
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative strong metal–support interactions (OMSI) of supported platinum-group metal catalysts†
Hailian
Tang‡
abc,
Yang
Su‡
a,
Yalin
Guo
ad,
Leilei
Zhang
a,
Tianbo
Li
ad,
Ketao
Zang
e,
Fei
Liu
a,
Lin
Li
a,
Jun
Luo
e,
Botao
Qiao
 *a and
Junhu
Wang
*ab
*a and
Junhu
Wang
*ab
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: bqiao@dicp.ac.cn
bMössbauer Effect Data Center, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
cCollege of Chemistry and Environmental Science, Hebei University, Baoding 071002, China
dUniversity of Chinese Academy of Sciences, Beijing 100049, China
eCenter for Electron Microscopy, Institute for New Energy Materials and Low-Carbon Technologies, School of Materials, Tianjin University of Technology, Tianjin 300384, China
First published on 12th July 2018
Abstract
Supported platinum-group metal (PGM) catalysts are widely used in many important industrial processes. Metal–support interaction is of great importance in tailoring their catalytic performance. Here, we report the first example of oxidative strong metal–support interactions (OMSIs) between PGM and hydroxyapatite (HAP) which can be extended to PGM and ZnO. It occurred under high-temperature oxidation conditions accompanied by the encapsulation of PGM by HAP and electron transfer between PGM and HAP. With this OMSI, the aggregation and leaching of PGMs were significantly inhibited, resulting in an excellent catalytic stability and much improved reusability of supported Pt and Pd catalysts, respectively. This is the first time to find that PGMs can manifest OMSI which benefits the stabilization of PGM catalysts under oxidative reaction conditions. This new type of SMSI not only contributed to a deeper understanding of SMSI but also provided a new way to develop new stable PGM catalysts.
Introduction
Platinum-group metals (PGMs) are widely used as catalysts in many commercial chemical processes such as automotive pollution control, petrochemical, pharmaceutical feedstocks and fuel cell power generation.1 To use them more efficiently, PGMs were usually finely dispersed on high-surface-area materials (referred as support). The support can therefore influence the metals' catalytic performance through the metal–support interaction between them.2–5 Specially, in the late 1970s Tauster and his co-workers observed a dramatic adsorption suppression of small molecules (such as CO and H2) over TiO2 supported PGMs after high-temperature reduction (HTR) and termed this phenomenon as strong metal–support interactions (SMSI).6,7 The discovery of SMSI triggered a hot research topic subsequently,7–12 owning to its significant influence on the structure and electric property of PGM catalysts and their catalytic performance.7,9,10 However, so far all studies concerning SMSI on PGM catalysts are focused on those occurred upon HTR; whether SMSI can occurred upon high-temperature oxidation (HTO) treatment remains undiscovered.Recently the topic of SMSI has re-attracted extensive interests, primarily due to discoveries13–17 on some new types of SMSI as well as their applications.18–20 For example, a new type of SMSI, i.e., HTO induced SMSI (referred as oxidative SMSI and denoted as OMSI hereafter) was found to occur between Au and ZnO-nanorod13 and Au and hydroxyapatite (HAP).14 The occurrence of OMSI was initially ascribed to the special property of Au that is different from that of PGMs because for a long time Au has not been found to manifest SMSI with TiO2. However, last year we discovered that the SMSI can indeed occur between Au and TiO2 with appropriate H2-treatments.15 This new finding suggested that there may be no intrinsic difference between Au and PGMs in manifesting SMSI, which in turn imply that PGMs may also manifest OMSI on suitable support and with appropriate treatments. If the latter is true, it would be highly significant in fundamental study because for decades people have believed that the HTR procedure is the prerequisite to the occurrence of SMSI.7,8,21 The occurrence of OMSI between PGM and supports can therefore change this viewpoint and make people re-recognize and re-understand the SMSI phenomenon and formation mechanism. Of more importance, the OMSI of PGMs would be very valuable in practical applications given that they are in many cases industrially used in oxidative conditions. For instance, automotive pollution control is one of the most important application areas of PGMs as more than half of the annually produced PGMs were consumed in preparing three-way catalysts (TWCs).22,23 The sintering of PGMs is still the major issue in regard to the catalyst deactivation.24,25 The SMSI effect can effectively stabilize the metal NPs and promote the durability of the catalysts.14,15,20 However, the stabilizing effect of the classical SMSI can, at least theoretically, not persist under HTO conditions (TWC's working condition) owning to the reverse of the SMSI26,27 while the OMSI might works well at this condition. The topic of SMSI re-attracted extensive interests recently due to some new discoveries13–17 on SMSI as well as their new applications.18–20 A new type of SMSI, i.e., HTO induced SMSI (referred as oxidative SMSI and denoted as OMSI hereafter) between Au and ZnO-nanorod13 and Au and hydroxyapatite (HAP)14 were recently discovered. This was initially considered to be due to the different properties of Au from those of PGMs because for a long time, Au has not been found to manifest H-SMSI with TiO2. However, in the last year we discovered that the H-SMSI can indeed occur between Au and TiO2 with appropriate H2-treatments.15 This new finding suggested that there may be no intrinsic difference between Au and PGMs in manifesting SMSI, which may in turn imply that PGMs can also manifest OMSI suitable support and appropriate treatments. If the latter is true, it would be highly significant in the fundamental study because for decades people have been believing that the HTR procedure is the prerequisite for the occurrence of SMSI.7,8,21 The OMSI between PGM and supports can therefore change this viewpoint and make people re-recognize and re-understand the SMSI phenomenon and formation mechanism. Of more importance, the OMSI of PGMs would be very valuable in practical applications given that they are in many cases industrially used in oxidative conditions. For instance, automotive pollution control is one of the most important application areas of PGMs as more than half of the annually produced PGMs were consumed in preparing three-way catalysts (TWCs).22,23 The sintering of PGMs is still the major issue concerning the catalyst deactivation.24,25 The SMSI effect can effectively stabilize the metal NPs and promote the durability of the catalysts.14,15,20 However, the stabilizing effect of the H-SMSI can, at least theoretically, not persist under HTO conditions (such as TWC's working conditions) owing to the reverse of the SMSI26,27 while the OMSI might work well at this condition.
