
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerforming DNA nanotechnology operations on a zebrafish†
Jian
Yang
*a,
Zhuojun
Meng
*b,
Qing
Liu
b,
Yasuhito
Shimada
 cd,
René C. L.
Olsthoorn
a,
Herman P.
Spaink
c,
Andreas
Herrmann‡
bef and
Alexander
Kros‡
a
cd,
René C. L.
Olsthoorn
a,
Herman P.
Spaink
c,
Andreas
Herrmann‡
bef and
Alexander
Kros‡
a
aSupramolecular & Biomaterials Chemistry, Leiden Institute of Chemistry, Leiden University, P.O. Box 9502, 2300 RA Leiden, The Netherlands. E-mail: a.kros@chem.leidenuniv.nl
bZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: a.herrmann@rug.nl
cInstitute of Biology, Leiden University, Leiden, The Netherlands
dDepartment of Integrative Pharmacology, Mie University Graduate School of Medicine, Mie, Japan. E-mail: shimada.yasuhito@mie-u.ac.jp
eDWI-Leibniz Institute for Interactive Materials, Forckenbeckstr. 50, 52056 Aachen, Germany
fInstitute of Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, 52074, Aachen, Germany
First published on 20th July 2018
Abstract
Nanoscale engineering of surfaces is becoming an indispensable technique to modify membranes and, thus cellular behaviour. Here, such membrane engineering related was explored on the surface of a living animal using DNA nanotechnology. We demonstrate the immobilization of oligonucleotides functionalized with a membrane anchor on 2 day old zebrafish. The protruding single-stranded DNA on the skin of zebrafish served as a handle for complementary DNAs, which allowed the attachment of small molecule cargo, liposomes and dynamic relabeling by DNA hybridization protocols. Robust anchoring of the oligonucleotides was proven as DNA-based amplification processes were successfully performed on the outer membrane of the zebrafish enabling the multiplication of surface functionalities from a single DNA-anchoring unit and the dramatic improvement of fluorescent labeling of these animals. As zebrafish are becoming an alternative to animal models in drug development, toxicology and nanoparticles characterization, we believe the platform presented here allows amalgamation of DNA nanotechnology tools with live animals and this opens up yet unexplored avenues like efficient bio-barcoding as well as in vivo tracking.
Introduction
Synthetic biology and cell surface engineering techniques in vitro and in vivo have resulted in novel tools for the development of membrane biology,1 offering promising membrane-based devices that may enable new types of artificial tissues,2,3 biosensors,4,5 drug delivery approaches,6,7 3D bio-printing and the study of lipid metabolism.8 To boost the development of these technologies, there is a growing need to enhance surface engineering techniques of membranes under in vitro and in vivo conditions with particular emphasis on exploiting artificial surface receptors9 and designing novel biomaterials guided by natural processes,10 such as self-assembling peptides, proteins and DNA oligonucleotides. Especially the latter class of biomacromolecules is very appealing to fabricate complex architectures because the sequence specific base pairing of oligonucleotides allows the prediction of the resulting structure based on the sequence composition; qualifying nucleic acids as indispensible building blocks in soft matter nanotechnology.11 In conjunction with advances in solid phase DNA synthesis methods,12 a plethora of programmed 2- and 3-dimensional self-assembled architectures can be achieved.13,14The facile chemical modification of oligonucleotides with hydrophobic anchors also permits the fabrication of DNA-based functional membranes.15 In the context of liposomes, DNA hybridization-induced vesicle aggregation,16 and fusion have been realized.17–19 Photoresponsive DNA–lipid assemblies, fabricated by either anchoring DNA with a azobenzene moiety20 or hybridization of a photosensitizer, mediated cargo release from liposomes.21 While these functions relied on simple DNA amphiphiles that were inserted in the membrane, vesicle deformation and even destruction of these containers was achieved with immobilizing and polymerizing more complex DNA origami structures.13,22 Further extension of these concepts led to a DNA-based atomistically determined molecular valve capable of controlling transport of small molecules across a biological membrane.23,24 However, what is still lacking in this burgeon field is the actual application of DNA nanotechnology in an in vivo environment.
In this contribution, we implement such membrane engineering related DNA nanotechnology on the surface of a living animal. We demonstrate that oligonucleotides functionalized with a membrane anchor can be stably immobilized on a zebrafish. Protruding single-stranded DNA strands in the exterior membrane of zebrafish were functionalized through hybridization by Watson–Crick base pairing employing complementary DNA sequences. In this way, small molecules and liposomes were guided and attached to the fish surface. The anchoring process can be designed to be reversible allowing exchange of surface functionalities by simple addition of the complementary DNA sequence. Finally, a DNA based amplification process was performed on the skin of zebrafish enabling the multiplication of surface functionalities from a single DNA anchoring unit.
