
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgrammable one-pot synthesis of heparin pentasaccharides enabling access to regiodefined sulfate derivatives†
Supriya
Dey
a and
Chi-Huey
Wong
 *ab
*ab
aDepartment of Chemistry, The Scripps Research Institute, 10550 N Torrey Pines Road, La Jolla 92037, USA
bThe Genomics Research Center, Academia Sinica, No. 128, Academia Road, Section 2, Taipei, Taiwan. E-mail: chwong@gate.sinica.edu.tw
First published on 3rd July 2018
Abstract
Heparin (H) and heparan sulfate (HS) belong to the glycosaminoglycan (GAG) family of oligosaccharides, and their sequences and sulfation patterns are known to regulate the functions of various proteins in biological processes. Among these, the 6-O-sulfation of HS/H contributes most significantly to the structural diversity and binding interactions. However, the synthesis of HS with defined sulfation patterns remains a major challenge. Herein, we report a highly efficient and programmable one-pot method for the synthesis of protected heparin pentasaccharides using thioglycoside building blocks with optimized relative reactivities to allow the selective deprotection and preparation of regiodefined sulfate derivatives.
Introduction
Heparin (H) and heparan sulfate (HS) are linear and highly dispersed polyanionic molecules of glycosaminoglycans (GAGs). They are highly sulfated and composed of D-N-acetylglucosamine (GlcNAc) and either D-glucuronic acid (GlcA) or L-iduronic acid (IdoA) linked by repeating 1,4-glycosidic linkages,1 and have been used as therapeutic agents for thrombosis and other cardiovascular diseases.2,3 They are often found on the cell surface, in the basement membrane, and in extracellular or tissue matrices.4,5 HS and heparin can be compared under the conventional definition as follows: (i) HS chains are typically composed of 50–250 disaccharide units with average molecular weights of 20–100 kDa, whereas heparin chains consist of repeating units with molecular weights ranging from 12 to 14 kDa;3 (ii) the average number of O-sulfate groups is more uniform and there is a higher level of sulfation (2.5–3 sulfate groups/disaccharide) in heparin than in HS which has a lower level of sulfation (0.5–1.5 sulfate groups/disaccharide); (iii) α-L-iduronates are predominantly present in heparin, whereas in HS, the uronates are the C-5 epimers of β-D-glucuronate; (iv) in heparin, D-glucosamine residues are mostly N-sulfated (80%), whereas in HS, they can be either N-acetylated or N-sulfated (30–60%). Overall, heparins are more sulfated and more charged than HS on the basis of structural properties.3c However, heparin and HS are less distinct than the conventional description implies, as isolated polysaccharides from some organisms were found to be hybrid structures.3dMore than 100 heparin-binding proteins have been identified and more are expected to be discovered.6 The biological properties of H/HS are mainly dictated by the sulfation pattern and the structure of the NS-domain,7 but the mechanisms are poorly understood due to their complex structural patterns and the heterogeneity of the oligosaccharides. However, the binding of antithrombin III with a minimal sequence of HSs has been well studied, showing that the repeating tri-sulfated disaccharide unit IdoA2S-(1,4)-GlcNS6S-(1,4) is responsible for the anticoagulant activity.8 Recently, a structure–function relationship study of 3-O-sulfated HS has been reported with an emphasis on the conformation of the pyranose and the 2-O-sulfated IdoA. It was shown that the skew boat 2So conformation of 2-O-sulfated IdoA, located at the reducing end of the GlcNS3S6, is essential for the anticoagulant activity.9 Thus, understanding the role of the sulfation pattern in H/HS can provide fundamental insights into the mechanism of molecular recognition and the activity of this class of molecule.10
The 6-O-sulfation of HS/H, especially that outside the NS-domains, contributes most significantly to the structural and functional diversity, and is the only modification step regulated by both the biosynthetic and post-synthetic processes.11 The implications of 6-O-sulfation in HS–protein interactions have been associated with many biological functions, such as anticoagulation, wound healing, embryonic development, inflammation and cancer progression.12,13 Most recently, it was shown that H/HS saccharides interact with β-amyloid fibrils associated with Alzheimer’s disease.14 Furthermore, addition of a single 6-O-sulfate group at the non-reducing end of the [IdoA2S-GlcNS]6 heparin oligosaccharide can switch its inhibitory properties, indicating the importance of the 6-O-sulfate group in HS/H chains.14a
The chemical synthesis of H/HS oligosaccharides is challenging due to the laborious and repetitive steps of protection, uncontrolled stereoselective glycosylation, deprotection, and multiple purification processes, which give low efficiencies and low yields.15 However, over the years, chemical and chemo-enzymatic16 methodologies have been reported for the synthesis of heparin-like oligosaccharides,17–20 and the heparin-based drugs fondaparinux21 and idraparinux.22 To overcome the disadvantages of multi-step oligosaccharide synthesis, we have developed a computer database, OptiMer, to store the relative reactivity values (RRVs) of many glycosyl donors/acceptors.23 The database stores the type of sugar core, locations of unprotected hydroxyl groups, the directing natures of protecting groups, pictures of the compounds, and names and references of the compounds and also predicts the product stereochemistry. For a targeted oligosaccharide, the program searches the database to find the best synthetic route. Using this database, various oligosaccharides including a heparin pentasaccharide were rapidly assembled without work-up of intermediates or purification processes.24 However, the overall yield in the one-pot synthesis of the heparin-like pentasaccharide was relatively low (20%) and this was not effective to access various regiodefined O-sulfates.24f Herein, we report an improved programmable one-pot method for the synthesis of a protected heparin pentasaccharide to allow access to various regiodefined O-sulfation patterns for the study of their binding and functions.
