
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic asymmetric synthesis of geminal-dicarboxylates†
Nisha
Mistry
 and
Stephen P.
Fletcher
*
and
Stephen P.
Fletcher
*
Department of Chemistry, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK. E-mail: stephen.fletcher@chem.ox.ac.uk
First published on 28th June 2018
Abstract
Stereogenic acetals, spiroacetals and ketals are well-studied stereochemical features that bear two heteroatoms at a common carbon atom. These stereocenters are normally found in cyclic structures while linear (or acyclic) analogues bearing two heteroatoms are rare. Chiral geminal-dicarboxylates are illustrative, there is no current way to access this class of compounds while controlling the stereochemistry at the carbon center bound to two oxygen atoms. Here we report a rhodium-catalysed asymmetric carboxylation of ester-containing allylic bromides to form stereogenic carbon centers bearing two different carboxylates with high yields and enantioselectivities. The products, which are surprisingly stable to a variety of acidic and basic conditions, can be manipulated with no loss of enantiomeric purity as demonstrated by ring closing metathesis reactions to form chiral lactones, which have been extensively used as building blocks in asymmetric synthesis.
Introduction
Stereocenters bearing two heteroatoms, including chiral acetals, spiroacetals and ketals (Fig. 1a), are some of the most prevalent structural motifs found in nature. These features are present in virtually all carbohydrate derivatives such as starch and cellulose, and in many small natural products including pheromones, steroids, and polyketides.1 The ability to control the stereochemistry of carbon centers featuring two heteroatoms is also important in the development of pharmaceuticals.1,2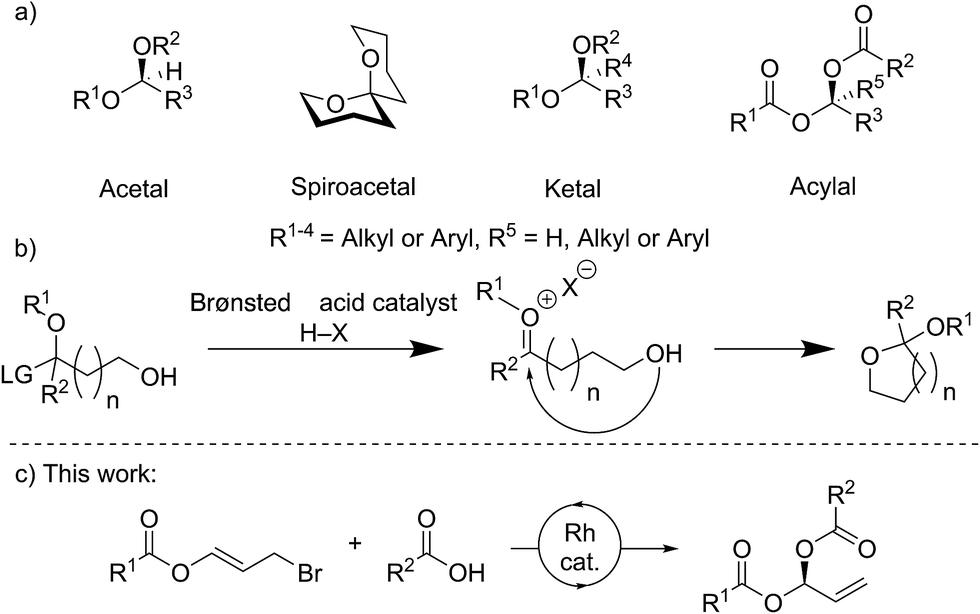 | ||
| Fig. 1 Common stereocenters bearing two heteroatoms and their catalytic asymmetric formation, including this work. (a) General structures for O,O-acetals, spiroacetals, ketals and dicarboxylates. (b) Formation of a bis-heteroatom bearing stereogenic center via an oxocarbenium ion intermediate using Brønsted acid catalysis. (c) This work: Rh catalyzed asymmetric carboxylation of heteroatom containing allyl bromides to form gem-dicarboxylates. | ||
For many years, stereocontrolled access to chiral acetal derivatives relied on derivatisation of chiral starting materials,3–8 metal mediated desymmetrisations,9–11 or kinetic or thermodynamically controlled cyclisation of carbonyl containing chiral-compounds.12–17 A recent approach to bis-heteroatom containing stereogenic centers involves catalysis with very sterically hindered chiral Brønsted acids. Though generally envisaged to occur via oxocarbenium ion or imine intermediates (Fig. 1b) which undergo stereoselective addition, some reactions likely proceed via single-step asynchronous pathways.18 This strategy has now been sufficiently developed to allow the synthesis of chiral N,N-,19–22N,O-,23N,S-24,25 and O,O-acetals.26–29 Methods for the stereocontrolled formation of analogous linear compounds are less common.19,20,23,24