Results and discussion
For surface anchoring of oligonucleotides, we employed lipid-modified DNA,25 consisting of a hydrophobic alkyl chain and an ethyne function attached to the nucleobase, i.e. at the 5-position of uracil (Scheme 1a). The incorporation of the hydrophobic building blocks was achieved using phosphoramidites during solid phase synthesis employing an automated DNA synthesizer by a previously established procedure.25 Due to this convenient incorporation method, multiple hydrophobic nucleotides can be introduced into the same oligonucleotide as desired, providing tuneable interaction with the phospholipid membrane. Here, we chose four lipid-modified deoxyuridine units attached either to the 3′- or to 5′-end of the oligonucleotide sequences, which are comprised of 18 or 28 nucleotides (Scheme 1b–d). They are abbreviated as UxTy, where x represents the number of lipid-modified uracils at the terminus while y denotes the overall number of nucleotides of the sequence (Scheme 1b–d). The four consecutive hydrophobic anchoring units guarantee stable incorporation into a phospholipid membrane of vesicles for at least 24 h as proven by a fluorometric assay26 (for details see ESI†).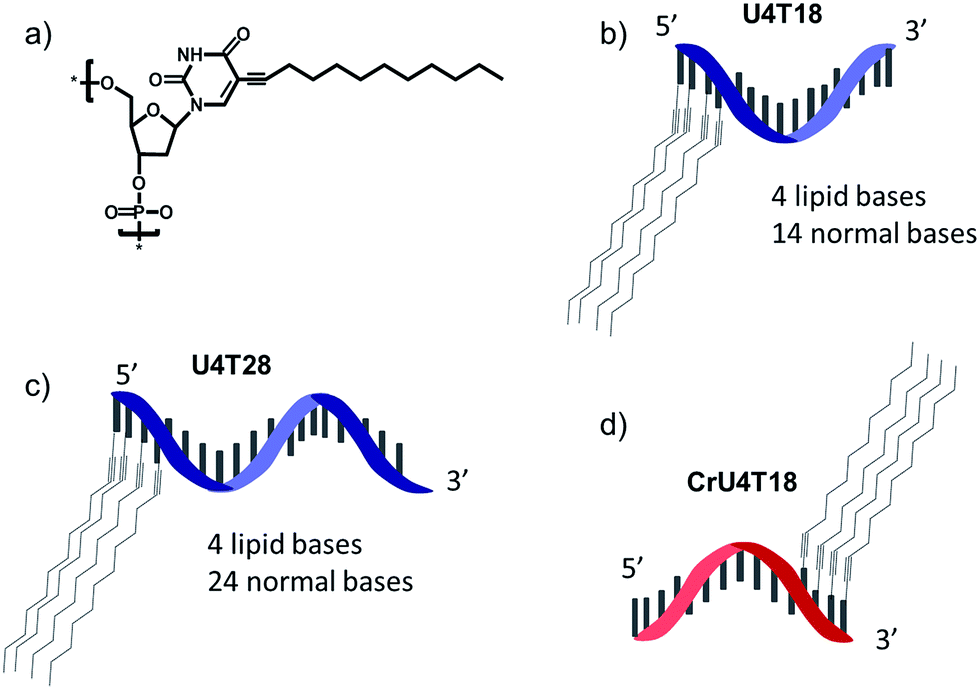 | ||
| Scheme 1 Schematic representation of structures: (a) chemical structure of lipid-modified deoxyuridine (dU). (b) U4T18, (c) U4T28 and (d) CrU4T18. | ||
DNA hybridization on a zebrafish surface and strand replacement
Prior to investigating the functionalization of zebrafish with a DNA tag, we performed studies to establish the anchoring of DNA amphiphiles in a phospholipid bilayer, subsequent hybridization and strand replacement. For that purpose, vesicles served as a model system (for details see ESI†). Previously, hybridization of DNA on a liposome surface was demonstrated,27 and sequence-specific DNA mediated liposome fusion can be detected by microRNA,28 however strand replacement has not been shown yet in animal systems. Furthermore, we investigated whether it is possible to insert the lipid-modified DNA sequences into the membrane of live zebrafish to function as an artificial receptor. This DNA-based receptor can then be used to modify the cell membrane with new functions, such as for immobilization of surface probes or other payloads for targeted delivery onto or through the lipid bilayer. For that purpose, 1 μM lipid–DNA U4T18 was utilized (Scheme 1b) and incubated with zebrafish embryos of 1 day post-fertilization (dpf) for 1 h. Previously the group of Irvine29 performed cell membrane insertion of oligonucleotides carrying diacyl-lipid (C18) or cholesterol anchors using an incubating time of 30 min. Because electrostatic repulsion might delay incorporation into the membrane, we prolonged the incubation time of lipid–DNA with the zebrafish membrane to 1 h.Subsequently, the 20-mer oligonucleotide C594 that contains the red emitting fluorophore ATTO594 and partially complementary to U4T18 was added to the lipid-modified surface anchor (Fig. 1, step 1). Fluorescence microscopy showed staining of the fish surface as evidenced by the red emission (Fig. 1b) indicating that U4T18 was successfully incorporated into the fish skin and that the protruding single-stranded DNA chain can selectively undergo sequence specific hybridization. Next, we investigated if it is possible to dynamically exchange the red label by a removal strand. This strategy of strand replacement was previously introduced in the context of a DNA fueled molecular machine.30 Here, we employed this strategy for the reversible and gentle labeling of the skin of a living animal. Therefore, 2 μM 20-mer of a DNA oligonucleotide that is fully complementary to C594 was introduced to peel off C594 from U4T18. The removal of C594 was proven by disappearance of red fluorescence on the fish surface (Fig. 1c). After removal of C594, U4T18 remained on the skin and kept the ability to hybridize with other complementary DNA sequences (Fig. 1c, step 2) such as C488, a 14-mer DNA oligonucleotide complementary to U4T18 and labeled with the green emitting fluorophore ATTO488. Clear evidence for the strand replacement was the green fluorescence observed on the exterior of the fish (Fig. 1d). Control experiments in which U4T18 lipid–DNA or 20-mer oligonucleotides were omitted or only incubated with C488 (ESI Fig. S1 and S2†) showed no (changes in) fluorescence.