Results and discussion
Key intermediate synthesis
Eighteen key intermediates (Fig. 1) were designed with improved RRVs for the one-pot synthesis to access the 6-O-sulfation patterns in heparin pentasaccharides. We used protecting groups to tune the reactivity of each building block to create significant differences in the RRVs. Regarding the case of the 2-aminosugars, the protecting group effect is in the following order: NHCbz > NHTroc > NPhth > N3 > NHAc.24b,e Thus, we designed the synthetic route with the following considerations: (i) highly stereoselective α-glycosylation influenced by the orthogonal protecting groups (N3, TBDPS, and OAc) by minimizing the electronic interactions with the developing positive charge in the transition state,25 (ii) use of the tert-butyldiphenyl silyl (TBDPS) protecting group at O6 to stabilize the oxocarbenium ion by through-space electron donation26 and to increase the RRV of donor 4, (iii) construction of stereoselective β-1,4-glycosidic linkages using the neighboring group participation effect of the O-2-benzoyl ester, (iv) selective installation of the 6-O-sulfate group with the aid of acetyl and levulinic protecting groups, (v) early-stage introduction of glucuronic acid at the disaccharide stage using 2,2,6,6-tetramethyl-1-piperidinyloxy/iodobenzene diacetate (TEMPO/BAIB)-mediated selective oxidation27 of the 6-hydroxyl group, or late-stage introduction of glucuronic acid using p-methoxy benzyl ether as the primary hydroxyl protecting group of 6, (vi) selective introduction of the N-sulfate groups using the N3 functionality, and (vii) use of benzyl ethers to mask the rest of the hydroxyl groups.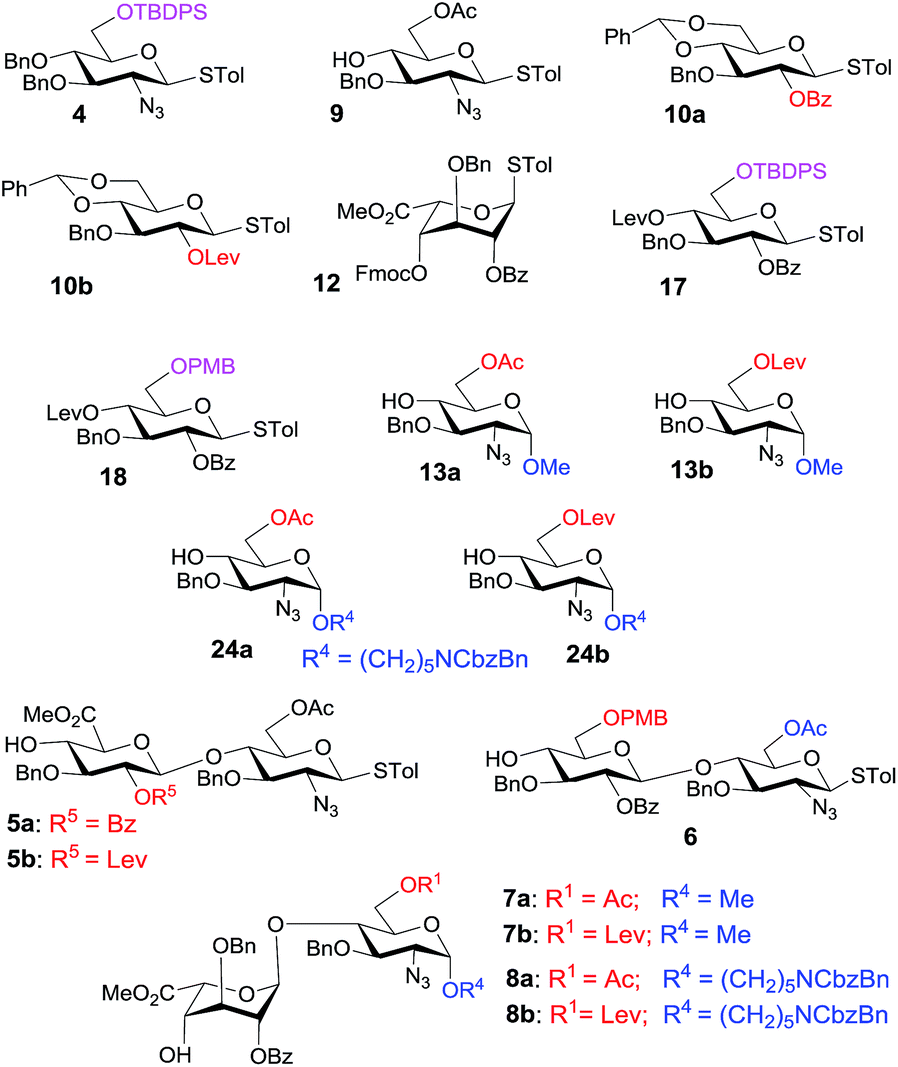 | ||
| Fig. 1 Monosaccharide and disaccharide building blocks. | ||
Our retrosynthetic analysis in Scheme 1 suggests that the pentasaccharides 1a and 1b could be generated from the fully protected pentasaccharides 3a and 3b, respectively, through sequential desilylation, 6-O-benzylation, saponification, O-sulfation, hydrogenolysis and N-sulfation (Scheme 1). The protected pentasaccharides 3a and 3b can be obtained from the programmable one-pot synthesis using the [1,2,2] approach with the building blocks shown in the scheme. Monosaccharides (4, 9 and 13) could be obtained from the D-glucosamine hydrochloride 16. The remaining thioglycoside donors 10 and 11 (building block B) can be synthesized from penta-O-acetyl-β-D-glucopyranoside 14. The iduronic acid unit 12 can be accessed from the known diacetone D-glucose 15,28 and the