A variety of rhodium-catalysed asymmetric allylation-type processes, including allylic substitutions, with oxygen, nitrogen and carbon nucleophiles have been reported to form allylic alcohols, amines, and tertiary or quaternary carbon stereogenic centers.30–39 Carboxylic acids may be used as nucleophiles, not only in rhodium-catalysed asymmetric reactions,40 but also in processes catalysed by iridium,41 palladium,42,43 and ruthenium.44 However, only limited examples of metal-catalysed asymmetric additions to make stereogenic centres bearing two heteroatoms are known, and no Rh-catalysed processes have been reported.45–48
Here we report the stereocontrolled synthesis of geminal-dicarboxylates (acylals) via a highly enantioselective rhodium-catalysed carboxylation of allyl bromide derivatives bearing ester groups. These allyl bromides have been used in copper-catalysed additions of Grignard reagents to give allylic esters.49 The linear products described here feature a stereogenic carbon center bearing two different carboxylates. Geminal-dicarboxylates are an understudied class of compounds with the exception of the diacetate and dipropionate derivatives, which can protect aldehydes and are important substrates for asymmetric Tsuji–Trost reactions.50,51
Methods for the synthesis of 1,1-diacetates include protic and Lewis acid catalysis,52,53 the action of I2,54 NBS,55 and various heterogeneous catalysts.56 However, none of these methods allow stereocontrol. As far as we are aware the only report of asymmetric induction in gem-dicarboxylate formation involved copper-catalysed allylic oxidation of an olefin in 23% yield and 10% ee.57
Results and discussion
Our standard reaction conditions involve 1.25 mol% [Rh(COD)(Cl)]2, 3 mol% of Ugi amine derived ligand A, 1 eq. of LiOt-Bu and THF at 40 °C. Using ester substituted allyl bromide 1a, easily prepared by mixing benzoyl bromide and acrolein at room temperature in CH2Cl2,58 we are able to add isobutyric acid to give 2a in good yield and excellent ee (82%, 96% ee, Table 1, entry 1).|
|
||||
|---|---|---|---|---|
| Entry | Variation from standard conditions | Reaction time | Yielda (%) | eeb (%) |
| a All yields are isolated yields. b Enantiomeric excesses determined by SFC using a chiral non-racemic stationary phase. c The reaction was stirred overnight. d Gram-scale reaction, carried out using 0.5 mol% [Rh(COD)(Cl)]2 and 1.2 mol% ligand. | ||||
| 1 | None | 1.5 h | 82 | 96 |
| 2 | No Rh | o/nc | 0 | — |
| 3 | No ligand | o/n | 76 | rac |
| 4 | (S)-BINAP instead of A | o/n | 69 | 45 |
| 5 | No LiOt-Bu | o/n | 0 | — |
| 6 | LiOMe instead of LiOt-Bu | 1 h | 85 | 96 |
| 7 | KOt-Bu instead of LiOt-Bu | o/n | 67 | 8 |
| 8 | Room temperature | 2 h | 89 | 94 |
| 9 | 60 °C | 1 h | 83 | 95 |
| 10 | 1 eq. isobutyric acid | 50 min | 82 | 96 |
| 11d | 4 mmol scale | 2 h | 82 | 95 |
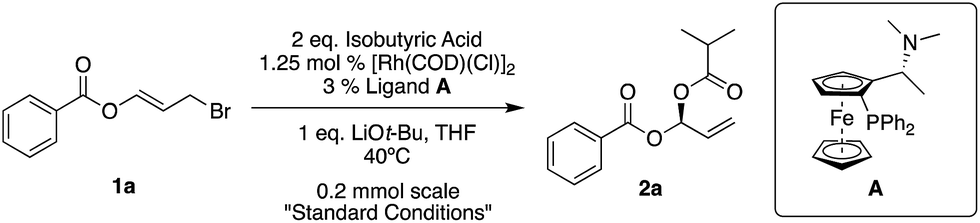
Pleasingly, we also observe complete regioselectivity for the SN2′ product over the SN2 product, which is a known challenge in allylic substitution reactions (see ESI† for further details).
If we remove either the rhodium source or base from the reaction, we obtain no product (Table 1, entries 2 and 5). A reaction without ligand A gave racemic product (Table 1, entry 3) and using (S)-BINAP instead of A gave 45% ee (Table 1, entry 4).
