
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visible light-promoted ring-opening functionalization of unstrained cycloalkanols via inert C–C bond scission†‡
Dongping
Wang
a,
Jincheng
Mao
 c and
Chen
Zhu
*ab
c and
Chen
Zhu
*ab
aKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, 199 Ren-Ai Road, Suzhou, Jiangsu 215123, China. E-mail: chzhu@suda.edu.cn
bKey Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
cState Key Laboratory of Oil and Gas Reservoir Geology and Exploitation, Southwest Petroleum University, Chengdu 610500, China
First published on 11th June 2018
Abstract
Described herein is a novel, useful, visible light-promoted ring-opening functionalization of unstrained cycloalkanols. Upon scission of an inert cyclic C–C σ-bond, a set of medium- and large-sized rings are readily brominated under mild reaction conditions to afford the corresponding distal bromo-substituted alkyl ketones that are hard to synthesize otherwise. The products are versatile building blocks, which are easily converted to other valuable molecules in one-step operation. This protocol is also applicable to the unprecedented ring-opening cyanation and alkynylation of unstrained cycloalkanols.
Introduction
C–C bond activation is always a challenging issue in synthetic chemistry. Over the past few decades, this area has received great attention relying on the cleavage of cyclic C–C bonds.1 Strained cycloalkanols such as cyclopropanols and cyclobutanols have emerged as privileged precursors for the preparation of β- and γ-substituted ketones through radical-mediated ring-opening functionalization.2,3 Recently, we have consecutively reported the ring-opening functionalization of cyclobutanols to construct various chemical bonds, e.g., C–F, C–Cl, C–N, C–S, C–C, etc., based on silver or manganese catalysis.4 However, the opening of unstrained rings (in particular, 5–7 and 12–15 membered rings) is always confronted with a formidable challenge. It is mainly attributed to the significantly decreased ring-strain energy (Scheme 1A).5 During our research, it was also found that the intramolecular dehydration took place in competition with a ring-opening pathway to suppress the reaction outcome. Therefore, an efficient catalytic protocol should be sought for the ring opening of unstrained cycloalkanols.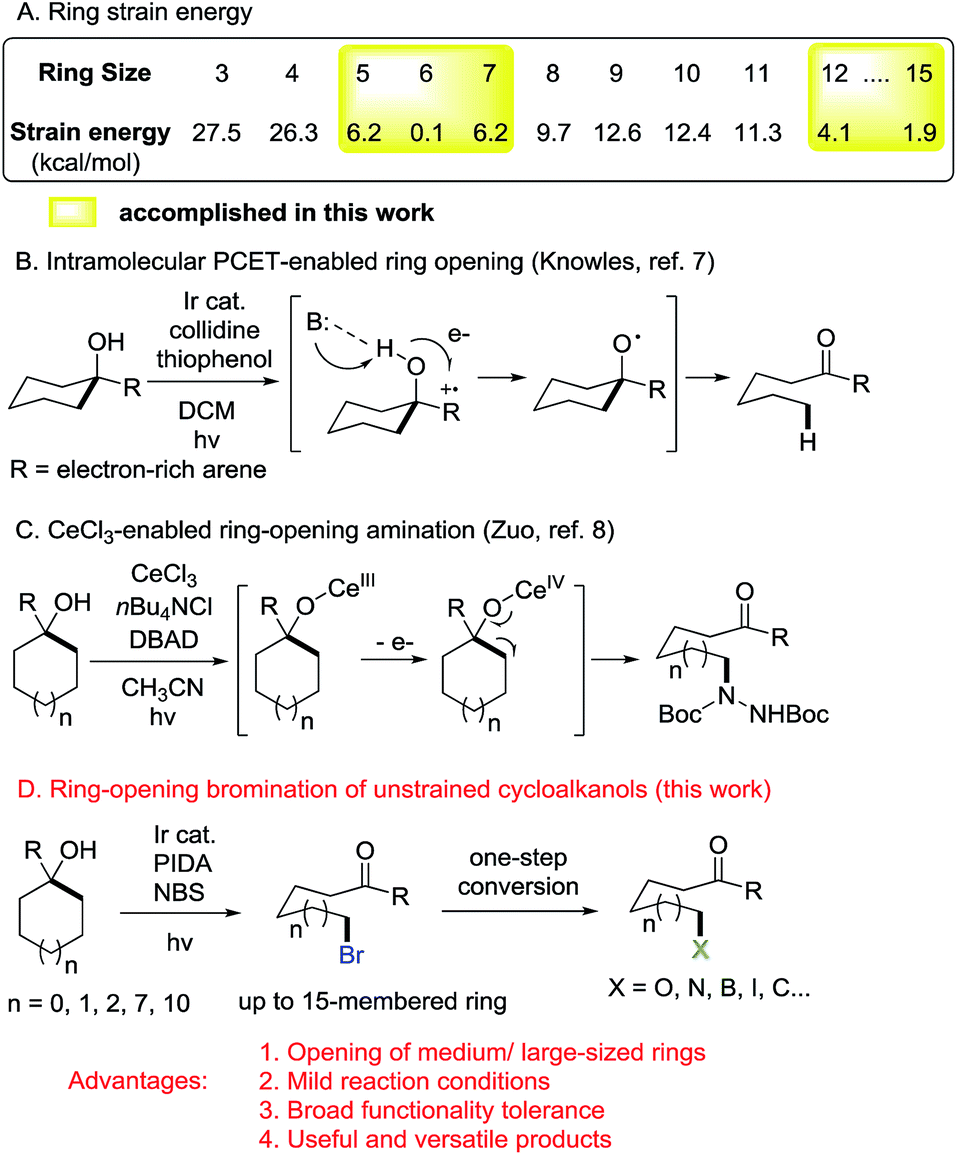 | ||
| Scheme 1 Ring-opening functionalization of unstrained cycloalkanols. | ||
Photoredox catalysis has proven to be a powerful tool for the mild generation of an alkyloxy radical that triggers the subsequent ring-opening reactions or other transformations.6 Recently, Knowles et al. applied intramolecular proton-coupled electron transfer (PCET) to the photocatalytic ring opening of unstrained cycloalkanols.7 In this remarkable process, however, electron-rich tertiary alcohols were required to generate the arene radical cation, a key intermediate for the intramolecular PCET process. This somewhat limited the practicality of the protocol (Scheme 1B). Later, Zuo et al. disclosed an elegant photocatalytic ring-opening amination of unstrained cycloalkanols using a cerium chloride complex (Scheme 1C).8 Beyond this, other types of ring-opening functionalization of unstrained cycloalkanols are still anticipated.