disaccharide building blocks (5, 6) could be obtained by NIS/TfOH-mediated glycosylation using the appropriate glycosyl donors and acceptors. We also synthesized the disaccharide acceptors 7a, 7b, 8a and 8b which contain the iduronic acid unit as building blocks for the synthesis of pentasaccharides with desirable 6-O-sulfation patterns. The syntheses of the monosaccharide building blocks (Fig. 1) are described in the ESI.†
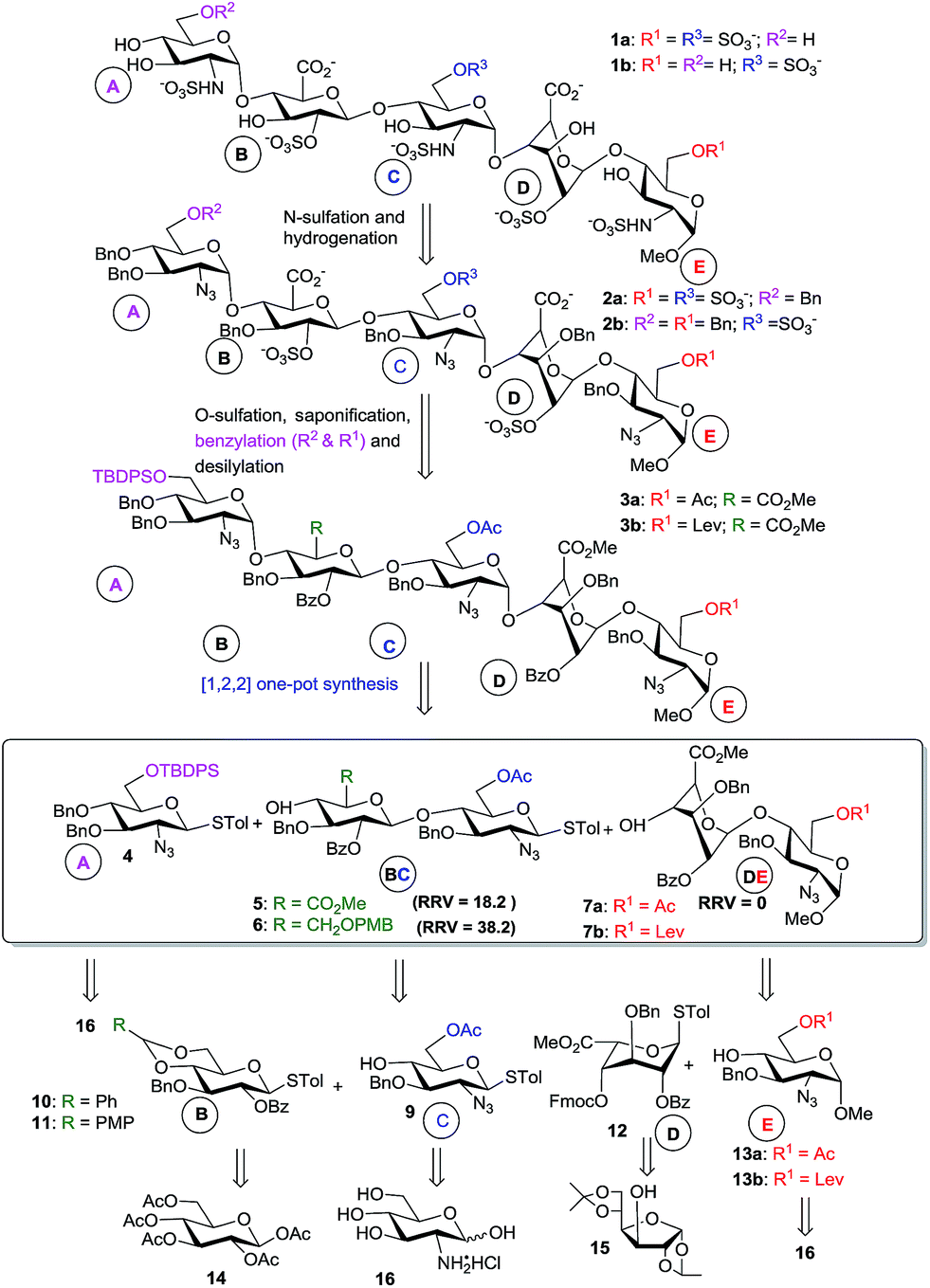 | ||
| Scheme 1 A retrosynthetic analysis of heparan pentasaccharide with regiodefined O-sulfation patterns. Fmoc = 9-fluorenylmethoxycarbonyl chloride, Ac = acetyl, Bn = benzyl, Bz = benzoyl, Lev = levulinyl, PMB = p-methoxybenzyl, TBDPS = tert-butyldiphenylsilyl, Me = methyl, Tol = 4-tolyl. | ||
Synthesis of disaccharides (BC and DE building blocks)
The synthesis of β-(1,4)-linked disaccharides (19 and 20) was performed using NIS/TfOH-mediated coupling of donors (17 and 18) with acceptor 9, respectively. Subsequently, removal of the levulinyl groups of 19 and 20 using hydrazine monohydrate in the presence of AcOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Py (2:3) afforded disaccharide acceptors 21 and 6 respectively, in excellent yields (Scheme 2). The TBDPS ether of 21 was readily deprotected using HF-Py to yield diol 22a. The primary hydroxyl group in 22a was selectively oxidized using 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) in the presence of BAIB. The crude acid underwent subsequent esterification with methyl iodide in the presence of KHCO3 to afford glucuronic acid acceptor 5a in 63% yield (Scheme 2). On the other hand, the 4,6-benzylidene protected glycosyl donor 10b was coupled with acceptor 9 in the presence of NIS/TMSOTf to furnish disaccharide 22b (Scheme 2). Hydrolysis of 4,6-O-benzylidene acetal using 80% AcOH led to the dihydroxy compound 23. Selective oxidation of the primary hydroxyl group of 23 with TEMPO/BAIB and subsequent esterification with methyl iodide in the presence of KHCO3 gave disaccharide 5b in 61% yield. For the synthesis of disaccharide IdoA-GlcN, the NIS/TfOH-mediated glycosylation of L-iduronic acid donor 12 and acceptors (13a and 13b), followed by the in situ removal of the Fmoc group using Et3N, was performed to furnish the disaccharides (building block DE) 7a and 7b, respectively, in excellent yields (Scheme 3). We also introduced an anomeric aminopentyl spacer to the disaccharide acceptors 8a and 8b through couplings of donor 12 and acceptors 24a and 24b, respectively (Scheme 3).