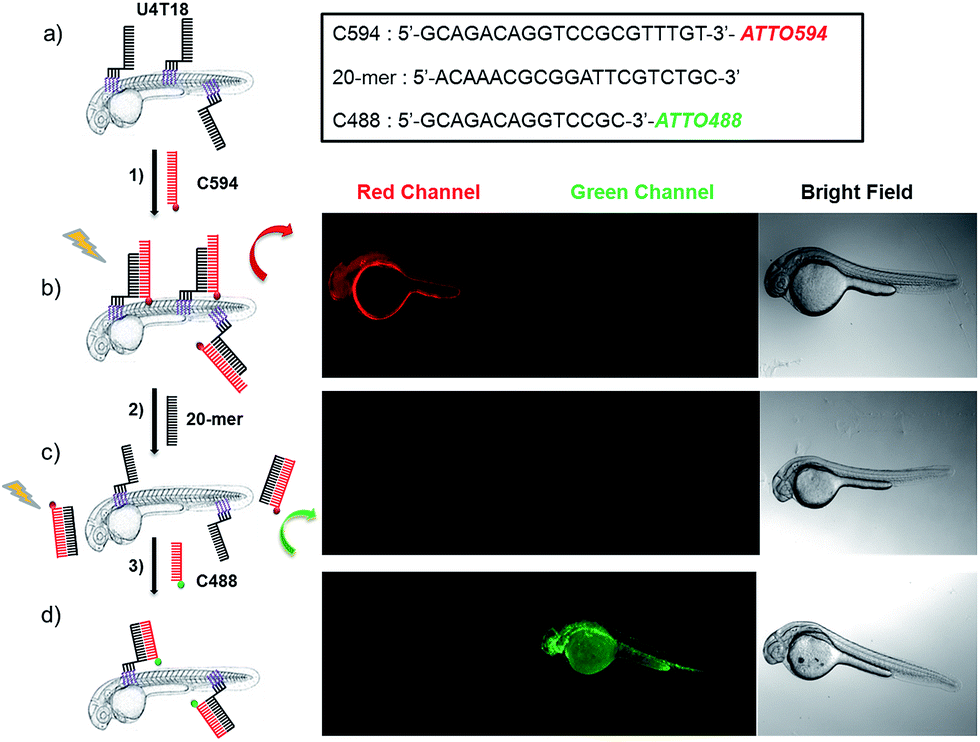 | ||
| Fig. 1 Fluorescent labeling and DNA replacement on the surface of zebrafish embryos. (a) Lipid DNA (U4T18) is anchored on the skin membrane of 1 dpf zebrafish embryos, and (b) hybridizes to 1 μM ATTO594 fluorescently labeled complementary DNA (C594) (step 1), resulting in red fluorescence on the zebrafish surface. (c) A 20-mer oligonucleotide replaces C594 by means of strand displacement (step 2), resulting in loss of fluorescence of the fish. (d) Addition of 1 μM ATTO488 fluorescently labeled complementary DNA (C488) results in hybridization with U4T18 (step 3) and the appearance of a green fluorescence at the zebrafish surface. The insert represents the sequences of C594, 20-mer and C488. Red channel = ATTO594; green channel = ATTO488. | ||
Loading larger containers to the zebrafish surface by Watson–Crick base pairing
After demonstrating that base pairing is a very efficient tool to attach small oligonucleotides to the live animal surface, we attempted to load larger cargo to the zebrafish membrane. Therefore, the phospholipid bilayer of liposomes of 120 nm in diameter was loaded with rhodamine-functionalized phospholipid (Rh-DHPE), which is characterized by a red emission. Likewise, the surface of the vesicles was decorated with lipid-modified DNA that is complementary to that on the zebrafish (Fig. 2a). Proof of successful loading of the fish surface by supramolecular bonds was provided by fluorescence microcopy showing characteristic red fluorescence originating from the liposomes, which are bound to the fish surface (Fig. 2b). This result opens the way for potential DNA-mediated delivery of liposomal cargo. These experiments demonstrate that the DNA hybridization overcomes the repulsive hydration forces between the lipid head groups and brings the two lipid bilayers with complementary DNA in close proximity to achieve surface docking (aggregation).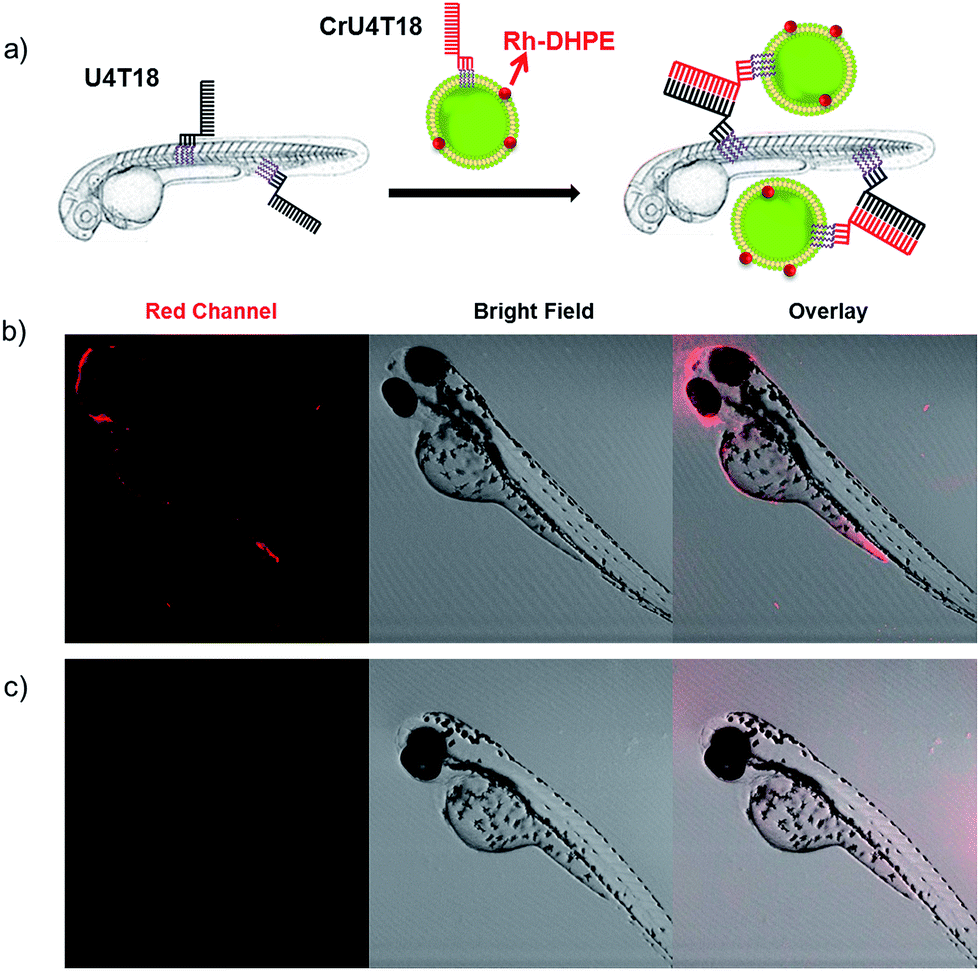 | ||
Fig. 2 DNA duplex formation between U4T18 and CrU4T18 decorated liposomes on the surface of zebrafish. (a) Schematic representation of liposomes docking on the surface of zebrafish embryos by lipid–DNA hybridization. Confocal images of 2 dpf zebrafish treated with (b) U4T18 for 1 h, followed by incubation with CrU4T18 decorated liposomes or (c) treated with 1 μM CrU4T18 decorated liposomes in absence of U4T18. The concentration of total lipids (DOPC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DOPE:Chol = 2:1:1 mol%) was 0.5 mM. Red channel: Rh-DHPE. :DOPE:Chol = 2:1:1 mol%) was 0.5 mM. Red channel: Rh-DHPE. | ||