Alkyl bromides are versatile building blocks in synthetic chemistry, which can be easily converted to other valuable molecules through nucleophilic substitution or cross coupling reactions. We considered that the ring-opening bromination of unstrained cycloalkanols would give rise to distally bromo-substituted alkyl ketones which are synthetically valuable but hard to synthesize otherwise. Herein, we provide support for this hypothesis (Scheme 1D). A set of medium- and large-sized rings are readily brominated through the mild cleavage of an inert cyclic C–C σ-bond with the assistance of visible-light irradiation. The newly formed C–Br bonds can function as privileged precursors for many other useful chemical bonds. Moreover, this protocol is also applicable to the unprecedented ring-opening cyanation and alkynylation of unstrained cycloalkanols.
Results and discussion
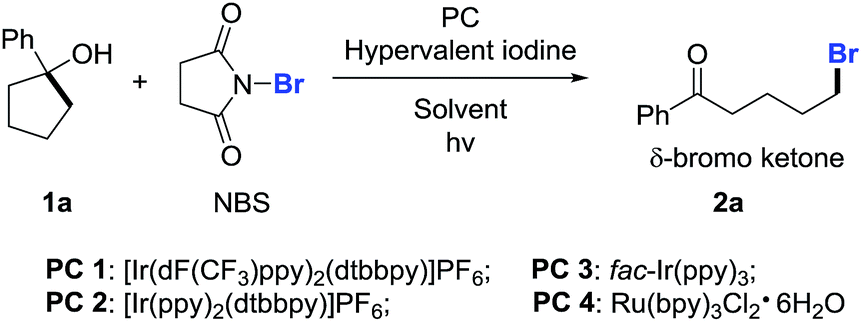
At the outset, we implemented the reaction parameter survey with the less strained 1-phenylcyclopentanol 1a as a model substrate and NBS as a bromine source (Table 1). It was found that the use of biphasic solvents was crucial to the reaction. In the presence of a photoredox catalyst (PC) and a hypervalent iodine reagent, the ring-opening bromination readily proceeded under visible-light irradiation, affording the desired δ-bromo alkyl ketone 2a. A brief evaluation of solvents indicated that CCl4/H2O delivered the best yield (entry 4), while the use of PhCF3/H2O also resulted in a comparable yield (entry 9). The yield was slightly improved to 76% by using PC 2 instead of PC 1 (entry 13). Other than PIDA, hypervalent iodine reagents such as BI-OH, IBX, and DMP were also suitable for the reaction, but led to lower yields (entries 16–18). The amount of H2O was important to the reaction outcome. Increasing the volume of H2O in biphasic solvents decreased the yield (entry 19), whereas using anhydrous CCl4 only led to trace amounts of product 2a (entry 20). Both PC and PIDA were not indispensable; removing either one still afforded the product in modest yields (entries 21 and 22). It might be suggested that the overall ring-opening process was the outcome of two different pathways. The reaction did not proceed in the absence of both PC and PIDA or in the dark (entries 23 and 24). Finally, the yield of 2a was further improved to 78% with the use of PhCF3/H2O as the mixed solvent (entry 25).|
|
||||
|---|---|---|---|---|
| Entry | Solvent (v/v 20/1) | Photoredox catalyst | Hypervalent iodine | Yield (%)b |
| a 1a (0.2 mmol), NBS (0.3 mmol, 1.5 equiv.), hypervalent iodine (0.4 mmol, 2.0 equiv.), and PC (0.006 mmol, 3 mol%) in mixed solvents (2.0 mL/0.1 mL) at rt, 14 W blue LED irradiation. b Yields of isolated products. c CCl4/H2O (2.0 mL/0.2 mL). d In the dark. | ||||
| 1 | DCM/H2O | PC 1 | PIDA | 22 |
| 2 | DCE/H2O | PC 1 | PIDA | 21 |
| 3 | CHCl3/H2O | PC 1 | PIDA | 15 |
| 4 | CCl4/H2O | PC 1 | PIDA | 74 |
| 5 | DMF/H2O | PC 1 | PIDA | 0 |