:Py (2:3) afforded disaccharide acceptors 21 and 6 respectively, in excellent yields (Scheme 2). The TBDPS ether of 21 was readily deprotected using HF-Py to yield diol 22a. The primary hydroxyl group in 22a was selectively oxidized using 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) in the presence of BAIB. The crude acid underwent subsequent esterification with methyl iodide in the presence of KHCO3 to afford glucuronic acid acceptor 5a in 63% yield (Scheme 2). On the other hand, the 4,6-benzylidene protected glycosyl donor 10b was coupled with acceptor 9 in the presence of NIS/TMSOTf to furnish disaccharide 22b (Scheme 2). Hydrolysis of 4,6-O-benzylidene acetal using 80% AcOH led to the dihydroxy compound 23. Selective oxidation of the primary hydroxyl group of 23 with TEMPO/BAIB and subsequent esterification with methyl iodide in the presence of KHCO3 gave disaccharide 5b in 61% yield. For the synthesis of disaccharide IdoA-GlcN, the NIS/TfOH-mediated glycosylation of L-iduronic acid donor 12 and acceptors (13a and 13b), followed by the in situ removal of the Fmoc group using Et3N, was performed to furnish the disaccharides (building block DE) 7a and 7b, respectively, in excellent yields (Scheme 3). We also introduced an anomeric aminopentyl spacer to the disaccharide acceptors 8a and 8b through couplings of donor 12 and acceptors 24a and 24b, respectively (Scheme 3).
 | ||
| Scheme 2 Reaction conditions: (a) NIS, TfOH, CH2Cl2, AW 300 MS, −45 °C to −30 °C, 2 h; 19: 76%, 20: 73%; (b) NH2NH2·H2O, AcOH:Py (2:3), rt, 2 h; 6: 90%, 21: 88% (c) HF-Py, Py, 0 °C to rt, 12 h, 81%; (d) (i) BAIB, TEMPO, CH2Cl2:H2O (2:1), 0 °C to rt, 2 h; (ii) MeI, KHCO3, DMF, rt, 4 h, 0 °C to rt, 5a: 63%, 5b: 61% (two steps); (e) (i) NIS, TMSOTf, CH2Cl2, AW 300 MS, −20 °C to 0 °C, 15 min; 22b: 89%. (f) 80% AcOH, 70 °C, 3 h; 23: 79%. | ||
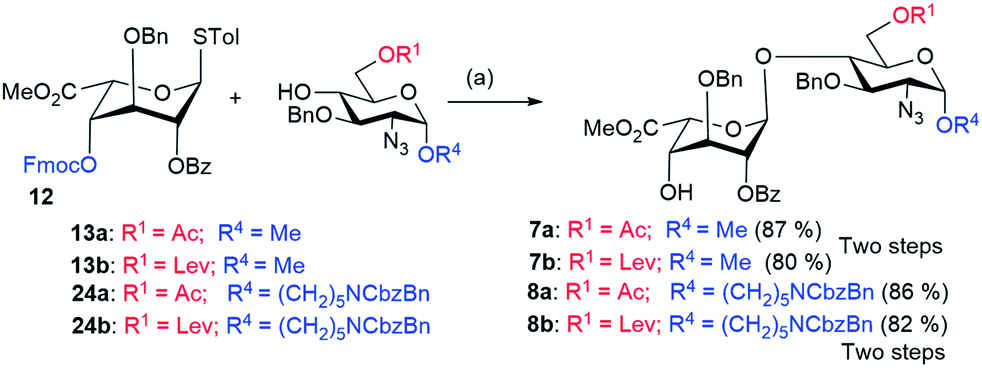 | ||
| Scheme 3 Reaction conditions: (a) (i) NIS, TfOH, CH2Cl2, AW 300 MS, −60 °C to −30 °C, 1 h; (ii) Et3N, rt, 1 h. | ||
One-pot synthesis
We wanted to test if glucuronic acid is better introduced as part of the disaccharide building block or through oxidation at the pentasaccharide stage. For the late stage oxidation to glucuronic acid, the disaccharide building block 6 was used in the one-pot synthesis of a pentasaccharide, followed by selective oxidation to an acid. Thus, the disaccharide building block 6 (RRV = 38.2) was used in the one-pot synthesis of the pentasaccharide 3c which was then subjected to selective deprotection, oxidation and esterification to generate pentasaccharide 3a in 42% yield. Similarly, another one-pot synthesis was performed using glucuronic acid containing disaccharide acceptor 5a (RRV = 18.2) to generate the pentasaccharide 3a in 54% yield (Scheme 