Nucleic acid mediated amplification process on live fish surface
To demonstrate the broad versatility of zebrafish surface engineering enabled by lipid–DNA, we performed a DNA-based amplification process on the animal, i.e. hybridization chain reaction (HCR). Previously, this method was employed for augmenting the signal during nucleic acid detection.31 Later, this technique was utilized for surface modification with DNA hydrogels.32 Here, we demonstrate that this supramolecular polymerization can be performed on the exterior of the living animal.Since initiation of HCR from a lipid membrane was not demonstrated before we first established a HCR protocol for decorating the outer surface of liposomes with a DNA layer (for details see ESI†). Then, the optimized DNA anchors and sequences were employed for modification of the fish surface (Fig. 3a). Compared to the previous experiments, for membrane anchoring U4T28, a 28 mer lipid–DNA with 4 modified lipid bases, was bound to zebrafish skin. Next, hairpin strands M1 (partially complementary to U4T28) and M2 (partially complementary to M1) were added.31 Hybridization of M1 to U4T28 results in liberation of its loop that subsequently can hybridize with M2. Opening of the M2 hairpin exposes a sequence that binds to a new M1 monomer from the solution. In turn, opening of the M1 hairpin exposes a sequence that can bind new M2. This effectively triggers the “supramolecular polymerization” of M1 and M2 with surface anchor U4T28 as initiator, leading to extended DNA on the zebra fish membrane. The DNA sequences used in this study are summarized in Table S1.† The realization of HCR was investigated on the membrane of 1 dpf zebrafish embryo. As shown schematically in Fig. 3b, U4T28 at a concentration of 1 μM was exposed to zebrafish embryos for 1 h, followed by the incubation with a mixture of 2 μM M1-FAM and 2 μM M2-Cy3 for 2 h. Both monomers were labeled with two different fluorophores (FAM and Cy3). Green and red fluorescence could be clearly observed due to progression of polymerization of M1-FAM and M2-Cy3 from the initiator (Fig. 3c).