As long as it is of high quality, it is possible to use LiOMe instead of LiOt-Bu (Table 1, entry 6), however when switching to non-Li bases, for example KOt-Bu, the ee drops significantly (8% ee, Table 1, entry 7).
Room temperature reactions proceed with excellent results but to maintain reaction component solubility, adequate stirring and reasonable reaction times (particularly when using other nucleophiles), 40 °C appears to be a suitable temperature (Table 1, entries 8 and 9). The reaction can easily be performed on a gram-scale while simultaneously reducing the amount of rhodium to 0.5 mol% [Rh(COD)(Cl)]2, and ligand to 1.2 mol% (Table 1, entry 11).
For reaction scope, aliphatic carboxylic acids including bulky tert-butyl (2b) and adamantyl groups (2c), provided the corresponding products in excellent enantioselectivities (>93% ee), however smaller nucleophiles such as formic and acetic acid (2e and 2f respectively), give lower yields and ee's. In addition to the reduced yield and ee, the reaction with formic acid also gave small amounts of the achiral dibenzoyloxy derivative of 2 and under some conditions 1a may decompose to benzoic acid, which then undergoes competitive Rh-catalysed carboxylation to the remaining 1a. We were not able to separate this by-product from 2f (Fig. 2).
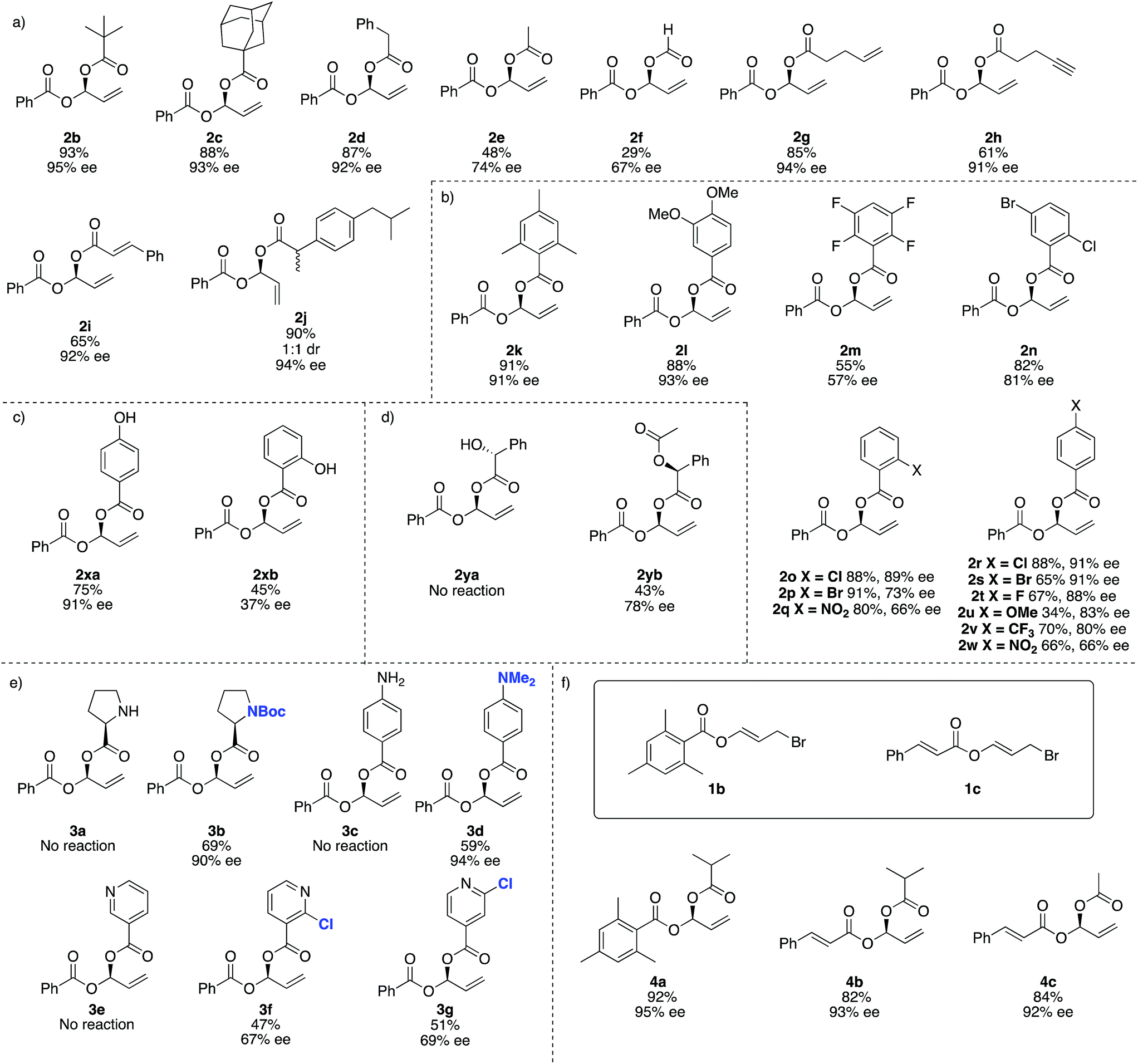 | ||
| Fig. 2 Reaction scope. (a) Scope of aliphatic carboxylic acids. (b) Scope of aromatic carboxylic acids. (c) 4-Hydroxybenzoic acid and 2-hydroxybenzoic acid give very different results. (d) Protection of free –OH groups may allow an otherwise problematic reaction to occur. (e) Nitrogen-containing and pyridyl substrates. (f) Using different carboxylic acid derived electrophiles. All reactions were carried out on a 0.4 mmol scale of the electrophile. All yields are isolated yields. Enantiomeric excesses determined by SFC using a chiral non-racemic stationary phase. | ||
This method is compatible with carboxylic acids that contain a terminal alkene (2g), terminal alkyne (2h) and an internal alkene (2i). When racemic ibuprofen is used as a nucleophile, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereoisomers is formed (both diastereomers having 94% ee, 2j).