4). Overall, the introduction of glucuronic acid at the disaccharide stage is more efficient than oxidation of the C-6 group to the carboxyl group after formation of the pentasaccharide motif. Next, to achieve the regioselective sulfation of partially protected heparin pentasaccharides, the unaltered donors (4 and 5a) and the disaccharide acceptor 7b with appropriate protecting groups were used to yield 3b in 48% yield.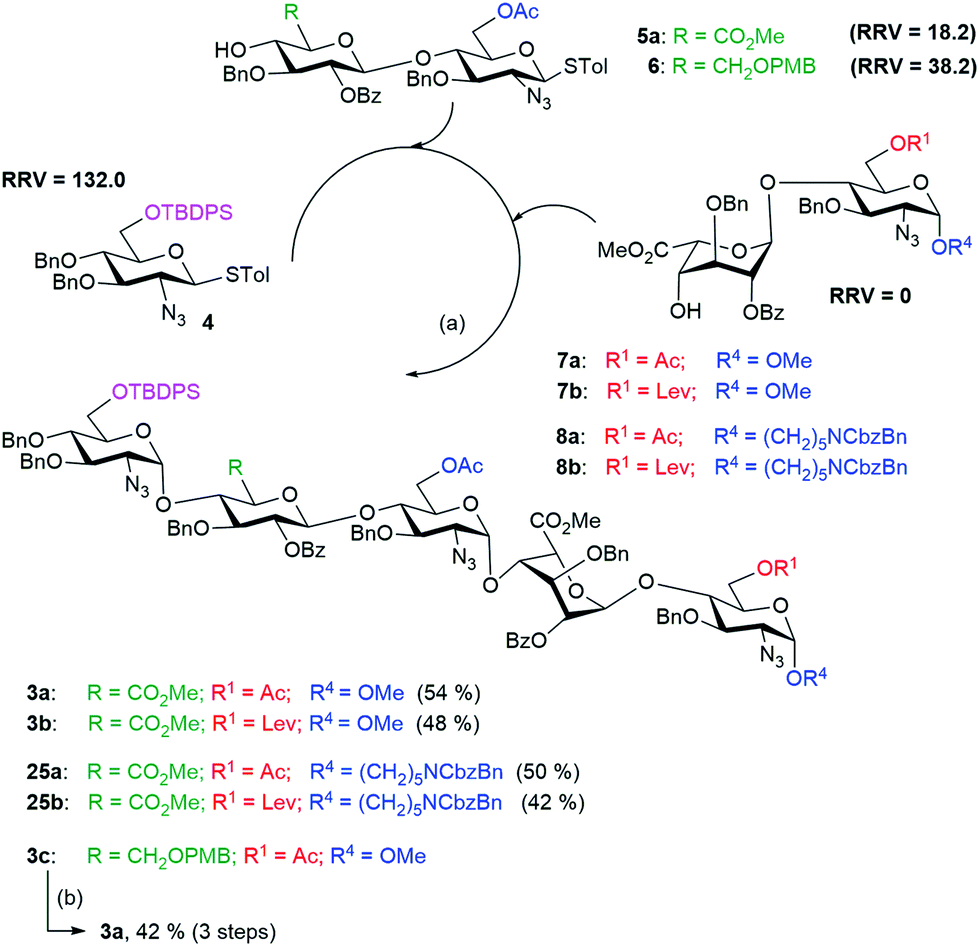 | ||
| Scheme 4 Reaction conditions: (a) NIS, TfOH, CH2Cl2, AW 300 MS, −45 °C to −25 °C; (b) (i) DDQ, CH2Cl2:H2O (10:1), rt, 1 h; (ii) BAIB, TEMPO, CH2Cl2:H2O (2:1), rt, 2 h; (iii) MeI, KHCO3, DMF, 0 °C to rt, 4 h. | ||
We also introduced an anomeric aminopentyl spacer to the disaccharide acceptors 8a and 8b for the one-pot synthesis of pentasaccharides 25a and 25b, respectively, using similar reaction conditions (Scheme 4) for the purpose of developing a glycan microarray.29 We confirmed the stereochemistry of the newly formed glycosidic linkages in the protected pentasaccharide using C–H coupling constants, measured from the 13C coupled HMQC experiment.30 In the case of pentasaccharide 3b, the 1JC–H coupling values were 1JC–H = 178.15 Hz (α), 176.06 Hz (α), 177.65 Hz (α), 172.56 Hz (α), and 163.3 Hz (β), indicating the presence of four α-linkages and one β-linkage.
Diversification of heparin pentasaccharide sulfates: access to regiodefined 6-O-sulfation
To generate the heparin pentasaccharides with regiodefined 6-O-sulfation patterns, we used the pentasaccharides 3a and 3b for further modification through chemical sulfation and deprotection. The installation of 6-O-SO3 failed in the presence of the TBDPS group, as the SO3 attacks the TBDPS ether groups and is inserted into the Si–O bond.31 So, we replaced the TBDPS protecting group with the benzyl ether group in order to introduce O-sulfate. The silyl groups in 3a and 3b were removed using HF-Py to generate compounds 26a and 26b respectively with free hydroxyl groups at the non-reducing end. Benzylation of 26a was attempted using a mixture of benzyl-2,2,2-trichloroacetimidate/TfOH or using NaH/BnBr. However, the decomposition of the starting material, hydrolysis of the methyl ester and acyl migration were observed under these reaction conditions. At this point, we came across the co-solvent promoted O-benzylation using Ag2O/BnBr reported by Hashimoto and co-workers.32 Thus, O-benzylations of 26a and 26b were performed under Hashimoto’s reaction conditions to furnish 27a and 27b, respectively, in excellent yields (Scheme 5). On the other hand, the 6-O-Lev ester in 27b was removed using hydrazine acetate in a mixture of MeOH and THF to give pentasaccharide 28 in a good yield. The co-solvent promoted O-benzylation of 28 with Ag2O/BnBr led to pentasaccharide 29 (Scheme 6).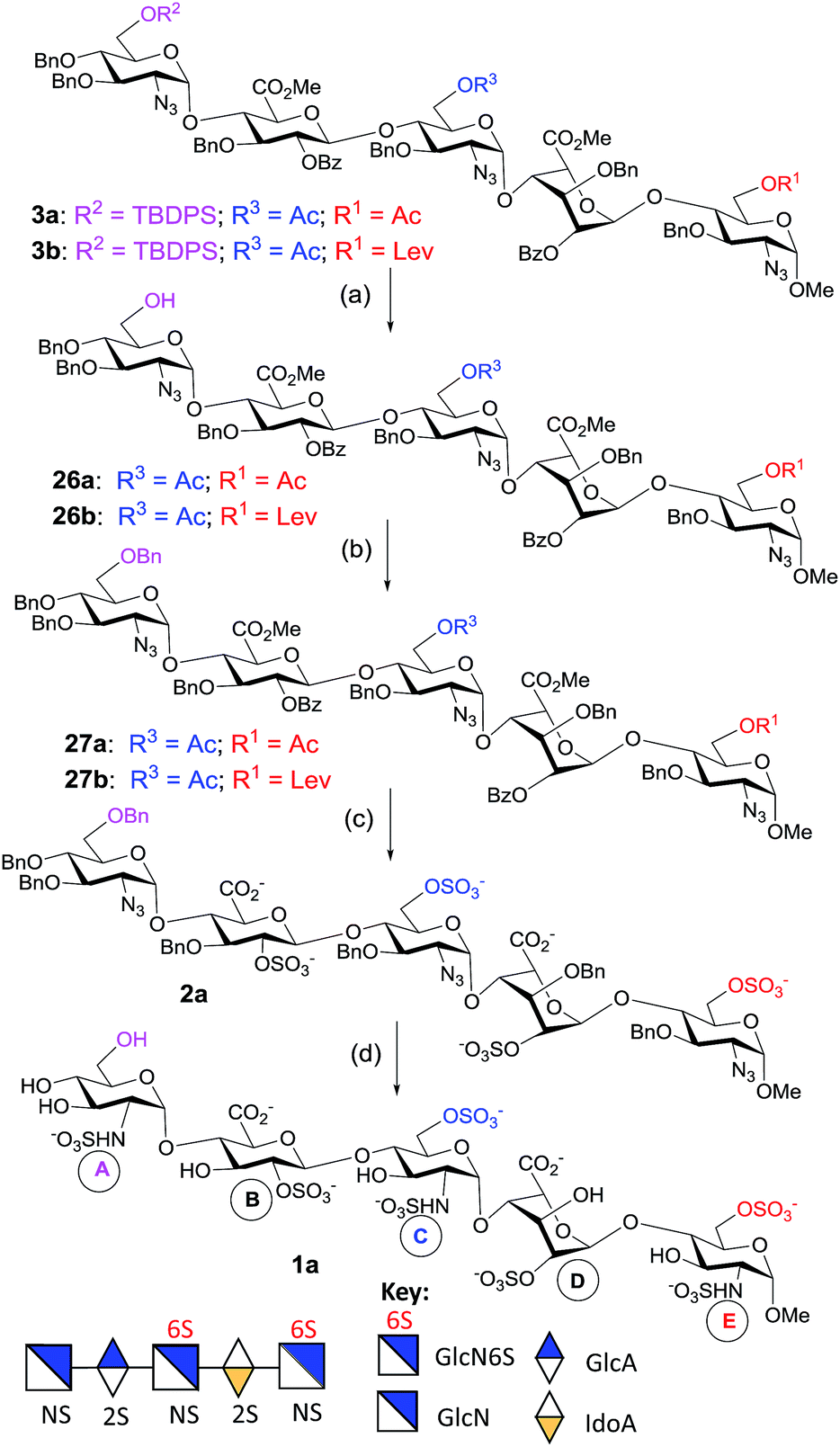 | ||