| 6 | DMSO/H2O | PC 1 | PIDA | 0 |
| 7 | Acetone/H2O | PC 1 | PIDA | <5 |
| 8 | Toluene/H2O | PC 1 | PIDA | <5 |
| 9 | PhCF3/H2O | PC 1 | PIDA | 68 |
| 10 | CH3CN/H2O | PC 1 | PIDA | 35 |
| 11 | THF/H2O | PC 1 | PIDA | 0 |
| 12 | MeOH/H2O | PC 1 | PIDA | 0 |
| 13 | CCl4/H2O | PC 2 | PIDA | 76 |
| 14 | CCl4/H2O | PC 3 | PIDA | 59 |
| 15 | CCl4/H2O | PC 4 | PIDA | 24 |
| 16 | CCl4/H2O | PC 2 | BI-OH | 70 |
| 17 | CCl4/H2O | PC 2 | IBX | 55 |
| 18 | CCl4/H2O | PC 2 | DMP | 65 |
| 19c | CCl4/H2O | PC 2 | PIDA | 70 |
| 20 | CCl4 | PC 2 | PIDA | <5 |
| 21 | CCl4/H2O | PC 2 | — | 53 |
| 22 | CCl4/H2O | — | PIDA | 45 |
| 23 | CCl4/H2O | — | — | 0 |
| 24d | CCl4/H2O | PC 2 | PIDA | 0 |
| 25 | PhCF3/H2O | PC 2 | PIDA | 78 |
With the optimized reaction conditions in hand, we set out to investigate the generality of the protocol (Scheme 2). First, a variety of cyclopentanols were tested. Generally, electron-deficient aryl substituents were preferred for the reaction, leading to δ-bromo alkyl ketones in synthetically useful yields. Halides (2b–2d), in particular bromides (2d), were well tolerated, providing a platform for the late-stage product manipulation. Positional change (para-, meta-, and ortho-) of substituents did not impede the transformation (2g–2i). Although highly electron-rich aryl compounds such as anisole were not suitable due to the electrophilic bromination of the aryl group, it could be overcome by mounting an additional electron-withdrawing group (2h). This method also allowed for the preparation of secondary bromides (2j). The ring opening of unsymmetric cycloalkanols proceeded in a regioselective manner. However, the synthesis of tertiary bromides failed under the current reaction conditions. The scope of cyclohexanols was investigated more extensively than that of cyclopentanols. Besides the functional groups already examined, other groups such as cyano (2q and 2t) and ester (2z) were also compatible to the reaction conditions. In addition to aryl-substituted cyclohexanols, alkyl-substituted cyclohexanols were apt to furnish the elusive distal-bromo dialkyl ketones (2w and 2x). Various substituents could be incorporated into the alkyl chain by using the functionalized cycloalkanol precursors (2y–2ab). In the presence of excess NBS, three-fold bromination took place on the toluene-substituted cycloalkanol to give product 2ac which was brominated at both benzylic and distal positions. The reaction with the cyclohexanol derived from 1,4-cyclohexandione afforded the unsaturated 1,4-diketone via dehydrobromination (2ad). Likewise, the ring-opening bromination of cycloheptanols bearing various functional groups also readily proceeded, affording the corresponding distal-bromo alkyl ketones in useful yields (2ae–2ak). Unfortunately, the ring opening of eight- and ten-membered cycloalkanols failed, leading to complex mixtures. The reason is unclear so far. Remarkably, the opening of large-sized rings such as cyclododecanols and cyclopentadecanols occurred efficiently, yielding a variety of distally brominated alkyl ketones that are hard to synthesize otherwise (2al–2ar).