:1 mixture of diastereoisomers is formed (both diastereomers having 94% ee, 2j).
Aromatic carboxylic acids generally work well as nucleophiles. 2,4,6-Trimethylbenzoic acid (2k) and 4,5-dimethoxybenzoic (2l) both gave high yields and ee's over 90%. 4-Chloro- (2r), 2-chloro- (2o) and 4-bromobenzoic acid (2s) also give good ee's. Interestingly the 2-bromo derivative (2p) was formed in excellent yield (91%) but the ee drops to only 73%. Increasing the electron withdrawing potential of ring substituents, for example having fluoro (2r), trifluoromethyl (2v), nitro (2q and 2w) or multiple halogens (2m and 2n) tends to decrease the yield and ee. Curiously 4-methoxybenzoic acid (2u) gives only a 34% yield and 83% ee, whereas 4,5-dimethoxybenzoic acid (2l) gave 88% yield and 93% ee.
For 4-hydroxybenzoic acid (2xa) we obtained good results (75%, 91% ee) but a hydroxy at the 2-position (2xb) is detrimental (45%, 37% ee), likely due to either chelation of 2xb to Rh during the reaction or hydrogen (or lithium) bonding of the 2-hydroxy group altering the nucleophilicity of the carboxylic acid. A free hydroxyl group in (R)-2-hydroxy-2-phenylacetic acid (used as a single enantiomer) entirely suppressed reactivity and no product 2ya is observed, but if acetylated (here the single S-enantiomer was arbitrarily used) 2yb can be obtained (43%, 78% ee).
Substrates containing free amino groups such as enantiomerically pure proline (3a) and 4-aminobenzoic acid (3c) give no product, but protected derivatives (single enantiomer Boc-proline and N,N-dimethyl-4-aminobenzoic acid) allowed synthesis of 3b and 3d in respectable yield with excellent enantioselectivity (>90% ee). 3b was obtained as a ‘single diastereoisomer’ which exists as a 3:1 mixture of rotamers at room temperature, as observed by NMR spectroscopy (see ESI† for NOESY spectra). Nicotinic acid did not give product 3e, but addition of a chloro group in the 2-position of the pyridyl ring allowed moderate yields and enantioselectivities to be achieved in formation of 3f and 3g. This observation is consistent with previous Rh-catalysed asymmetric processes,59 and overall these experiments suggest that many other carboxylic acid bearing heterocycles and heteroatoms would be compatible with this method if appropriate protecting group strategies are used.
We then examined different allylic bromides 1b and 1c58 with isobutyric acid which gave 4a and 4b with good yields and excellent ee's. We note that acetic acid in combination with 1a gave 48% yield and 74% ee however with 1c much better results (84%, 92% ee) are observed.
To investigate the stability of the gem-dicarboxylates, we subjected 2a to various conditions for 1 hour at room temperature. 2a is remarkably stable to a range of conditions; in aqueous acidic solutions of up to 3 M HCl, there is a negligible loss in the yield and ee of 2a (Fig. 3a, entries 1 and 2). In aqueous basic solutions of up to 2 M NaOH or KOH we see some loss of yield, there is no change in ee (Fig. 3a, entries 1–4). Unsurprisingly, the products were unstable to methanolic basic solutions in combination with potassium salts, and under these conditions complete decomposition was observed (Fig. 3a, entries 5 and 6). Using Na2CO3 and MeOH trace decomposition was observed after 1 h, and complete decomposition occurs overnight (Fig. 3).
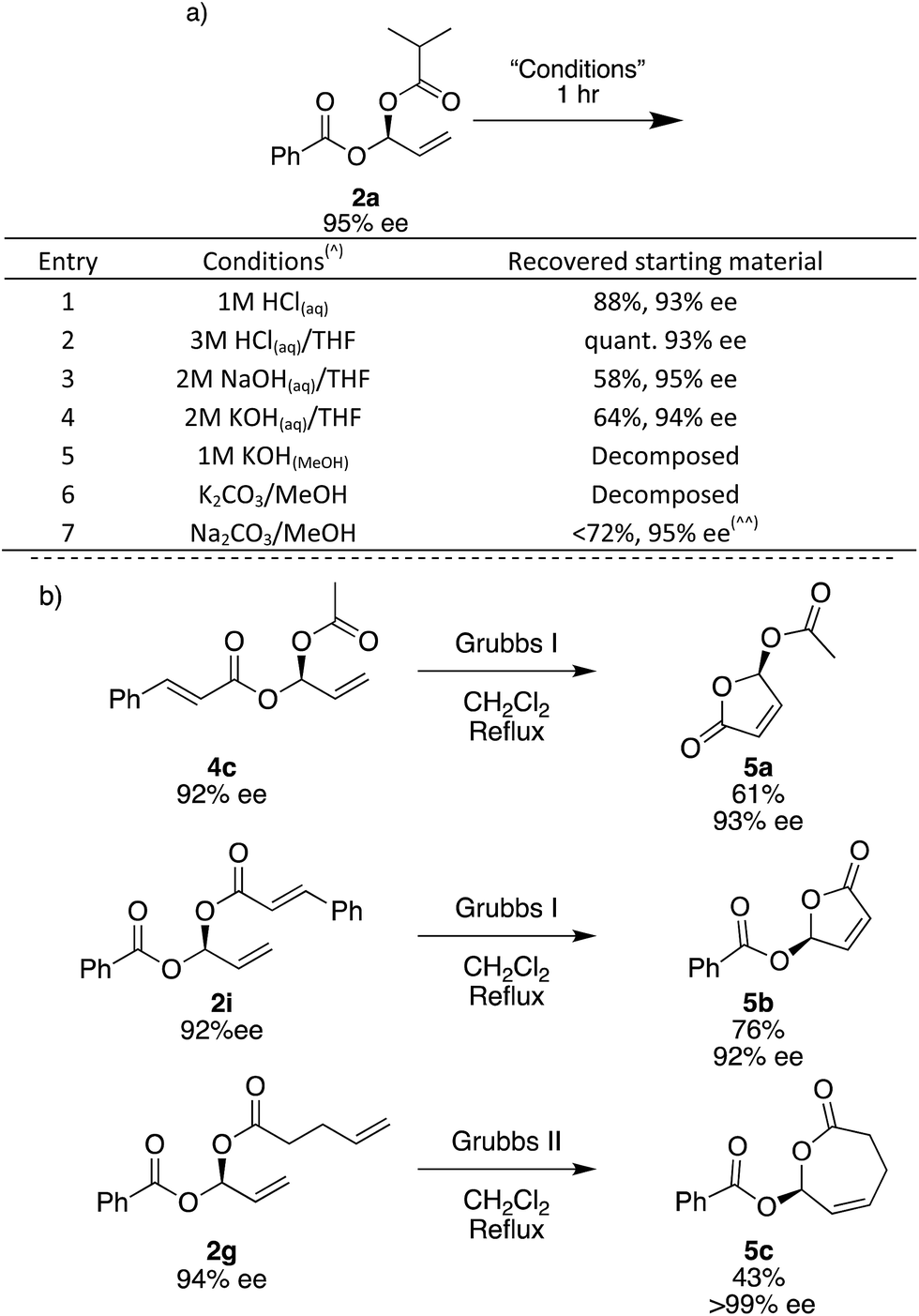 | ||
| Fig. 3 Stability and derivatization of gem-dicarboxylates. (a) (^) 25 mg of 2a stirred in 1 mL of a 1:1 mixture of solvents for 1 hour. (^^) Complete decomposition observed overnight. (aq) – queous, (MeOH) – methanolic solution. (b) Ring closing metathesis of gem-dicarboxylates to lactones. Isolated yields. Enantiomeric excesses determined by SFC. | ||
The gem-dicarboxylates shown here have not previously been described in the literature and since there has been no good way to access these chiral compounds before, their chemistry has not yet been explored. In order to demonstrate if the gem-dicarboxylates may be useful we briefly examined their conversion to other species using ring-closing metathesis. The synthesis of cyclic small-ring esters is of considerable interest as they frequently appear in natural products and show important biological activity.60 Asymmetric γ-butenolides have been widely studied but only acetyl esters in the γ-position have been reported, and asymmetry is normally induced using enzymes.61–64
Using the terminal alkene formed during the Rh-catalysed addition, we are able to access 5- and 7-membered lactones. Treatment of 4c and 2i with the 1st generation Grubbs catalyst in refluxing CH2Cl2 overnight gave 5-membered lactones 5a and 5b, which have a γ-stereogenic center. We assign the absolute stereochemistry of all gem-dicarboxylate products here, including diastereomeric mixture 2j, and single diastereoisomers 2yb and 3b, by comparing the optical rotation of lactone 5a with that quoted in the literature.62 This is the first report of an asymmetric synthesis of compound 5b.65 Using electrophile 1c specifically, the combination of Rh-catalysed carboxylation followed by RCM has the potential to give access to a range of new chiral γ-butenolides. Attempts to use the Grubbs I catalyst to form a 7-membered ring did not give the desired 5c, but the 2nd generation Grubbs catalyst gave 43% yield and 5c was obtained as a solid with >99% ee.
Conclusions