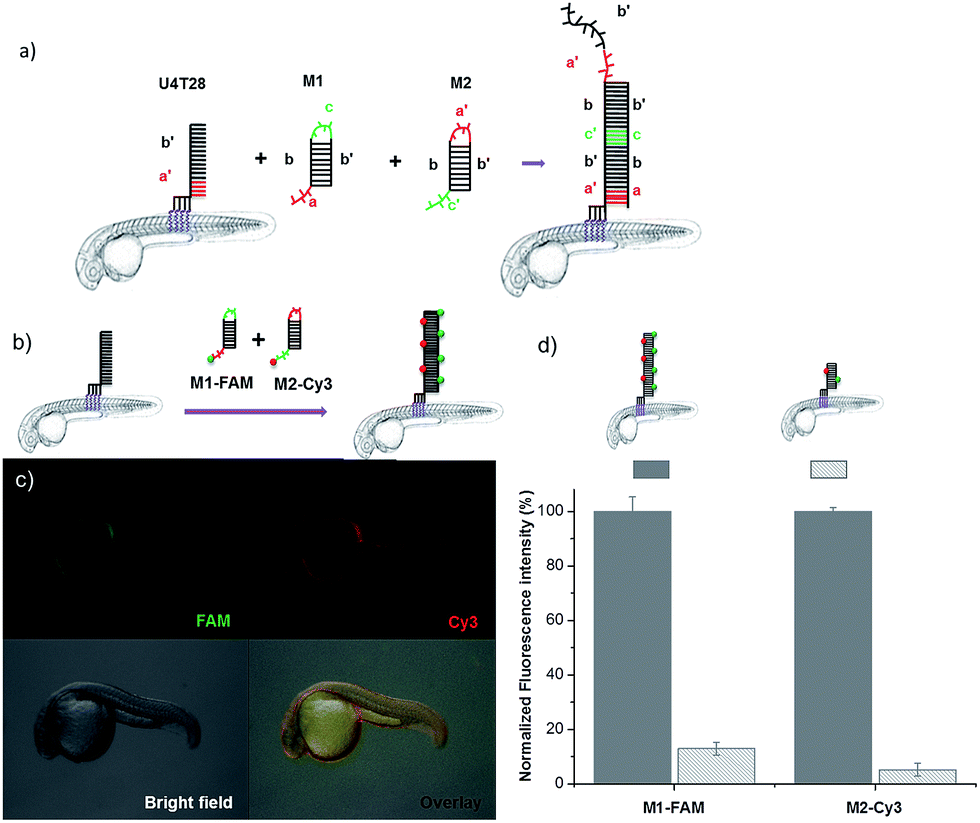 | ||
| Fig. 3 DNA hybridization chain reaction (HCR) on the surface of zebrafish embryos. (a) a′, b′, and c′ are regions that are complementary to regions a, b, and c, respectively. Hairpin M1 can be unfolded by hybridization with initiator U4T28, resulting in growing DNA strands. (b) Addition of 6-FAM fluorescently labeled M1 (M1-FAM) and Cy3 fluorescently labeled M2 (M2-Cy3) to the U4T28 pre-treated zebrafish results in DNA HCR and concomitant increase in fluorescence. (c) Fluorescence images of 1 dpf zebrafish embryos after incubation with U4T28 for 1 h and subsequent exposure to 1 μM M1-FAM and M2-Cy3 for 1 h. Green channel: 6-FAM; red channel: Cy3. (d) Normalized fluorescence intensity of attached DNA on the surface of zebrafish embryos. Fluorescence intensities of images (c) and ESI Fig. S3a† were calculated by Image J and plotted as a percentage relative to the fluorescence of M1-FAM or M2-Cy3 of Fig. 3c. The intensities of Fig. 3c were set to 100%. | ||
In the absence of a HCR (see ESI Fig. S3a† for experimental details), the fluorescent signals of M1-FAM and M2-Cy3 were 10 and 20 times lower, respectively, than those obtained in the presence of HCR (Fig. 3d). Also, when the monomer M1-FAM was omitted the HCR could not proceed and consequently no fluorescence could be detected on the fish surface (ESI Fig. S3b†).