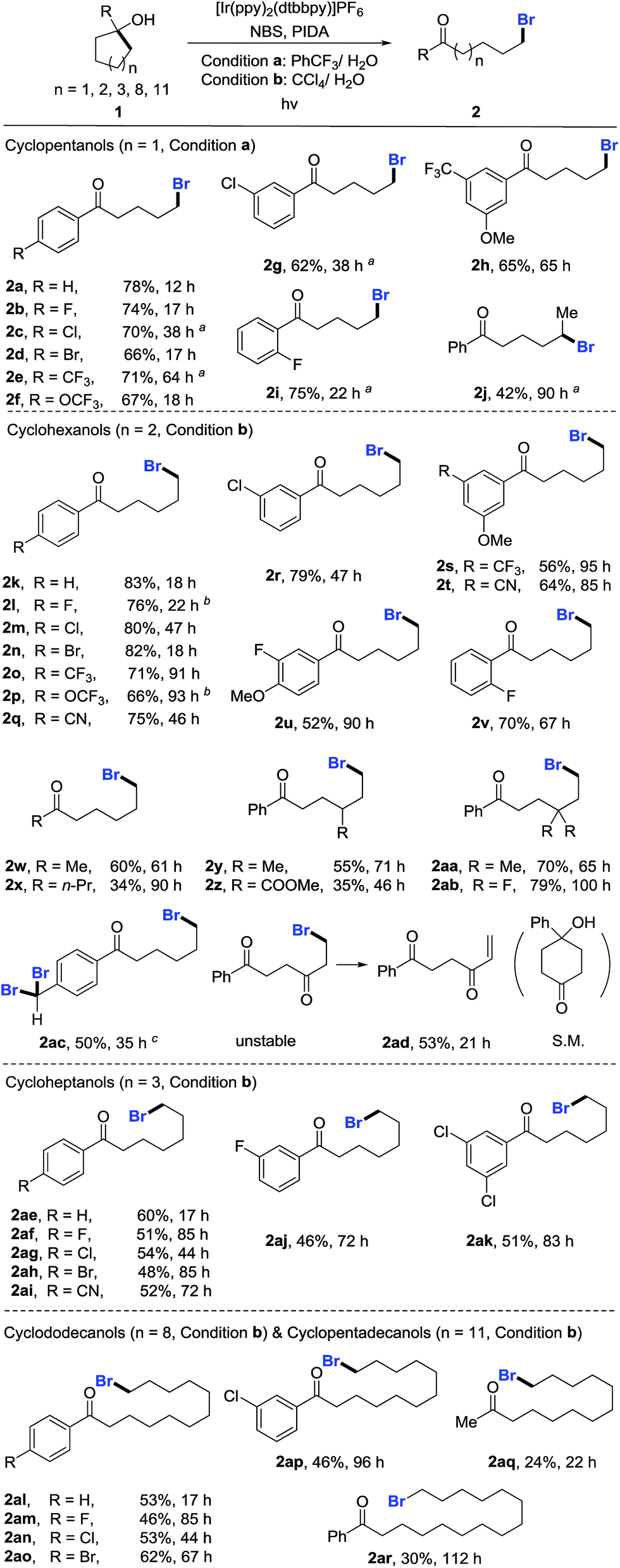 | ||
| Scheme 2 Scope of unstrained cycloalkanols. Reaction conditions: 1 (0.2 mmol), NBS (0.3 mmol, 1.5 equiv.), PIDA (0.4 mmol, 2.0 equiv.), and PC (0.006 mmol, 3 mol%) in mixed solvents (2.0 mL/0.1 mL) at rt, 14 W blue LED irradiation. Yields of isolated products are given. aWith condition b. b[Ir(dF(CF3)ppy)2(dtbbpy)]PF6 as PC. cNBS (0.8 mmol, 4.0 equiv.). | ||
To manifest the utility of the protocol, the products were converted to other valuable molecules via nucleophilic substitution or cross-coupling in one or two steps (Scheme 3). First, the bromide in 2a was easily replaced by azide and iodide, forming new C–N3 and C–I bonds in good yields (3 and 4).9 Then, the ketone in 2a was reduced to alcohol that intramolecularly attacked the alkyl bromide, leading to tetrahydropyran 5 in two steps with high yield.10 Alternatively, the C–Br bond was readily converted to C–B and C–C bonds via transition-metal catalyzed cross-coupling reactions, affording synthetically useful building blocks or molecules (6 and 7).11
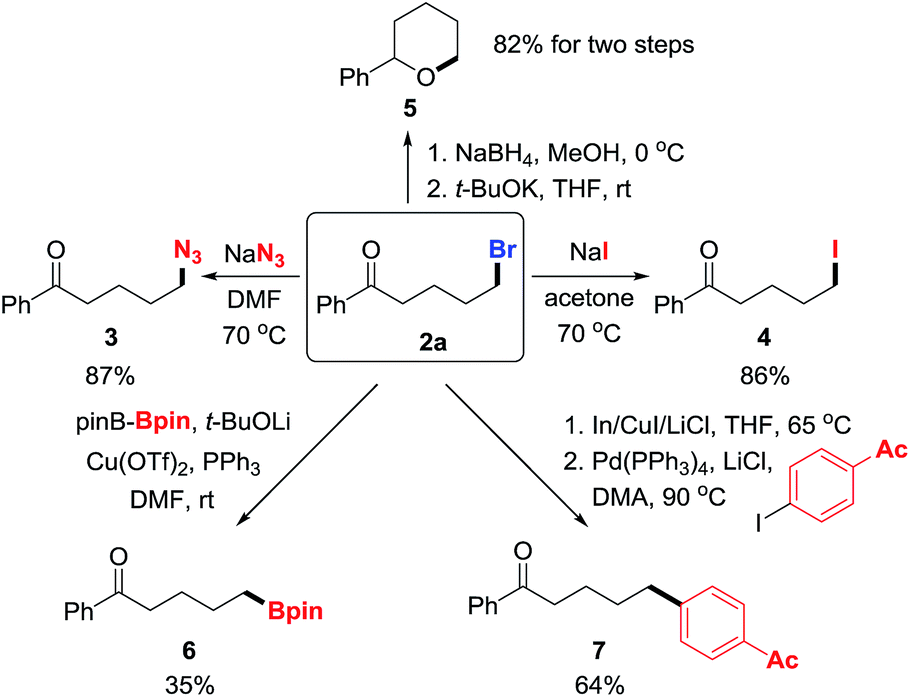 | ||
| Scheme 3 Transformation of products to other useful molecules. | ||
Haloperidol is a marketed antipsychotic drug extensively used to treat schizophrenia and Tourette's syndrome. After gaining a portfolio of distal-bromo alkyl bromides as precursors, we accomplished the preparation of haloperidol analogues in high yields (8a–8d, Scheme 4).12 Testing of their biological activities is ongoing.