In conclusion, we have developed a method to form chiral gem-dicarboxylates by Rh-catalysed asymmetric carboxylation. Many different carboxylic acid nucleophiles can be used to give novel gem-dicarboxylates in good yields with high enantioselectivity. The products are remarkably stable to a variety of acidic and basic conditions. We have demonstrated that the products can be used to form other chiral acetal derivatives using the terminal alkene formed in the reaction in subsequent RCM reactions to access novel cyclic products including valuable γ-butenolides. We anticipate that these gem-dicarboxylates may have many other potential uses and now that we have described an efficient synthesis, this chemistry can now be explored more fully.More generally, linear products, chiral by virtue of a stereogenic carbon bearing two differentiated hetereoatoms, can be obtained by metal catalysed asymmetric additions to appropriately substituted electrophiles. The ease of synthesis and stability of the products suggests that a broad range of new chemical species may be accessible by developing strategies to form stereogenic carbon centers featuring different combinations of hetereoatoms.
Experimental procedures
General procedure for asymmetric carboxylation reaction to give chiral gem-dicarboxylates
In a flame-dried 10 mL round bottomed flask [Rh(COD)(Cl)]2 (2.5 mg, 0.0050 mmol, 0.0125 eq.), ligand A (5.3 mg, 0.012 mmol, 0.030 eq.) and LiOt-Bu (32 mg, 0.40 mmol, 1.0 eq.) were stirred in THF (2 mL) at 60 °C for 30 min. The reaction was cooled to 40 °C then a solution of the allylic bromide (1a–c, 0.40 mmol, 1.0 eq.) and the carboxylic acid (0.80 mmol, 2.0 eq.) in THF (1.5 mL) was added via syringe and the flask rinsed with THF (0.5 mL). The resulting mixture was then stirred at 40 °C until the reaction was complete by TLC. SiO2 was added and the solvent was then carefully evaporated. The resulting solid was directly loaded onto a chromatographic column and eluted with Et2O/pentane to afford the products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Engineering and Physical Sciences Research Council.Notes and references
- J. E. Aho, P. M. Pihko and T. K. Rissa, Chem. Rev., 2005, 105, 4406–4440 CrossRef PubMed
.
- Y. Qi and S. Ma, ChemMedChem, 2011, 6, 399–409 CrossRef PubMed
- H. Matsutani, S. Ichikawa, J. Yaruva, T. Kusumoto and T. Hiyama, J. Am. Chem. Soc., 1997, 119, 4541 CrossRef
- M. D. Spantulescu, M. A. Boudreau and J. C. Vederas, Org. Lett., 2009, 11, 645–648 CrossRef PubMed
- S. D. Rychnovsky and B. M. Bax, Tetrahedron Lett., 2000, 41, 3593–3596 CrossRef
- M. Uchiyama, M. Oka, S. Harai and A. Ohta, Tetrahedron Lett., 2001, 42, 1931–1934 CrossRef
- P. H. S. Paioti, J. M. Ketcham and A. Aponick, Org. Lett., 2014, 16, 5320–5323 CrossRef PubMed
- S. G. Davies and L. M. A. R. B. Correia, Chem. Commun., 1996, 1803–1804 RSC
- G. S. Weatherhead, J. H. Houser, J. G. Ford, J. Y. Jamieson, R. R. Schrock and A. H. Hoveyda, Tetrahedron Lett., 2000, 41, 9553–9559 CrossRef
- H. Frauenrath, S. Reim and A. Wiesner, Tetrahedron: Asymmetry, 1998, 9, 1103–1106 CrossRef
- S. D. Burke, N. Müller and C. M. Beaudry, Org. Lett., 1999, 1, 1827–1829 CrossRef PubMed
- I. Paterson, D. Y.-K. Chen, M. J. Coster, J. L. Acea, J. Bach, K. R. Gibson, L. E. Keown, R. M. Oballa, T. Trieselmann and D. J. Wallace, Angew. Chem., Int. Ed., 2001, 40, 4055–4060 CrossRef PubMed
- M. Ball, M. J. Gaunt, D. F. Hook, A. S. Jessiman, S. Kawahara, P. Orsini, A. Scolaro, A. C. Talbot, H. R. Tanner, S. Yamanoi and S. V. Ley, Angew. Chem., Int. Ed., 2005, 44, 5433–5438 CrossRef PubMed
- A. B. Smith, V. A. Doughty, Q. Lin, L. Zhuang, M. D. McBriar, A. M. Boldi, W. H. Moser, N. Murase, K. Nakayama and M. Sobukawa, Angew. Chem., Int. Ed., 2001, 40, 191–195 CrossRef
- M. J. Gaunt, D. F. Hook, H. R. Tanner and S. V. Ley, Org. Lett., 2003, 5, 4815–4818 CrossRef PubMed
- S. Favre, S. Gerber-Lemaire and P. Vogel, Org. Lett., 2007, 9, 5107–5110 CrossRef PubMed
- S. Favre, P. Vogel and S. Gerber-Lemaire, Molecules, 2008, 13, 2570–2600 CrossRef PubMed
- Y. Y. Khomutnyk, A. J. Argüelles, G. A. Winschel, Z. Sun, P. M. Zimmerman and P. Nagorny, J. Am. Chem. Soc., 2016, 138, 444–456 CrossRef PubMed
- Y. Liang, E. B. Rowland, G. B. Rowland, J. A. Perman and J. C. Antilla, Chem. Commun., 2007, 4477–4479 RSC
- G. B. Rowland, H. Zhang, E. B. Rowland, S. Chennamadhavuni, Y. Wang and J. C. Antilla, J. Am. Chem. Soc., 2005, 127, 15696–15697 CrossRef PubMed
- X. Cheng, S. Vellalath, R. Goddard and B. List, J. Am. Chem. Soc., 2008, 130, 15786–15787 CrossRef PubMed
- M. Rueping, A. P. Antonchick, E. Sugiono and K. Grenader, Angew. Chem., Int. Ed., 2009, 48, 908–910 CrossRef PubMed
- G. Li, F. R. Fronczek and J. C. Antilla, J. Am. Chem. Soc., 2008, 130, 12216–12217 CrossRef PubMed
- G. K. Ingle, M. G. Mormino, L. Wojtas and J. C. Antilla, Org. Lett., 2011, 13, 4822–4825 CrossRef PubMed
- F. D. Toste, S. Biswas, K. Kubota, M. S. Sigman, M. Orlandi, M. Turberg and D. Miles, Angew. Chem., Int. Ed., 2017, 57, 589–593 Search PubMed
- I. Čorić, S. Vellalath and B. List, J. Am. Chem. Soc., 2010, 132, 8536–8537 CrossRef PubMed
- I. Čorić and B. List, Nature, 2012, 483, 315–319 CrossRef PubMed
- P. Nagorny, Z. Sun and G. A. Winschel, Synlett, 2013, 24, 661–665 CrossRef
- Z. Sun, G. A. Winschel, A. Borovika and P. Nagorny, J. Am. Chem. Soc., 2012, 134, 8074–8077 CrossRef PubMed
- Y. Zhou and B. Breit, Chem.–Eur. J., 2017, 23, 18156–18160 CrossRef PubMed
- C. Li and B. Breit, Chem.–Eur. J., 2016, 22, 14655–14663 CrossRef PubMed
- A. Lumbroso, P. Koschker, N. R. Vautravers and B. Breit, J. Am. Chem. Soc., 2011, 133, 2386–2389 CrossRef PubMed