The signal increase by HCR was also clearly demonstrated by an experiment involving non-fluorescent M2 and M1-FAM (Fig. 4). In case of HCR approximately 10-fold stronger green fluorescence was observed on the surface of 2 dpf zebrafish embryos (Fig. 4a) compared to labeling with a single fluorophore per anchor unit (Fig. 4b).
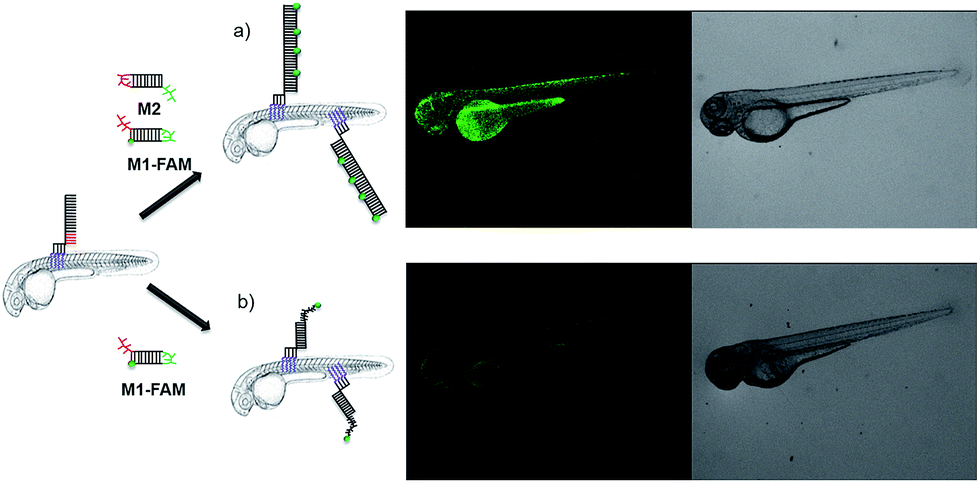 | ||
| Fig. 4 In vivo DNA HCR enhances the fluorescence intensity of labeling. (a) Fluorescence images of 2 dpf zebrafish embryos that were first decorated with U4T28, followed by 3 times washing with egg water, incubation with M1-FAM for 1 h and M2 for another 1 h. (b) Only FAM fluorescent labeled M1 (M1-FAM) was added after anchoring of U4T28 on the fish and washing three times with egg water. Green channel: M1-FAM. | ||
Experimental section
Materials
Cholesterol (Chol), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) were purchased from Avanti Polar Lipids (Alabaster, USA) (purity >99%) and used without further purification. Headgroup-labeled phospholipid, Lissamine rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (triethylammonium salt) (Rh-DHPE) and N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoeth-anolamine (triethylammonium salt) (NBD-DHPE) were purchased from Invitrogen (Amsterdam, Netherlands), and used as received (Scheme S1†). Tricaine and PTHK polysulfone membrane filters with a NMWL of 100.000 Da were purchased from Sigma-Aldrich. The DNA-dye conjugates C488, C594, M1-FAM and M2-Cy3 (Table S1†) were purchased from Biomers.net GmbH (Ulm, Germany). Triton X-100 (10% in water) was purchased from Nota company, United States. Anhydrous CHCl3 was acquired from Acros Organics (Geel, Belgium) and stored over molecular sieves. Preparation of liposomes was performed in double deionized water (Super Q Millipore system).Preparation and characterization lipid–DNA liposomes
An appropriate amount of freeze-dried lipid–DNA was mixed with DOPC:DOPE:Chol (50:25:25 mol% in chloroform) to reach a total of 0.1 mM lipid with 0.5 mol% lipid–DNA. Afterwards, chloroform was removed by evaporation under nitrogen and then under vacuum overnight. The dried lipid:lipid–DNA mixture was dissolved in an aqueous PBS buffer (150 mM NaCl, 15 mM K2HPO4, 5 mM KH2PO4) by 5 cycles of vortexing and freeze–thawing. Subsequently, the sample was dispersed by extruding 21 times, using an extruder and 100 nm polycarbonate membranes (Whatman), to obtain large unilamellar vesicles (LUVs), after which the liposomes with lipid–DNA were incubated at 50 °C for 1 h. All lipid–DNA liposomes were used within one day and had an average diameter of around 120 nm as determined by DLS (ALV/CGS-3 ALV-Laser Vertriebsgesellschaft m-b.H., Langen, Germany). The molar ratio between lipid and U4T18 was 500:1. The Final lipid concentration was 0.45 mM.