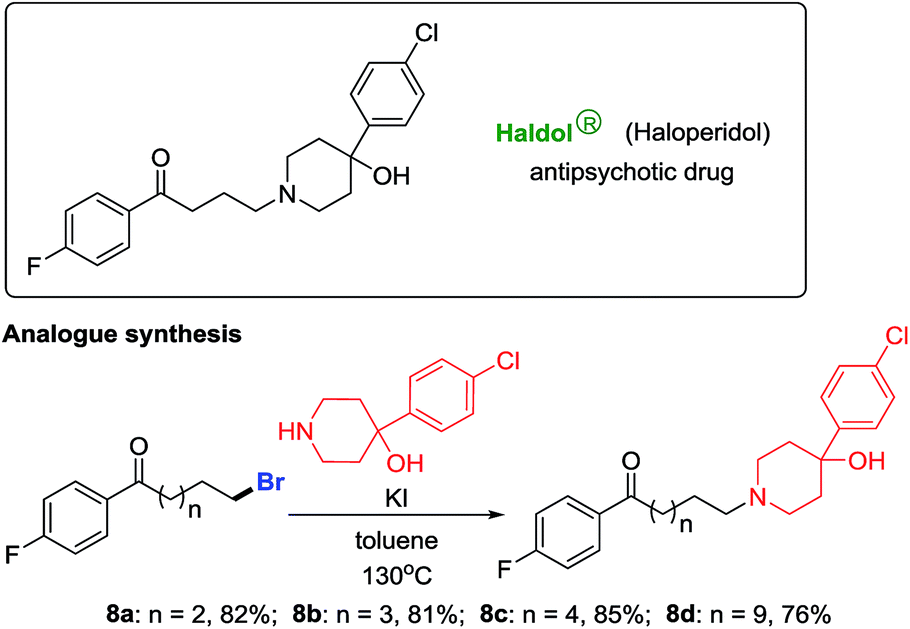 | ||
| Scheme 4 Production of haloperidol analogues. | ||
According to the experimental results, the plausible mechanism is depicted (Scheme 5, top). Although the detailed process for the generation of cycloalkoxy radical I is not fully understood, the radical I might be formed via two pathways. First, Stern–Volmer studies disclose that the excited state of IrIII catalyst PC 2 could be oxidatively quenched by NBS to generate the IrIV complex (see the ESI†). However, the oxidation potential of this IrIV complex (E1/2IV/III = 1.21 V in MeCN vs. SCE) is insufficient to oxidize cycloalkanol 1 (Ep/2 > 2.0 V in MeCN vs. SCE) to alkoxy radical Ivia the SET process (for the cyclic voltammetry studies, see the ESI†). Thus, we postulate an intermolecular proton-coupled electron transfer (PCET) process for the generation of alkoxy radical I in the presence of an IrIV complex and weak base such as a succinimide anion (path a).13 Alternatively, homolysis of the O–I bond in situ formed by the reaction of cycloalkanol 1 with PIDA might also lead to the alkoxy radical I (path b).14 In either way, the visible-light irradiation is indispensable. The subsequent β-C scission of I leads to the ring-opened alkyl radical II, which is intercepted by NBS to give the final product 2. Notably, CBr4 is also a competent bromine source in lieu of NBS to afford the same brominated product, suggesting the formation of intermediate II during the reaction (Scheme 5, bottom).
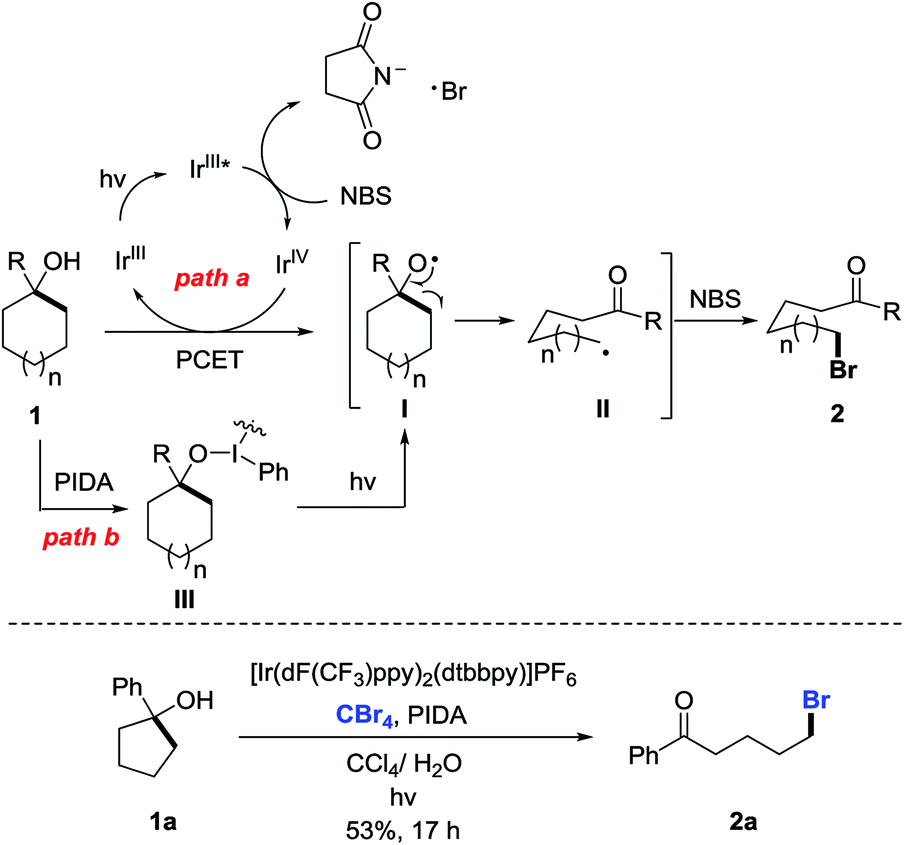 | ||
| Scheme 5 Plausible pathways of ring-opening bromination. | ||
This protocol is also applicable to the challenging ring-opening cyanation and alkynylation of unstrained cycloalkanols.15 Under the similar reaction conditions, 1-phenyl cyclododecanol was readily converted to the distally cyano- or alkynyl-substituted alkyl ketones in synthetically useful yields (9 and 10), providing a non-trivial approach to construct remote C–C bonds (Scheme 6).
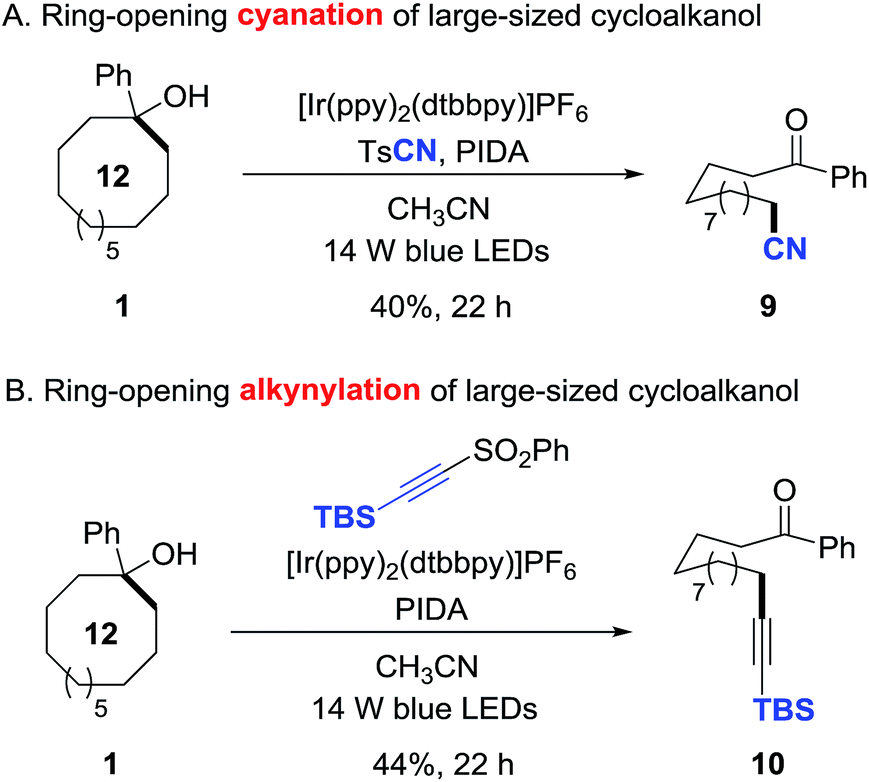 | ||
| Scheme 6 Ring-opening cyanation and alkynylation of unstrained cycloalkanols. | ||
Conclusions
We have described a novel and efficient visible light-enabled ring-opening functionalization of unstrained cycloalkanols via the mild cleavage of cyclic C–C σ-bonds. Ring opening of a variety of cyclopentanols, cyclohexanols, cycloheptanols, cyclododecanols, and cyclopentadecanols readily proceeded to furnish the distally brominated alkyl ketones that often function as precursors of complex molecules in organic and medicinal synthesis. As shown above, this method provides important feedstocks for the production of haloperidol analogues. The newly formed C–Br bonds in products are easily transformed into other valuable chemical bonds, thus illustrating the utility of this method. This protocol is also applicable to the elusive ring-opening cyanation and alkynylation of unstrained cycloalkanols, providing an unusual strategy for the construction of remote C–C bonds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. Z. is grateful for the financial support from Soochow University, the National Natural Science Foundation of China (Grant no. 21722205), the Project of Scientific and Technologic Infrastructure of Suzhou (SZS201708), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- For selected reviews, see: (a) A. K. Sadan, R. K. Saini and W. E. Billups, Chem. Rev., 2003, 103, 1539 CrossRef PubMed; (b) T. Seiser and N. Cramer, Org. Biomol. Chem., 2009, 7, 2835 RSC; (c) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef PubMed; (d) A. Flores-Gaspar and R. Martin, Synthesis, 2013, 45, 563 CrossRef; (e) L. Souillart, E. Parker and N. Cramer, Top. Curr. Chem., 2014, 346, 163 CrossRef PubMed; (f) T. Xu, A. Dermenci and G. B. Dong, Top. Curr. Chem., 2014, 346, 233 CrossRef PubMed; (g) I. Marek, A. Masarwa, P. O. Delaye and M. Leibeling, Angew. Chem., Int. Ed., 2015, 54, 414 Search PubMed; (h) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410 CrossRef PubMed.
- For special reviews on radical-mediated ring opening of cycloalkanols, see: (a) R. Ren and C. Zhu, Synlett, 2016, 27, 1139 CrossRef; (b) X. Wu and C. Zhu, Chem. Rec., 2018 DOI:10.1002/tcr.201700090.
- For selected examples, see: (a) J. Rocek and A. E. Radkowsky, J. Am. Chem. Soc., 1968, 90, 2986 CrossRef; (b) K. Meyer and J. Rocek, J. Am. Chem. Soc., 1972, 94, 1209 CrossRef; (c) S. Tsunoi, I. Ryu, Y. Tamura, S. Yamasaki and N. Sonoda, Synlett, 1994, 1009 CrossRef; (d) N. I. Kapustina, L. L. Sokova, V. D. Makhaev, L. A. Petrava and G. I. Nikishin, Russ. Chem. Bull., 1999, 48, 2080 CrossRef; (e) B. M. Casey, C. A. Eakin and R. A. Flowers II, Tetrahedron Lett., 2009, 50, 1264 CrossRef PubMed; (f) Y.-F. Wang and S. Chiba, J. Am. Chem. Soc., 2009, 131, 12570 CrossRef PubMed; (g) S. Wang, L. N. Guo, H. Wang and X. H. Duan, Org. Lett., 2015, 17, 4798 CrossRef PubMed; (h) S. Bloom, D. D. Bume, C. R. Pitts and T. Lectka, Chem.–Eur. J., 2015, 21, 8060 CrossRef PubMed; (i) S. Ren, C. Feng and T.-P. Loh, Org. Biomol. Chem., 2015, 13, 5105 RSC; (j) N. Ishida, S. Okumura, Y. Nakanishi and M. Murakami, Chem. Lett., 2015, 44, 821 CrossRef; (k) F.-Q. Huang, J. Xie, J.-G. Sun, Y.-W. Wang, X. Dong, L.-W. Qi and B. Zhang, Org. Lett., 2016, 18, 684 CrossRef PubMed; (l) K. Jia, F. Zhang, H. Huang and Y. Chen, J. Am. Chem. Soc., 2016, 138, 1514 CrossRef PubMed; (m) Q. Tian, B. Chen and G. Zhang, Green Chem., 2016, 18, 6236 RSC; (n) R. Zhao, Y. Yao, D. Zhu, D. Chang, Y. Liu and L. Shi, Org. Lett., 2018, 20, 1228 CrossRef PubMed.