- F. A. Cruz, Y. Zhu, Q. D. Tercenio, Z. Shen and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 10641–10644 CrossRef PubMed
- Q. A. Chen, Z. Chen and V. M. Dong, J. Am. Chem. Soc., 2015, 137, 8392–8395 CrossRef PubMed
- P. A. Evans, E. A. Clizbe, M. J. Lawler and S. Oliver, Chem. Sci., 2012, 3, 1835 RSC
- P. A. Evans, S. Oliver and J. Chae, J. Am. Chem. Soc., 2012, 134, 19314–19317 CrossRef PubMed
- B. W. H. Turnbull and P. A. Evans, J. Am. Chem. Soc., 2015, 137, 6156–6159 CrossRef PubMed
- B. W. H. Turnbull, S. Oliver and P. A. Evans, J. Am. Chem. Soc., 2015, 137, 15374–15377 CrossRef PubMed
- M. Sidera and S. P. Fletcher, Nat. Chem., 2015, 7, 935–939 CrossRef PubMed
- P. Koschker and B. Breit, Acc. Chem. Res., 2016, 49, 1524–1536 CrossRef PubMed
- M. Gärtner, S. Mader, K. Seehafer and G. Helmchen, J. Am. Chem. Soc., 2011, 133, 2072–2075 CrossRef PubMed
- J. S. Cannon, S. F. Kirsch and L. E. Overman, J. Am. Chem. Soc., 2010, 132, 15185–15191 CrossRef PubMed
- D. J. Covell and M. C. White, Angew. Chem., Int. Ed., 2008, 47, 6448–6451 CrossRef PubMed
- N. Kanbayashi and K. Onitsuka, J. Am. Chem. Soc., 2010, 132, 1206–1207 CrossRef PubMed
- M. Kim, S. Kang and Y. H. Rhee, Angew. Chem., Int. Ed., 2016, 55, 9733–9737 CrossRef PubMed
- S. H. Jang, H. W. Kim, W. Jeong, D. Moon and Y. H. Rhee, Org. Lett., 2018, 20, 1248–1251 CrossRef PubMed
- B. M. Trost, E. Gnanamani and C.-I. I. J. Hung, Angew. Chem., Int. Ed., 2017, 56, 10451–10456 CrossRef PubMed
- J. Y. Hamilton, S. L. Rössler and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 8082–8085 CrossRef PubMed
- K. Geurts, S. P. Fletcher and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 15572–15573 CrossRef PubMed
- B. M. Trost and C. B. Lee, J. Am. Chem. Soc., 2001, 123, 3671–3686 CrossRef PubMed
- B. M. Trost, J. T. Masters and A. C. Burns, Angew. Chem., Int. Ed., 2013, 52, 2260–2264 CrossRef PubMed
- F. Freeman and E. M. Karchefski, J. Chem. Eng. Data, 1977, 22, 355–357 CrossRef
- J. K. Michie and J. A. Miller, Synthesis, 1981, 824 CrossRef
- J. M. Thomas, Angew. Chem., Int. Ed., 1988, 27, 1673–1691 CrossRef
- B. Karimi, H. Seradj and G. R. Ebrahimian, Synlett, 2000, 623–624 Search PubMed
- F. Zhang, H. Liu, Q.-J. Zhang, Y.-F. Zhao and F.-L. Yang, Synth. Commun., 2010, 40, 3240–3250 CrossRef
- B. Zhang, S.-F. Zhu and Q.-L. Zhou, Tetrahedron Lett., 2013, 54, 2665–2668 CrossRef
- M. Lombardo, S. Morganti and C. Trombini, J. Org. Chem., 2003, 68, 997–1006 CrossRef PubMed
- P. Schäfer, T. Palacin, M. Sidera and S. P. Fletcher, Nat. Commun., 2017, 8, 15762 CrossRef PubMed
- B. Mao, M. Fañanás-Mastral and B. L. Feringa, Chem. Rev., 2017, 117, 10502–10566 CrossRef PubMed
- N. G. Turrini, M. Hall and K. Faber, Adv. Synth. Catal., 2015, 357, 1861–1871 CrossRef
- J. Brinksma, H. van der Deen, A. van Oeveren and B. L. Feringa, J. Chem. Soc., Perkin Trans. 1, 1998, 0, 4159–4164 RSC
- H. Van Der Deen, R. P. Hof, A. Van Oeveren, B. L. Feringa and R. M. Kellogg, Tetrahedron Lett., 1994, 35, 8441–8444 CrossRef
- H. Van Der Deen, A. D. Cuiper, R. P. Hof, A. Van Oeveren, B. L. Feringa and R. M. Kellogg, J. Am. Chem. Soc., 1996, 118, 3801–3803 CrossRef
- B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 3090–3100 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01786g |
| This journal is © The Royal Society of Chemistry 2018 |