Zebrafish strain, husbandry, and egg collection
Wildtype zebrafish were used in this study. Livestock was maintained and handled according to the guidelines from http://zfin.org. Fertilization was performed by natural spawning at the beginning of the light period, and eggs were raised at 28 °C. All experimental procedures were conducted in compliance with the directives of the animal welfare committee of Leiden University.Microscopy images
Zebrafishes were seeded in a glass bottom flask with egg water. After incubation for 1 h with lipid–DNA, they were washed three times and then incubated with other DNA oligonucleotides for 1 h. For live imaging, zebrafish embryos were anaesthetized with 0.003% tricaine and mounted on 0.6% low-melting agarose. Fluorescent images were acquired using a Leica TCS SP8 confocal laser scanning microscope (Leica Microsystems, Wetzlar, Germany) and merged with Leica application suite advanced fluorescence software (Leica Microsystems) or ImageJ software (National Institutes of Health, Bethesda, MD, USA). A Leica MZ16FA stereo microscope was used for stereo images. Images were adjusted for brightness and contrast using ImageJ. The wavelength settings for C488 were Ex/Em: 495/520 nm (Ex laser: 488 nm), for C594 Ex/Em: 601/627 nm (Ex laser: 532 nm), for M1-FAM Ex/Em: 494/518 nm (Ex laser: 488 nm), and M2-Cy3 Ex/Em: 550/570 nm (Ex laser: 532 nm).Discussion and conclusion
Previously, oligonucleotides were covalently attached to live cells by metabolic oligosaccharide engineering allowing the introduction of orthogonal chemical handles on the cell surface for DNA anchoring without dependence on endogenous receptors.33 Besides oligosaccharides, cell-surface proteins were exploited for the chemical modification of cells with DNA.34,35 Similarly, cell-surface proteins were decorated with DNA by non-covalent interactions.36 An alternative strategy for introducing artificial DNA receptors on live cell surfaces represents the utilization of oligonucleotides carrying hydrophobic membrane anchors, as described in this study.37 Based on such an anchoring strategy, Watson–Crick base pairing was exploited for the programmed synthesis of three-dimensional tissues.14 The examples above demonstrate that anchored DNA in a lipid bilayer developed into a powerful tool for realizing exciting functionalities in the context of synthetic and natural membranes, even including live cells. Similar work from Qu et al. also showed that RGD-functionalized DNA can program on-chip cell adhesion, allowing regulation of cell behavior.38 On the other hand, DNA nanostructures were employed in higher organisms in the context of functional in vivo imaging39 and for the targeted delivery of siRNA.40 To the best of our knowledge, there is no experiment about DNA-based membrane engineering in a living animal involving a wide variety of functions yet.Here, we demonstrated that lipid–DNA sequences with four anchoring units could be readily incorporated in the surface layer of zebrafish embryos. The single-stranded DNA present on the surface can be functionalized by Watson–Crick base pairing enabling the sequence specific functionalization of the live animal with small molecules or larger cargos, for example, liposomal systems. The payloads connected by the supramolecular tether DNA can be reversibly removed employing a removal strand, which represents a very mild stimulus only requiring the addition of a DNA sequence not affecting the viability of the fish. Finally, we successfully demonstrated the performance of a DNA mediated amplification process on the fish skin. The hybridization chain reaction allows attachment of multiple moieties on a single anchored DNA strand allowing multiplication of cargoes or signals on the surface. Moreover, it was shown that surface modification of model membranes in form of liposomes by various DNA nanotechnology procedures could be easily transferred to the live animal. This allows establishment of DNA based surface functionalization procedures and their facile and fast implementation in zebrafish. Due to the broad application of zebrafish as animal model in drug development, toxicology and nanoparticles characterization,41 we believe that the platform presented here combines for the first time the advantages of DNA nanotechnology tools with live animals and opens up yet unexplored new avenues like for example efficient bio-barcoding as well as in vivo tracking to name a few.
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Leiden University and approved by the Animal Ethics Committee of Leiden University. Zebrafish livestock was maintained and handled according to the guidelines from http://zfin.org. Fertilization was performed by natural spawning at the beginning of the light period, and eggs were raised at 28 °C. All experimental procedures were conducted in compliance with the directives of the animal welfare committee of Leiden University.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by funds of China Scholarship Council (CSC) to Z. M., J. Y. and Q. L. A. K acknowledges the Netherlands scientific research organization (NWO) for the financial support via a VICI grant (No. 724.014.001). J. Y. and Z. M. contributed equally to this work. A. H. gratefully acknowledges support from the European Union (ERC Advanced Grant SUPRABIOTICS, No. 694610) and the Zernike Institute for Advanced Materials.References
- C. T. Saeui, M. P. Mathew and L. Liu, et al. , J. Funct. Biomater., 2015, 6, 454–485 CrossRef PubMed.
- E. Cukierman, R. Pankov and D. R. Stevens, et al. , Science, 2001, 294, 1708–1712 CrossRef PubMed.
- A. Revzin, P. Rajagopalan and A. W. Tilles, et al. , Langmuir, 2004, 20, 2999–3005 CrossRef PubMed.
- J. B. Shear, H. A. Fishman and N. L. Allbritton, et al. , Science, 1995, 267, 74–77 CrossRef PubMed.
- T. H. Rider, M. S. Petrovick and F. E. Nargi, et al. , Science, 2003, 301, 213–215 CrossRef PubMed.
- J. Yang, Y. Shimada and R. C. L. Olsthoorn, et al. , ACS Nano, 2016, 10, 7428–7435 CrossRef PubMed.
- J. Yang, A. Bahreman and G. Daudey, et al. , ACS Cent. Sci., 2016, 2, 621–630 CrossRef PubMed.