| Scheme 5 Synthesis of GlcNS-GlcA2S-GlcNS6S-IdoA2S-GlcNS6S-OMe (1a). Reaction conditions: (a) HF-Py, Py, 0 °C to rt, 12 h, 26a: 83%, 26b: 85%; (b) BnBr, Ag2O, n-Hex:CH2Cl2 (4:1), MS 4 Å, 70 °C, sealed tube, 12 h, 27a: 82%, 27b: 78%; (c) (i) 1 M LiOH, H2O2, THF, −5 °C to rt, 8 h; (ii) 4 M NaOH, MeOH, rt, 18 h; (iii) SO3–Et3N, DMF, 55 °C, 12 h, 75%; (d) (i) H2, 20% Pd(OH)2/C, MeOH, 36 h, rt; (ii) SO3–Py, 1 M NaOH, pH – 9.5, H2O, rt, 38 h, 40% (5 steps). | ||
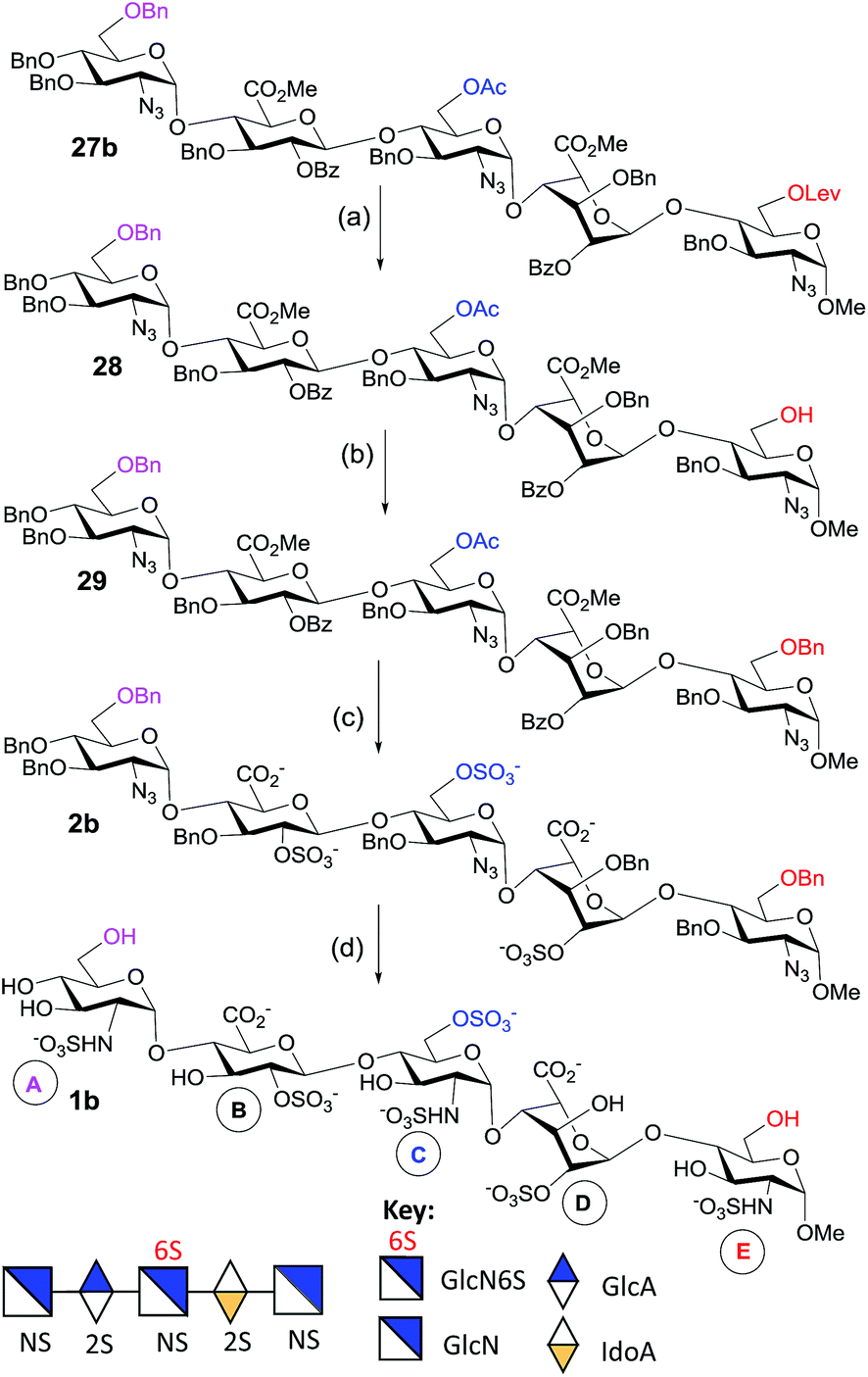 | ||
| Scheme 6 Synthesis of GlcNS-GlcA2S-GlcNS6S-IdoA2S-GlcNS-OMe (1b). Reaction conditions: (a) NH2NH2·AcOH, THF:MeOH (1:1), 0 °C to rt, 2 h, 85%; (b) BnBr, Ag2O, n-Hex:CH2Cl2 (4:1), MS 4 Å, 70 °C, sealed tube, 12 h, 81%; (c) (i) 1 M LiOH, H2O2, THF, −5 °C to rt, 8 h; (ii) 4 M NaOH, MeOH, rt, 18 h; (iii) SO3–Et3N, DMF, 55 °C, 12 h, 77%; (d) (i) H2, 20% Pd(OH)2/C, MeOH, 36 h, rt; (ii) SO3–Py, 1 M NaOH, pH – 9.5, H2O, rt, 38 h, 47% (5 steps). | ||
Saponification of pentasaccharides 27a, 27b and 29 using LiOH/H2O2 in THF, followed by NaOH in methanol, removed all ester functionalities.33 To generate the GlcNS-GlcA2S-GlcNS6S-IdoA2S-GlcNS6S-OMe motif, the crude tetraol derivative was treated with SO3–Et3N at 55 °C for 12 h to install two 6-O-sulfate and two 2-O-sulfate groups to generate 2a (Scheme 5). Similarly, O-sulfation of the crude triol generated pentasaccharide GlcNS-GlcA2S-GlcNS6S-IdoA2S-GlcNS-OMe 2b (Scheme 6).
The O-sulfated pentasaccharides 2a and 2b were converted to the protecting group free pentasaccharides in a two-step sequence: (i) catalytic hydrogenation in the presence of Pd(OH)2/C removed all benzyl ethers and reduced all azides to amines and (ii) selective N-sulfation was performed using SO3–pyridine at pH 9.5 with 1 M NaOH (aq). Finally, the crude O- and N-sulfated product was purified using column chromatography with Sephadex G-25 using water as the eluent, and the desired fractions were pooled and passed through an ion exchange column of Dowex 50WX8Na+. The pure fractions were lyophilized to give the N-sulfated pentasaccharides 1a (Scheme 5) and 1b (Scheme 6), respectively.
Conclusions
In summary, we have developed a new programmable one-pot strategy for the synthesis of protected heparin pentasaccharides, which can be selectively deprotected to enable the regioselective introduction of the sulfate groups. Using a set of designed thioglycoside building blocks with well-defined RRVs, a highly efficient synthesis of heparin pentasaccharide was achieved with improved overall yields. This method was applied to generate heparin pentasaccharides with mono 6-O-sulfation (ring C) in 1b and with double 6-O-sulfation (rings C and E) of 1a. The highly efficient and stereoselective formation of α-glycosidic linkages between the building blocks was achieved by a proper tuning of the protecting groups at the 6-hydroxyl positions during the one-pot synthesis. The use of silyl and acetyl groups at O-6 positions enhances the α-selectivity by remote participation, and the use of the tert-butyldiphenylsilyl group at O-6 of the thioglycoside increases its reactivity (RRV) which seems to be the driving force for the high-yielding one-pot synthesis. The glucuronic acid unit is better introduced at the disaccharide stage as a building block than at the pentasaccharide stage through oxidation, and the selection of building blocks with significant differences in RRVs is important to eliminate the problem of self-coupling. This work provides a new set of building blocks and illustrates the programmable one-pot synthesis of complex heparin pentasaccharides with regiodefined sulfate patterns. Currently, we are using this strategy for the synthesis of other GAG derivatives to facilitate the structure–activity study of this class of molecule.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was funded by the Academia Sinica and by the National Institutes of Health (AI072155) and the National Science Foundation (1664283).References
- F. E. Poulain and H. J. Yost, Development, 2015, 142, 3456 CrossRef PubMed
; L. Fu, M. Suflita and R. J. Linhardt, Adv. Drug Delivery Rev., 2016, 97, 237 CrossRef PubMed
- I. Ahmed, A. Majeed and R. Powell, Postgrad. Med. J., 2007, 83, 575 CrossRef PubMed
-
(a) D. Xu and J. Esko, Annu. Rev. Biochem., 2014, 83, 129 CrossRef PubMed
-
R. J. Linhardt and T. Toida, in Carbohydrates in Drug design, ed. Z. B. Witczak and K. A. Nieforth, Marcel Dekker, New York, 1997, ch. 4, p. 277 Search PubMed
-
(a) E. I. Oduah, R. J. Linhardt and S. T. Sharfstein, Pharmaceuticals, 2016, 9, 38 CrossRef PubMed
-
(a) A. Ori, M. C. Wilkinson and D. G. Fernig, Front. Biosci., 2008, 13, 4309 CrossRef PubMed
-
(a) A. Imberty, H. Lortat-Jacob and S. Perez, Carbohydr. Res., 2007, 342, 430 CrossRef PubMed
-
(a) X. Zhang, V. Pagadala, H. M. Jester, A. M. Lim, T. Q. Pham, A. M. P. Goulas, J. Liu and R. J. Linhardt, Chem. Sci., 2017, 8, 7932 RSC
- Z. Wang, P.-H. Hsieh, Y. Xu, D. Thieker, E. J. E. Chai, S. Xie, B. Cooley, R. J. Woods, L. Chi and J. Liu, J. Am. Chem. Soc., 2017, 139, 5249 CrossRef PubMed
-
(a) C. I. Gama, S. E. Tully, N. Sotogaku, P. M. Clark, M. Rawat, N. Vaidehi, W. A. Gaddard III, A. Nishi and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2006, 2, 467 CrossRef PubMed
-
(a) E. Avizienyte, C. L. Cole, G. Rushton, G. J. Miller, A. Bugatti, M. Presta, J. M. Gardiner and G. C. Jayson, PLoS One, 2016, 11, 1 CrossRef PubMed
- M. Petitou, B. Casu and U. Lindahl, Biochimie, 2003, 85, 83 CrossRef PubMed
-
(a) V. N. Patel, K. M. Likar, S. Zisman-Rozen, S. N. Cowherd, K. S. Lassiter, I. Sher, E. A. Yates, J. E. Turnbull, D. Ron and M. P. Hoffman, J. Biol. Chem., 2008, 283, 9308 CrossRef PubMed
-
(a) K. L. Stewart, E. Hughes, E. A. Yates, G. R. Akien, T.-Y. Huang, M. A. Lima, T. R. Rudd, M. Guerrini, S.-C. Hung, S. E. Radford and D. A. Middleton, J. Am. Chem. Soc., 2016, 138, 8328 CrossRef PubMed
- S. U. Hansen, G. J. Miller, M. J. Cliff, G. C. Jayson and J. M. Gardiner, Chem. Sci., 2015, 6, 6158 RSC
-
(a) W. Lu, C. Zong, P. Chopra, L. E. Pepi, Y. Xu, I. J. Amster, J. Liu and G.-J. Boons, Angew. Chem., Int. Ed., 2018, 57, 1 CrossRef
-
(a) J.-C. Jacquinet, M. Petitou, P. Duchaussoy, I. Lederman, J. Choay, G. Torri and P. Sinaÿ, Carbohydr. Res., 1984, 130, 221 CrossRef
-
(a) H. N. Yu, J.-I. Furukawa, T. Ikeda and C.-H. Wong, Org. Lett., 2004, 6, 723 CrossRef PubMed
-
(a) S. Arungundram, K. Al-Mafraji, J. Asong, F. E. Leach III, I. J. Amster, A. Venot, J. E. Turnbull and G.-J. Boons, J. Am. Chem. Soc., 2009, 131, 17394 CrossRef PubMed
-
(a) Z. Wang, Y. Xu, B. Yang, G. Tiruchinapally, B. Sun, R. Liu, S. Dulaney, J. Liu and X. Huang, Chem.–Eur. J., 2010, 16, 8365 CrossRef PubMed
-
(a) C.-H. Chang, L. S. Lico, T.-Y. Huang, S. Y. Lin, C.-L. Chang, S. D. Arco and S.-C. Hung, Angew. Chem., Int. Ed., 2014, 53, 9876 CrossRef PubMed
- G. Łopatkiewicz, S. Buda and J. Mlynarski, J. Org. Chem., 2017, 82, 12701 CrossRef PubMed
- Z. Zhang, I. R. Ollmann, X.-S. Ye, R. Wischnat, T. Baasov and C.-H. Wong, J. Am. Chem. Soc., 1999, 121, 734 CrossRef
-
(a) J.-C. Lee, W. A. Greenberg and C.-H. Wong, Nat. Protoc., 2006, 1, 3143 CrossRef PubMed
-
(a) C. M. Pedersen, L. U. Nordstrøm and M. Bols, J. Am. Chem. Soc., 2007, 129, 9222 CrossRef PubMed
- A. Imamura, N. Matsuzawa, S. Sakai, T. Udagawa, S. Nakashima, H. Ando, H. Ishida and M. Kiso, J. Org. Chem., 2016, 81, 9086 CrossRef PubMed
- L. J. van den Bos, J. D. C. Codée, J. C. van der Toorn, T. J. Boltje, J. H. van Boom, H. S. Overkleeft and G. A. van der Marel, Org. Lett., 2004, 6, 2165 CrossRef PubMed
- J. Tatai, G. Osztrovszky, M. Kajtár-Peredy and P. Fügedi, Carbohydr. Res., 2008, 343, 596 CrossRef PubMed
-
(a) P.-H. Liang, C.-Y. Wu, W. A. Greenberg and C.-H. Wong, Curr. Opin. Chem. Biol., 2008, 12, 86 CrossRef PubMed
-
(a) K. Bock, I. Lundt and C. Pederson, Tetrahedron Lett., 1973, 13, 1037 CrossRef
- A. Richter and D. Klemm, Cellulose, 2003, 10, 133 CrossRef
- L. Wang, Y. Hashidoko and M. Hashimoto, J. Org. Chem., 2016, 81, 4464 CrossRef PubMed
- H. Lucas, J. E. M. Basten, T. G. van Dinther, D. G. Meuleman, S. F. van Aelst and C. A. A. van Boeckel, Tetrahedron, 1990, 46, 8207 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01743c |
| This journal is © The Royal Society of Chemistry 2018 |