- (a) H. Zhao, X. Fan, J. Yu and C. Zhu, J. Am. Chem. Soc., 2015, 137, 3490 CrossRef PubMed; (b) X. Fan, H. Zhao and C. Zhu, Acta Chim. Sin., 2015, 73, 979 Search PubMed; (c) H. Yan and C. Zhu, Sci. China: Chem., 2017, 60, 214 CrossRef; (d) J. Yu, H. Zhao, S. Liang, X. Bao and C. Zhu, Org. Biomol. Chem., 2015, 13, 7924 RSC; (e) X. Fan, H. Zhao, J. Yu, X. Bao and C. Zhu, Org. Chem. Front., 2016, 3, 227 RSC; (f) L. Huan and C. Zhu, Org. Chem. Front., 2016, 3, 1467 RSC; (g) R. Ren, H. Zhao, L. Huan and C. Zhu, Angew. Chem., Int. Ed., 2015, 54, 12692 CrossRef PubMed; (h) D. Wang, R. Ren and C. Zhu, J. Org. Chem., 2016, 81, 8043 CrossRef PubMed; (i) R. Ren, Z. Wu, Y. Xu and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 2866 CrossRef PubMed; (j) R. Ren, Z. Wu and C. Zhu, Chem. Commun., 2016, 52, 8160 RSC; (k) M. Wang, Z. Wu and C. Zhu, Org. Chem. Front., 2017, 4, 427 RSC.
- P. R. Khoury, J. D. Goddard and W. Tam, Tetrahedron, 2004, 60, 8103 CrossRef.
- For selected reviews on photoredox catalysis, see: (a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527 CrossRef PubMed; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (c) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828 CrossRef PubMed; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef PubMed; (e) J. Xuan, Z. G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632 CrossRef PubMed.
- H. G. Yayla, H. Wang, K. T. Tarantino, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2016, 138, 10794 CrossRef PubMed.
- J.-J. Guo, A. Hu, Y. Chen, J. Sun, H. Tang and Z. Zuo, Angew. Chem., Int. Ed., 2016, 55, 15319 CrossRef PubMed.
- K. Ngu, D. S. Weinstein, W. Liu, C. Langevine, D. W. Combs, S. Zhuang, X. Chen, C. S. Madsen, T. W. Harper, S. Ahmad and J. A. Robl, Bioorg. Med. Chem. Lett., 2011, 21, 4141 CrossRef PubMed.
- R. Mansueto, V. Mallardo, F. M. Perna, A. Salomone and V. Capriati, Chem. Commun., 2013, 49, 10160 RSC.
- (a) Z.-L. Shen, K. K. K. Goh, C. H. A. Wong, Y.-S. Yang, Y.-C. Lai, H.-L. Cheong and T.-P. Loh, Chem. Commun., 2011, 47, 4778 RSC; (b) M.-Y. Liu, S.-B. Hong, W. Zhang and W. Deng, Chin. Chem. Lett., 2015, 26, 373 CrossRef.
- M. A. Iorio, T. P. Reymer and V. Frigeni, J. Med. Chem., 1987, 30, 1906 CrossRef PubMed.
- (a) J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961 CrossRef PubMed; (b) D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016 CrossRef PubMed.
- (a) J. I. Concepción, C. G. Francisco, R. Hernández, J. A. Salazar and E. Suárez, Tetrahedron Lett., 1984, 25, 1953 CrossRef; (b) A. Martín, J. A. Salazar and E. Suárez, Tetrahedron Lett., 1995, 36, 4489 CrossRef; (c) A. Martín, J. A. Salazar and E. Suárez, J. Org. Chem., 1996, 61, 3999 CrossRef; (d) A. Martín, I. Pérez-Martín and E. Suárez, Org. Lett., 2005, 7, 2027 CrossRef PubMed.
- The ring-opening cyanation and alkynylation of cyclobutanols have been reported in ref. 3g, 3l, and 4i. However, the reaction with unstrained medium- or large-sized cycloalkanols remains unknown..
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01763h |
| ‡ Dedicated to Professor Takahiko Akiyama on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry 2018 |