- G. S. Han, L. O'Hara and G. M. Carman, et al. , J. Biol. Chem., 2008, 283, 20433–20442 CrossRef PubMed.
- M. Mrksich, Curr. Opin. Chem. Biol., 2002, 6, 794–797 CrossRef PubMed.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef PubMed.
- N. C. Seeman, Nature, 2003, 421, 427–431 CrossRef PubMed.
- M. H. Caruthers, Science, 1985, 230, 281–285 CrossRef PubMed.
- S. Kocabey, S. Kempter and J. List, et al. , ACS Nano, 2015, 9, 3530–3539 CrossRef PubMed.
- M. E. Todhunter, N. Y. Jee and A. J. Hughes, et al. , Nat. Methods, 2015, 12, 975–981 CrossRef PubMed.
- C. Yoshina-Ishii and S. G. Boxer, J. Am. Chem. Soc., 2003, 125, 3696–3697 CrossRef PubMed.
- U. Jakobsen, A. C. Simonsen and S. Vogel, J. Am. Chem. Soc., 2008, 130, 10462–10463 CrossRef PubMed.
- G. Stengel, R. Zahn and F. Höök, J. Am. Chem. Soc., 2007, 129, 9584–9585 CrossRef PubMed.
- O. Ries, P. M. G. Loffler and S. Vogel, Org. Biomol. Chem., 2015, 13, 9673–9680 RSC.
- P. M. G. Loffler, O. Ries and A. Rabe, et al. , Angew. Chem., Int. Ed., 2017, 56, 13228–13231 CrossRef PubMed.
- S. Hernández-Ainsa, M. Ricci and L. Hilton, et al. , Nano Lett., 2016, 16, 4462–4466 CrossRef PubMed.
- A. Rodríguez-Pulido, A. I. Kondrachuk and D. K. Prusty, et al. , Angew. Chem., Int. Ed., 2013, 52, 1008–1012 CrossRef PubMed.
- A. Czogalla, D. J. Kauert and H. G. Franquelim, et al. , Angew. Chem., 2015, 127, 6601–6605 CrossRef.
- J. R. Burns, A. Seifert and N. Fertig, et al. , Nat. Nanotechnol., 2016, 11, 152–156 CrossRef PubMed.
- A. Rodriguez-Pulido, A. I. Kondrachuk and D. K. Prusty, et al. , Angew. Chem., Int. Ed., 2013, 52, 1008–1012 CrossRef PubMed.
- M. Anaya, M. Kwak and A. J. Musser, et al. , Chem.–Eur. J., 2010, 16, 12852–12859 CrossRef PubMed.
- A. Herrmann, Z. Meng and J. Yang, et al. , Chem.–Eur. J., 2017, 23, 9391–9396 CrossRef PubMed.
- A. Kurz, A. Bunge and A.-K. Windeck, et al. , Angew. Chem., Int. Ed., 2006, 45, 4440–4444 CrossRef PubMed.
- C. Jumeaux, O. Wahlsten and S. Block, et al. , ChemBioChem, 2018, 19, 434–438 CrossRef PubMed.
- H. Liu, B. Kwong and D. J. Irvine, Angew. Chem., Int. Ed., 2011, 50, 7052–7055 CrossRef PubMed.
- B. Yurke, A. J. Turberfield and A. P. Mills, et al. , Nature, 2000, 406, 605–608 CrossRef PubMed.
- R. M. Dirks and N. A. Pierce, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15275–15278 CrossRef PubMed.
- J. S. Kahn, A. Trifonov and A. Cecconello, et al. , Nano Lett., 2015, 15, 7773–7778 CrossRef PubMed.
- R. A. Chandra, E. S. Douglas and R. A. Mathies, et al. , Angew. Chem., Int. Ed., 2006, 45, 896–901 CrossRef PubMed.
- S. C. Hsiao, B. J. Shum and H. Onoe, et al. , Langmuir, 2009, 25, 6985–6991 CrossRef PubMed.
- W. Zhao, W. Loh and I. A. Droujinine, et al. , FASEB J., 2011, 25, 3045–3056 CrossRef PubMed.
- R. C. Bailey, G. A. Kwong and C. G. Radu, et al. , J. Am. Chem. Soc., 2007, 129, 1959–1967 CrossRef PubMed.
- N. S. Selden, M. E. Todhunter and N. Y. Jee, et al. , J. Am. Chem. Soc., 2012, 134, 765–768 CrossRef PubMed.
- X. Qu, S. Wang and Z. Ge, et al. , J. Am. Chem. Soc., 2017, 139, 10176–10179 CrossRef PubMed.
- D. Bhatia, S. Surana and S. Chakraborty, et al. , Nat. Commun., 2011, 2, 339 CrossRef PubMed.
- H. Lee, A. K. Lytton-Jean and Y. Chen, et al. , Nat. Nanotechnol., 2012, 7, 389–393 CrossRef PubMed.
- L. Evensen, P. L. Johansen and G. Koster, et al. , Nanoscale, 2016, 8, 862–877 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01771a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |