition. The two possible pathways were evaluated using DFT and the free energy profile for the reactions are shown in Fig. 2C. The Diels–Alder transition state TS2 is lower in energy than the nucleophilic addition transition state TS1 by 1.9 kcal mol−1 (Fig. 2B, C and 3). For phthalazine 14 and aryne 2, the nucleophilic addition is more favorable by 5.2 kcal mol−1 (Fig. 2D, E, and 3). These results are in good agreement with the experimental observation that the first Diels–Alder reaction is favored over the nucleophilic addition pathway, but the second Diels–Alder reaction is outcompeted by nucleophilic addition. Both nucleophilic addition pathways are overall somewhat exergonic, whereas both Diels–Alder pathways are strongly exergonic, with very low barriers for dinitrogen extrusion from the Diels–Alder adducts.10,11
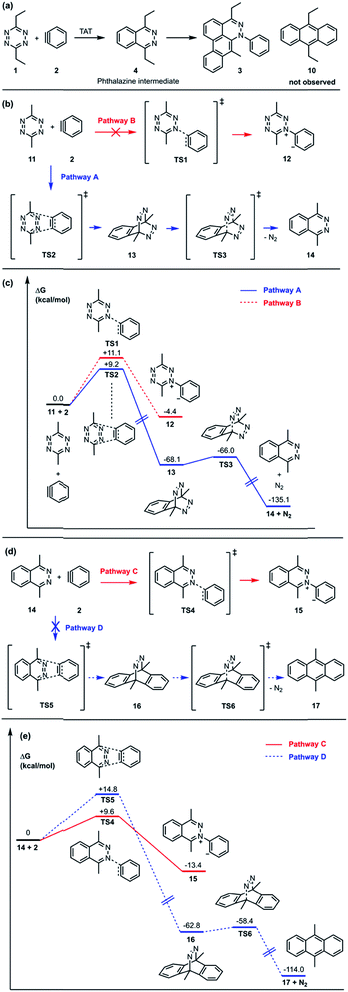 |
| | Fig. 2 (a) The reported TAT reaction and isolated phthalazine intermediate 4. (b) Diels–Alder reaction (pathway A) vs. nucleophilic addition (pathway B) from tetrazine 11 and aryne 2. (c) Free energy profiles of pathways A and B proceeding from tetrazine 11 and aryne 2 computed at the M06-2X/6-311+G(d,p)/SMD (CH2Cl2)//M06-2X/6-31G(d) level of theory. (d) Nucleophilic addition (pathway C) vs. Diels–Alder reaction (pathway D) from phtalazine 14 and aryne 2. (e) Free energy profiles of pathways C and D proceeding from phthalazine 14 and aryne 2 computed at the M06-2X/6-311+G(d,p)/SMD (CH2Cl2)//M06-2X/6-31G(d) level of theory. Gibbs free energies are reported in kcal mol−1. | |
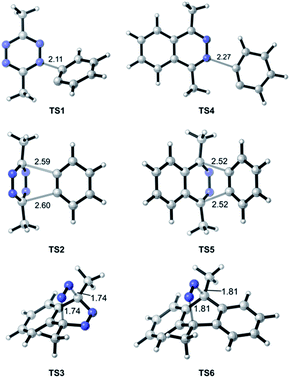 |
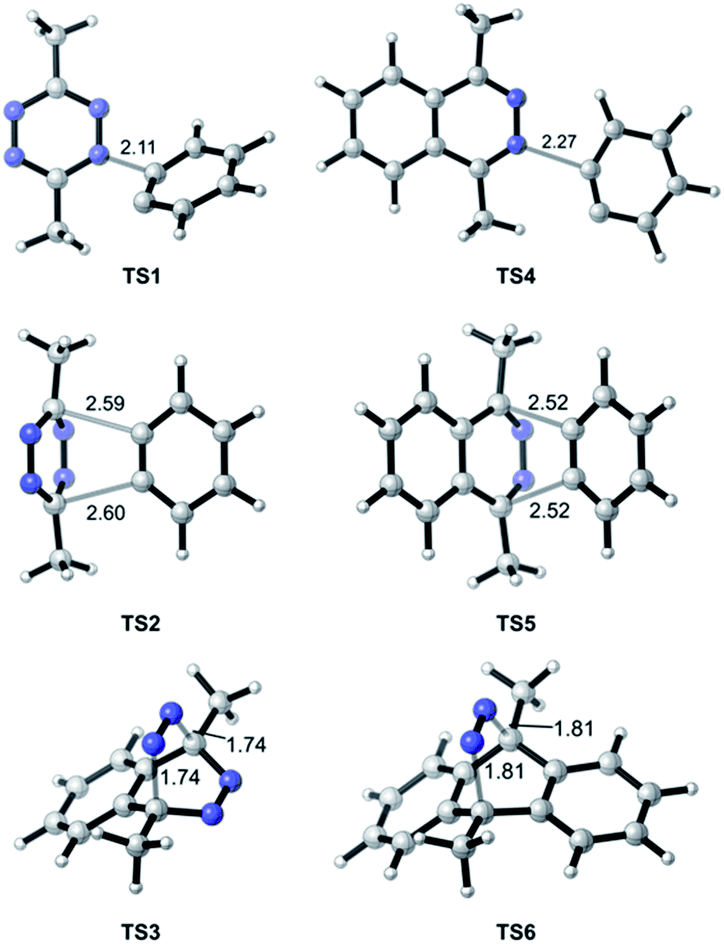
| | Fig. 3 Optimized geometries of transition states along pathways A–D. Interatomic distances are shown in ångströms. | |
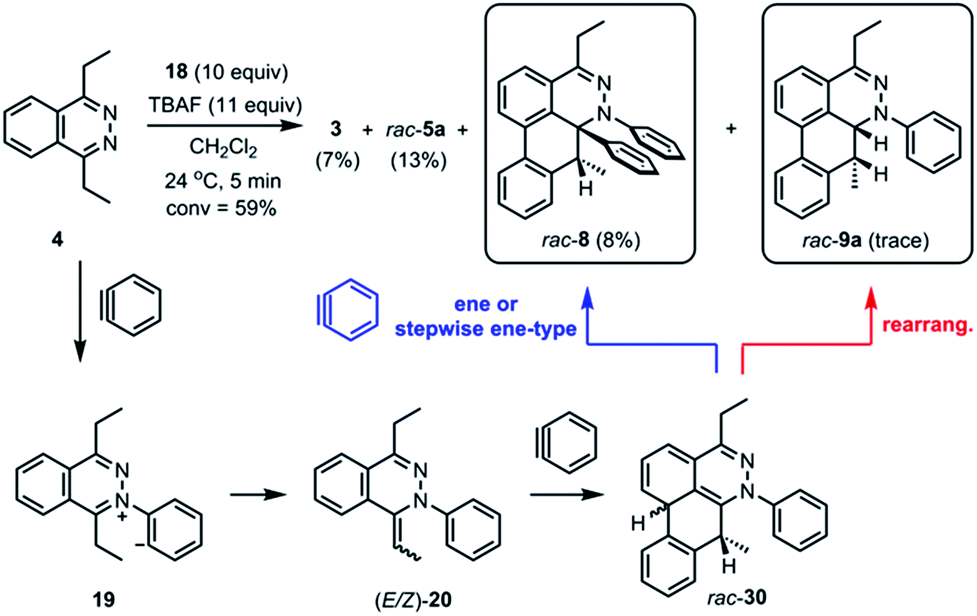
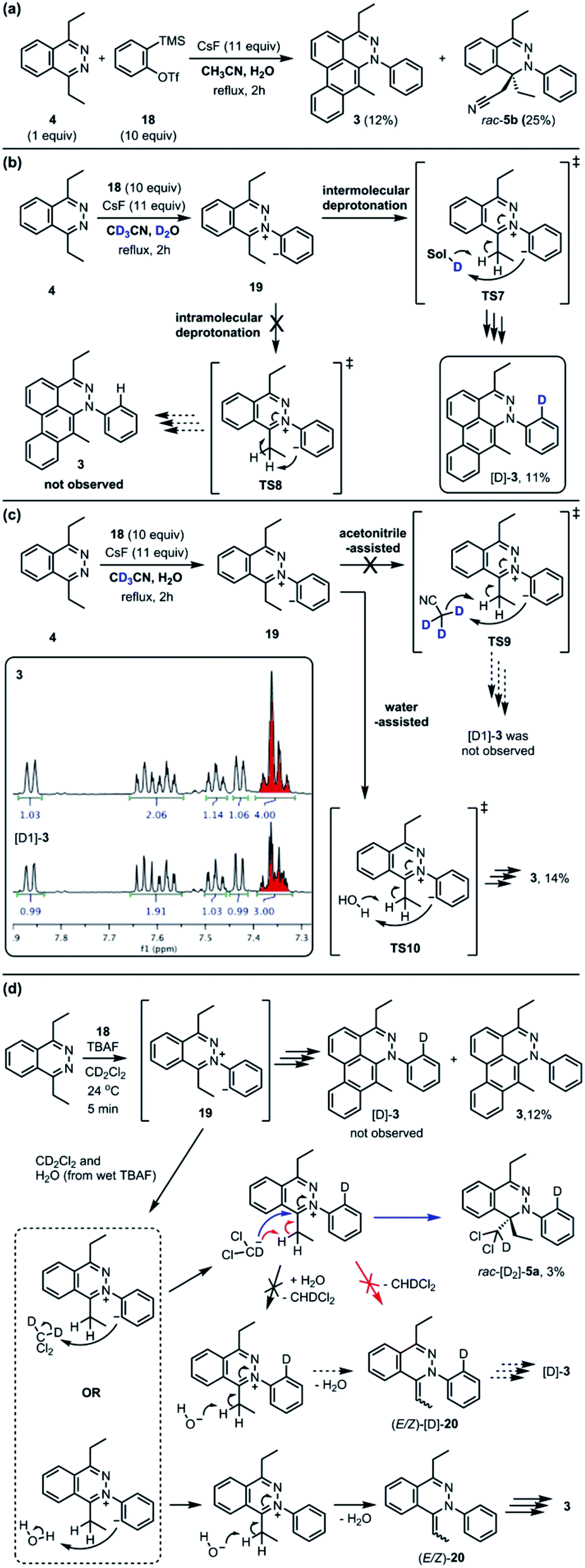
The standard TAT reaction is conducted with 1, 11 equivalents of tetrabutylammonium fluoride (TBAF) in THF, and 10 equivalents of 2-(trimethylsilyl)phenyl trifluoromethanesulfonate 18 in CH2Cl2 at 24 °C, to produce product 3 in 44% yield within 5 minutes.1 The intermediate 4 is isolable from the TAT reaction when conducted with an excess of tetrazine 1. The reaction between 18 and the intermediate 4 under the standard TAT reaction condition afforded the desired product 3 and rac-5b in 7% and 13% yield, respectively (Scheme 1).3 Here, using anhydrous CsF instead of TBAF, which has a low concentration of water and is considered a wet fluoride source, we conducted the reaction in CH3CN under reflux. Higher conversion and higher yield of both 3 and rac-5b were observed (Fig. 4A). The source of low mass balance for the TAT sequence is due to competing aryne and tetrazine decomposition pathways under the reaction conditions. To discriminate between an intra- versus intermolecular proton transfer pathway, the reaction was performed in CD3CN and D2O under reflux (Fig. 4B). Only deuterated dibenzo[de,g]cinnoline [D]-3 was isolated (11% yield), as analyzed by 1H NMR experiment. The result indicates that the intermediate 19 most likely did not proceed through an intramolecular pathway but instead through an intermolecular proton transfer pathway, assisted by a fluoride, a hydroxide, conjugate base of CH3CN, or a phenyl anion of another benzyne adduct. To further probe solvent participation in the proton transfer step, experiments were conducted in CD3CN and H2O (Fig. 4C). According to the result shown in Fig. 4B, the intramolecular pathway is unlikely to be favored. Two possibilities arise for the intermolecular pathway, a water-mediated or CH3CN-mediated proton transfer, assuming H2O can only serve as the proton source and CD3CN can only provide deuterium during the proton transfer step. After this reaction, only non-deuterated 3 was observed in 14% yield, which ruled out proton transfer with the conjugate base of CH3CN, supporting a water-mediated proton transfer pathway. These results account for why only 3 and rac-[D2]-5a were found and [D]-3 was not observed in our previous crossover experiments performed in CD2Cl2 (Fig. 4D).3 When it comes to the stepwise mechanism in Fig. 4D, after deuterium transfer from CD2Cl2 to the intermediate 19, the deuterated cationic intermediate is formed. According to the isolation of 3 and rac-[D2]-5a, the cationic intermediate may only react with dichloromethanide-d via 1,2-addition to provide rac-[D2]-5a but may not undergo further deprotonation assisted by either dichloromethanide-d or hydroxide to afford (E/Z)-[D]-20 because [D]-3 was not detected. A plausible pathway to account for formation of 3 may involve a water-mediated proton transfer to give (E/Z)-20. As shown above, water is an important proton source for the TAT reaction, and this result is consistent with the observation that no desired product from the TAT reaction is observed with anhydrous tetrabutylammonium difluorotriphenylsilicate (TBAT).1
 |
| | Scheme 1 Ene or stepwise ene-type reaction and rearrangement of the intermediate rac-30. | |
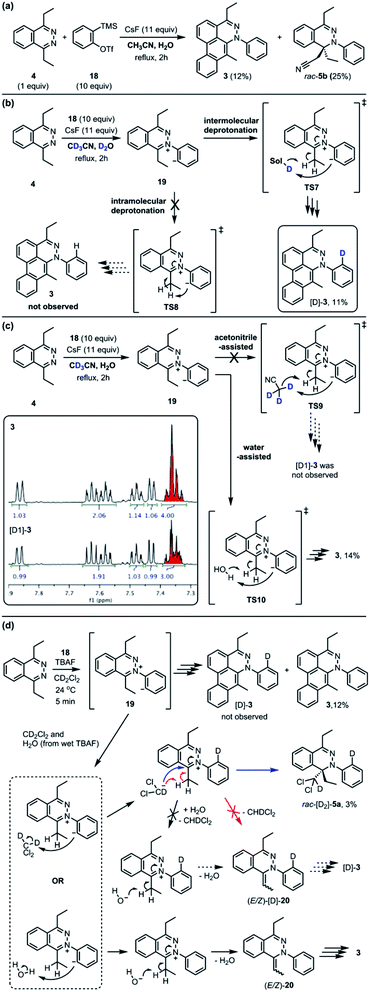 |
| | Fig. 4 Crossover experiment to determine intra-, acetonitrile-assisted inter-, or water-assisted intermolecular proton transfer. (a) The reaction was conducted in the mixture of 1:1 ratio of CH3CN/H2O. (b) The mixture of 1:1 ratio of CD3CN/D2O. (c) The mixture of 1:1 ratio of CD3CN/H2O. (d) Importance of water in the previous crossover experiment.3 | |
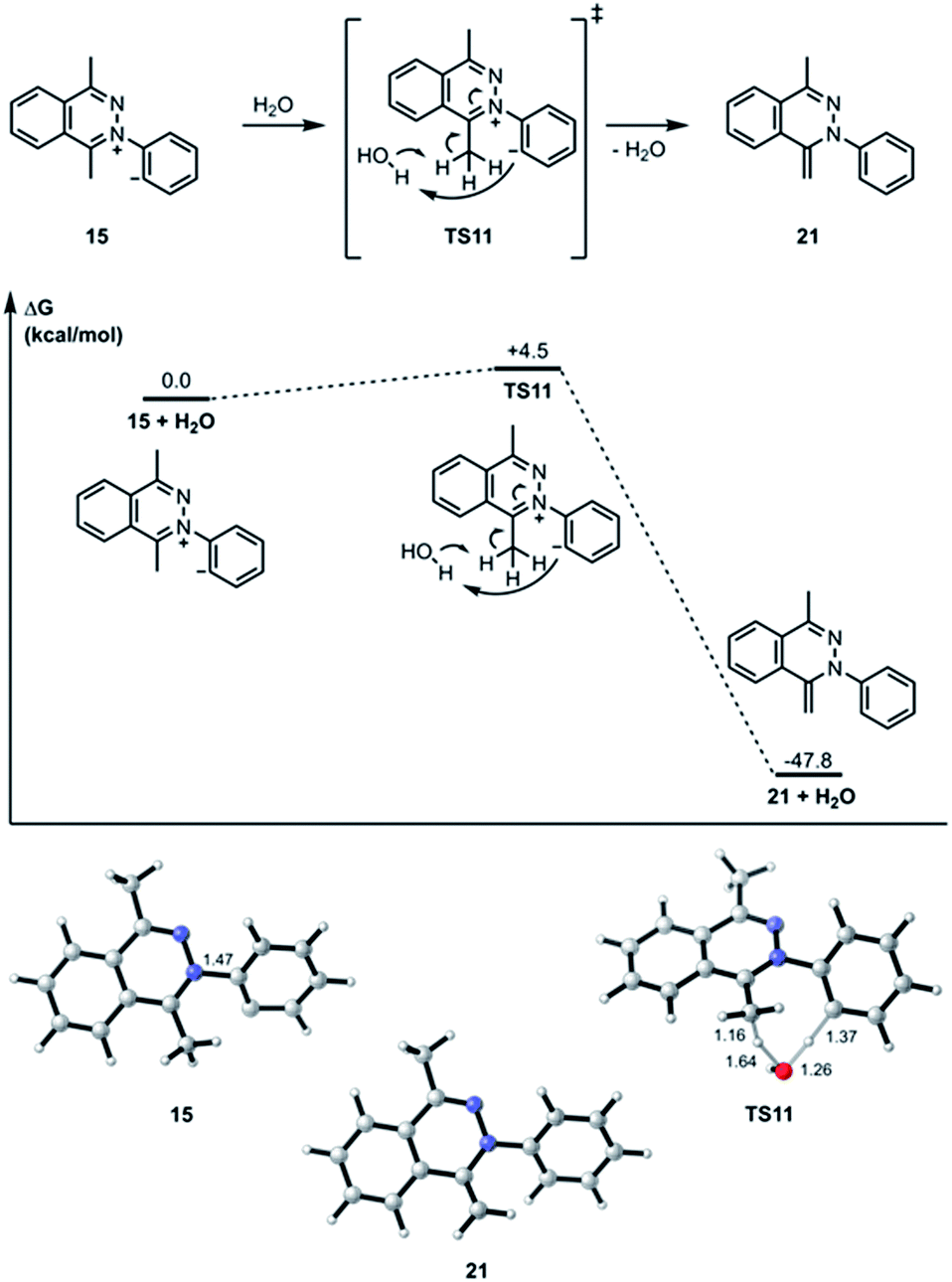
The transition state structure (TS11) for the concerted water-assisted proton transfer is shown in Fig. 5. The transition state for the direct intramolecular transfer of proton was not located. It is possible that due to the requirement that the σ C–H bond being broken in the proton abstraction must be in plane with the π system of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, only a proton abstraction from the top or bottom face of the molecule would have been favorable. Due to the geometrical constraints of the molecular structure, however, such a top- or bottom-face proton abstraction by the anionic carbon is extremely difficult to achieve.
C bond, only a proton abstraction from the top or bottom face of the molecule would have been favorable. Due to the geometrical constraints of the molecular structure, however, such a top- or bottom-face proton abstraction by the anionic carbon is extremely difficult to achieve.
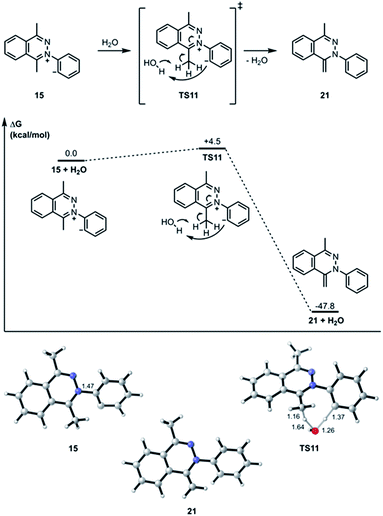 |
| | Fig. 5 Optimized geometries of intermediates, products, and transition states for the water-assisted proton transfer pathway computed at the M06-2X/6-311+G(d,p)/SMD (CH2Cl2)//M06-2X/6-31G(d) level of theory. Gibbs free energies of activation are calculated with respect to isolated reactants and reported in kcal mol−1. Interatomic distances are shown in ångströms. | |
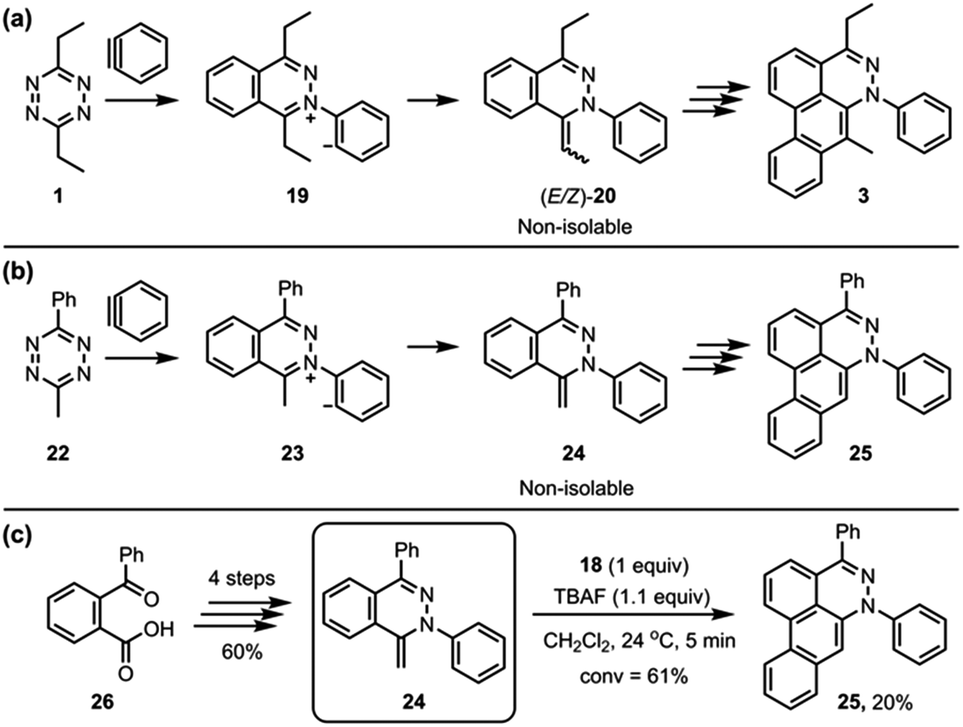
To simplify the analysis of possible alkylidene intermediate, 3-methyl-6-phenyl-1,2,4,5-tetrazine (24) was employed (Fig. 6B); a phenyl group at the 6-position was expected to block the further side reaction and also stabilize the tetrazine ring, and a methyl group at the 3-position would afford a single methylidene intermediate 24 while 3,6-diethyltetrazine 1 could afford (E/Z)-20 as an isomeric mixture (Fig. 6A). The aryne reaction of newly employed tetrazine 22 also did not stop at the intermediate 24, instead proceeding to afford the dibenzo[de,g]cinnoline 25. Even though 24 was not isolated, we were able to synthesize 24 through an alternate route (Fig. 6C).12 TBAF (1.1 equiv.) was subjected to a solution containing 24 (1.0 equiv.) and 18 (1.0 equiv.) in CH2Cl2, which afforded the desired product 25 in 20% yield. This result supports the notion that a methylidene intermediate, such as 24 or (E/Z)-20, might be the key intermediate in the third benzyne addition step of the TAT reaction.
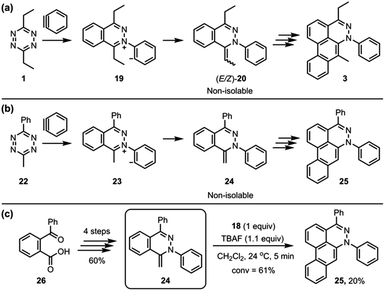 |
| | Fig. 6 (a) The TAT reaction of 3,6-diethyltetrazine 1. (b) Alternative probe 24 to the intermediates (E/Z)-20 of the TAT reaction. (c) The formation of 25 from the reaction of 24 and aryne. | |
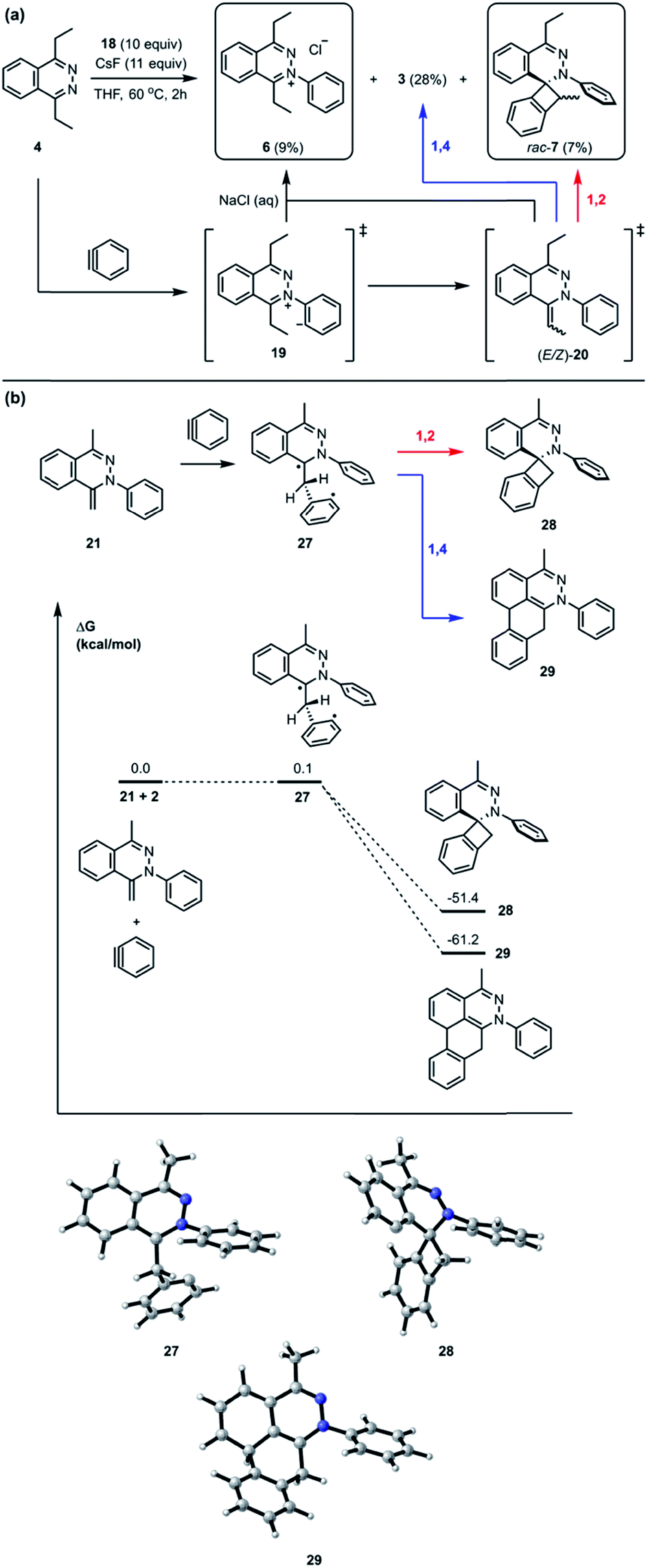
Interestingly, the reaction between 4 and 18 with CsF in THF at 60 °C for 2 hours followed by treatment with saturated brine solution afforded the trapped intermediate 6 in 9% yield (Fig. 7A). There are two possible routes leading to the formation of 6. One possibility is that the intermediate 19 is quenched by an external proton source such as a solvent molecule. The other possibility is that the intermediate 19 undergoes an intermolecular proton transfer to yield neutral intermediates (E/Z)-20, followed by tautomerization, to furnish hydrazonium salt 6. The intermolecular pathway was ruled out in Fig. 4B. The [2 + 2] cycloaddition product rac-7 was obtained as a mixture of inseparable diastereomers in 7% yield.13 The relatively smaller amounts of the [2 + 2] cycloaddition product compared to [4 + 2] product 3 indicate that the [4 + 2] mode is experimentally more favorable than the [2 + 2] mode. Calculations support the idea that the intermediate 21 can react with another equivalent of benzyne in either a formal [2 + 2] or [4 + 2] cycloaddition to give 28 and 29, respectively (Fig. 7B). We performed calculations to elucidate whether the formal [2 + 2] and [4 + 2] cycloadditions occur via stepwise or concerted mechanisms, and whether they proceed through zwitterionic or diradical intermediates. A stepwise [2 + 2] pathway would be expected in light of the Woodward–Hoffmann rules, while a stepwise [4 + 2] is also likely for this system because aromaticity would need to be broken to form the σ C–C bond on one side but not the other, leading to very different energetic penalties. Indeed, transition states for concerted [2 + 2] or [4 + 2] cycloadditions were not found for this system. While all of the possible closed-shell zwitterionic intermediates for stepwise [2 + 2] and [4 + 2] pathways were found to be unstable, we located the common diradical intermediate 27 expected along stepwise pathways of both the formal [2 + 2] and [4 + 2] cycloadditions between 21 and 2, with a free energy of +0.1 kcal mol−1. Gibbs free energies of the [2 + 2] adduct 28 and [4 + 2] adduct 29 indicate that these formal cycloadditions are strongly exergonic.
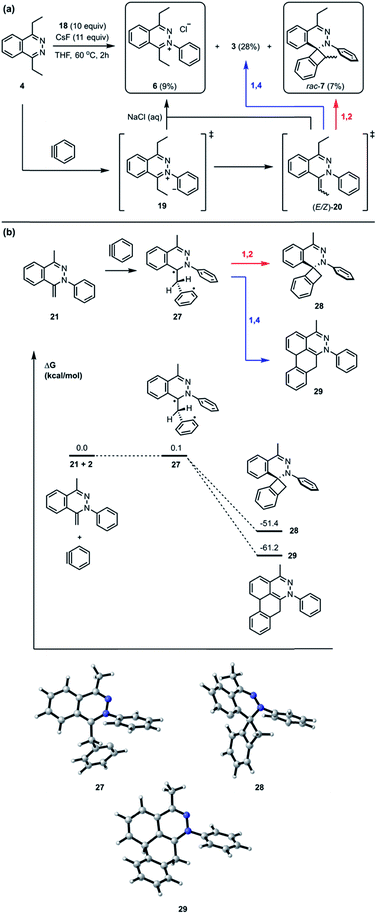 |
| | Fig. 7 (a) The trapped intermediate salt 6 and competitive cycloaddition between [2 + 2] and [4 + 2]. (b) Optimized geometries of intermediates and product structures for [2 + 2] and [4 + 2] cycloaddition pathways from the intermediate 21 and aryne 2 computed at the M06-2X/6-311+G(d,p)/SMD (CH2Cl2)//M06-2X/6-31G(d) level of theory. Gibbs free energies are calculated with respect to isolated reactants, and reported in kcal mol−1. | |
During the analysis of the standard TAT reaction in Scheme 1, 8% of rac-8 was isolated and characterized by HMBC NMR experiments, and its relative stereochemistry was determined by NOESY NMR experiments (Fig. 8). We propose two possible mechanisms for the synthesis of rac-8 consisting of a concerted ene-reaction or stepwise ene-type reaction of rac-30.14 In both mechanisms, the two possible pathways support the existence of a currently non-isolable intermediate rac-30.
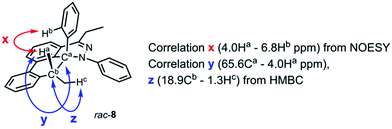 |
| | Fig. 8 Representative NMR correlation assignments of rac-8. | |
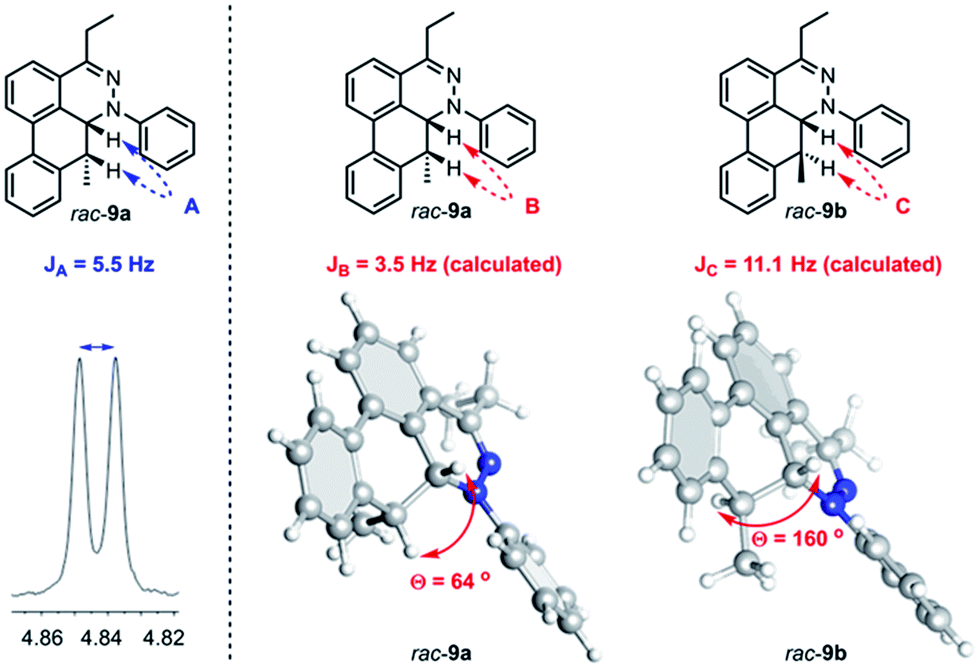
Further observation of the rearranged intermediate rac-9a indirectly supports that the intermediate rac-30 is in the course of the TAT reaction. The structure of rac-9a was characterized by 1H NMR and HRMS spectroscopy. The J coupling constant of two vicinal protons (JA) was determined to be 5.5 Hz (Fig. 9). Based on the results from the B3LYP/IGLO-III//HF/6-31G(d) calculations, cis isomer rac-9a possessed 64° dihedral angles and a J coupling of 3.5 Hz between the two vicinal protons. Larger dihedral angles of 160° for the trans isomer (rac-9b) resulted in a JC of 11.1 Hz, which is quite different from the observed value.
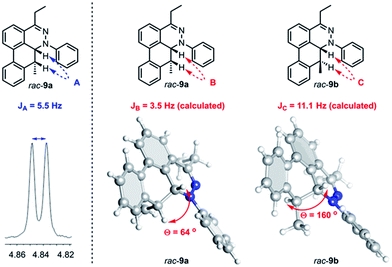 |
| | Fig. 9 Determination of stereochemistry of rac-9a. J coupling constant of A from 1H NMR spectrum (left) and calculated J coupling constant and degree between two protons on cis intermediate rac-9a and trans intermediate rac-9b (right). | |
Conclusions
We have provided a detailed mechanism of the reaction between a tetrazine and arynes. During the course of the reaction, three aryne addition steps occur consecutively with each step displaying a distinct reactivity mode with benzyne. By isolating and trapping the key intermediates, each of these distinct aryne reactivity modes as well as alternative side product pathways were elucidated. DFT calculations of transition states of two competitive reactions, further [4 + 2] cycloaddition and nucleophilic addition, support the assertion that [4 + 2] cycloaddition has higher activation energy barrier, and thus, the formation of anthracene is supposed to be unfavorable. The computational and experimental study for three plausible proton transfer pathways were performed. This analysis showed that the intermolecular water-assisted proton transfer is more favorable than the intramolecular proton transfer. The dibenzocinnoline 25 was observed during the reaction of an alternative intermediate 24 with an aryne via [4 + 2] cycloaddition. The intermediate 19 undergoes further formal [2 + 2] and [4 + 2] cycloaddition, with both the [2 + 2] cycloaddition product rac-7 and the [4 + 2] product 3 being observed. In addition, the plausibility of non-isolable intermediates rac-30 was confirmed by the isolation and characterization of the ene or the stepwise ene-type product rac-8 and rearranged product rac-9a. The observed intermediates and byproducts are consistent with the overall proposed mechanism for the TAT reaction. We anticipate that the study of arynes–tetrazine-engaged reactivity modes will serve as guidance for future explorations of aryne chemistry, and especially in the context of intermolecular multiple consecutive aryne addition reactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by funding from the University of Pennsylvania, NIH Grant GM118510 (to D. M. C.) and NSF Grant CHE-1361104 (to K. N. H.). Instruments supported by the NSF and NIH including HRMS (NIH RR-023444). We thank Dr George Furst and Dr Rakesh Kohli for assistance in obtaining the high resolution NMR and mass spectral data, respectively.
Notes and references
- S.-E. Suh, S. A. Barros and D. M. Chenoweth, Chem. Sci., 2015, 6, 5128–5132 RSC.
- J. Yang, M. R. Karver, W. Li, S. Sahu and N. K. Devaraj, Angew. Chem., Int. Ed., 2012, 51, 5222–5225 CrossRef PubMed.
- S.-E. Suh and D. M. Chenoweth, Org. Lett., 2016, 18, 4080–4083 CrossRef PubMed.
-
(a) C. M. Gampe and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3766–3778 CrossRef PubMed;
(b) R. Sanz, Org. Prep. Proced. Int., 2008, 40, 215–291 CrossRef;
(c) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550–3577 CrossRef PubMed;
(d) C. Wu and F. Shi, Asian J. Org. Chem., 2013, 2, 116–125 CrossRef;
(e)
H. Yoshida, Compr. Org. Synth., 2nd edn, 2014, vol. 4, pp. 517–579 Search PubMed;
(f) S. Yoshida and T. Hosoya, Chem. Lett., 2015, 44, 1450–1460 CrossRef;
(g) J. A. Garcia-Lopez and M. F. Greaney, Chem. Soc. Rev., 2016, 45, 6766–6798 RSC.
-
(a) A. V. Dubrovskiy, N. A. Markina and R. C. Larock, Org. Biomol. Chem., 2013, 11, 191–218 RSC;
(b) K. Okuma, Heterocycles, 2012, 85, 515–544 CrossRef.
-
(a) S. S. Bhojgude, A. Bhunia and A. T. Biju, Acc. Chem. Res., 2016, 49, 1658–1670 CrossRef PubMed;
(b) S. S. Bhojgude and A. T. Biju, Angew. Chem., Int. Ed., 2012, 51, 1520–1522 CrossRef PubMed;
(c) A. Biju and A. Bhunia, Synlett, 2014, 25, 608–614 CrossRef;
(d) H. Yoshida and K. Takaki, Heterocycles, 2012, 85, 1333–1349 CrossRef.
- J. Sauer and G. Heinrichs, Tetrahedron Lett., 1966, 7, 4979–4984 CrossRef.
- G. Seitz, R. Hoferichter and R. Mohr, Angew. Chem., Int. Ed., 1987, 26, 332–334 CrossRef.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- For prior computational investigation into nucleophilic additions to arynes and heterocyclic arynes, see
(a) P. H.-Y. Cheong, R. S. Paton, S. M. Bronner, G.-Y. J. Im, N. K. Garg and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 1267–1269 CrossRef PubMed;
(b) G.-Y. J. Im, S. M. Bronner, A. E. Goetz, R. S. Paton, P. H.-Y. Cheong, K. N. Houk and N. K. Garg, J. Am. Chem. Soc., 2010, 132, 17933–17944 CrossRef PubMed;
(c) A. E. Goetz, S. M. Bronner, J. D. Cisneros, J. M. Melamed, R. S. Paton, K. N. Houk and N. K. Garg, Angew. Chem., Int. Ed., 2012, 51, 2758–2762 CrossRef PubMed;
(d) S. M. Bronner, J. L. Mackey, K. N. Houk and N. K. Garg, J. Am. Chem. Soc., 2012, 134, 13966–13969 CrossRef PubMed;
(e) J. M. Medina, J. L. Mackey, N. K. Garg and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 15798–15805 CrossRef PubMed.
- For a study of Diels–Alder reactions of tetrazines with alkenes with extrusion of dinitrogen, see: L. Tork, G. Jimenez-Oses, C. Doubleday, F. Liu and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 4749–4758 CrossRef PubMed.
-
(a) I. G. Ovchinnikova, I. V. Samartseva and E. A. Pavlova, Zh. Org. Khim., 1984, 20, 2248–2249 Search PubMed;
(b) M. M. Hemdan, S. M. Taha, A. M. Gabr and M. Y. Elkady, J. Chem. Res., 2010, 34, 102–105 CrossRef.
- L. Castedo, E. Guitian, C. Saá, R. Suau and J. M. Saá, Tetrahedron Lett., 1983, 24, 2107–2108 CrossRef.
- H. X. Siyang, X. R. Wu, H. L. Liu, X. Y. Wu and P. N. Liu, J. Org. Chem., 2014, 79, 1505–1510 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, characterization data, and NMR spectra of all new compounds. See DOI: 10.1039/c8sc01796d |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Shuming
Chen
a,
Shuming
Chen