In this work, we report our discovery that PGMs (Pd and Pt) can manifest OMSI with both HAP and the ZnO-nanorod support wherein the PGM NPs were encapsulated by the support thus the metal leaching and aggregation were effectively inhibited, resulting in an excellent reaction stability of Pt/HAP in CO oxidation in a simulated practical application conditions and a much-improved reusability of Pd/HAP in Suzuki cross-coupling, a liquid phase reaction. This is the first time to discover that PGMs can form OMSI which not only contributed to a deeper understanding of the SMSI effect, but also provided a new way to fabricate stable PGM catalysts in diverse application conditions.
Results and discussion
Preparation of HAP supported PGM catalysts
The HAP was synthesized by a precipitation method28 and further calcined at 400 °C before used as a support. The Pd and Pt species were introduced onto the HAP by a facile adsorption method29,30 with metal loading of 0.22 and 0.61 wt%, respectively, and denoted as Pd/HAP-fresh and Pt/HAP-fresh, respectively. The fresh samples were subsequently oxidized at 500 °C under 10 vol% O2/He flow and denoted as M/HAP-O, where M represents Pd and Pt. For comparison, the samples of M/HAP-O were further reduced under 10 vol% H2/He flow at 250 or 500 °C and denoted as M/HAP-OH. Details of the synthesis procedure of HAP, preparation methods and thermal treating protocols (Scheme S1†) of M/HAP samples are presented in the Experimental section in the ESI.†Characterization of PGM dispersion
The XRD patterns of the Pd/HAP and Pt/HAP samples are shown in Fig. S1†. It shows that the patterns of both the series of catalysts are exactly the same as that of HAP; that is, no diffuse diffraction peaks of the supported metal or metal oxide phase were observed. This result suggested that either the Pd or Pt species are highly dispersed on the support of HAP under various redox treatments, demonstrating a good stability which is probably due to a strong interaction with the support, or the metal loading amount is too low to be detected by XRD. To confirm the high dispersion of Pd and Pt species, the samples were further examined by electron microscopy. Representative high-angle annular dark-field scanning transmission electron microscopy (HADDF-STEM) images of various Pd/HAP and Pt/HAP samples are presented in Fig. S2†. It reveals clearly that on all samples the Pd and Pt NPs are highly dispersed with a size of <2 nm.Identification of OMSI
There are four basic characteristics concerning the SMSI,13–15i.e., (i) the significant suppression of small-molecule adsorption (such as CO and H2), (ii) electron transfer between metals and the support, (iii) mass transport from the support to form the encapsulation of metal NPs, and (iv) a reversal of the preceding phenomena upon reversal treatment.The suppression of chemisorption of small molecules over these samples was firstly explored as it is the essential characteristic of SMSI. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements were performed since it is sensitive to adsorption properties of the metal surface when an appropriate probe molecule was selected.31,32 As shown in Fig. 1a, for the sample of Pd/HAP-fresh, two peaks at 2088 and 1920 cm−1 were observed and attributed to linear- and bridge-bonded CO on Pd species, respectively.33,34 However, no CO adsorption peak was observed for the sample of Pd/HAP-O, suggesting a complete suppression of the adsorption after calcination with 10 v% O2/He at elevated temperatures. It's worth noting that this suppression should not be, at least not majorly, caused by the oxidation of Pd, since CO adsorption can be detected on the oxidised Pd species.35,36 To confirm this, a control experiment on the Pd/HAP catalyst calcined at lower temperatures (300 and 400 °C) under 10 v% O2/He were performed and CO adsorption on oxidized Pd was clearly observed (as shown in Fig. S3†). Similar results were obtained on Pt/HAP samples, as shown in Fig. 1b; the CO adsorption peaks on Pt species at 2106, 2084, 2058 and 1828 cm−1 over the Pt/HAP-fresh sample, which were separately ascribed to linear CO adsorption on PtOx NPs, Ptδ+ single atoms, Pt0 NPs and bridged CO adsorption on Pt species,37–40 were not observed after oxidation. This dramatic suppression of chemisorption on small metal NPs (<2 nm, Fig. S2b and S2e†) after HTO is identical to the feature that is observed on HAP supported Au NPs due to the physical coverage of the metal NPs surface by a thin layer of HAP.14 However, in this work it is difficult to directly verify the encapsulation layer by the high-resolution transmission electron microscopy (HRTEM) due to the ultrasmall metal NPs. We therefore performed a HTR treatment of the fresh samples at a higher temperature (800 °C) with a longer time (5 h) before calcination to cause the aggregation of metal NPs. Then the reduced samples were subjected to the same calcination procedure and denoted as Pd/HAP-RO and Pt/HAP-RO, respectively. As expected, on these samples the size of some metal NPs increased significantly (Fig. S4†). Notably, the HRTEM images (Fig. 2) revealed clearly that both Pd and Pt NPs were covered by a thin layer, which should originated from the migration of HAP species onto them.14
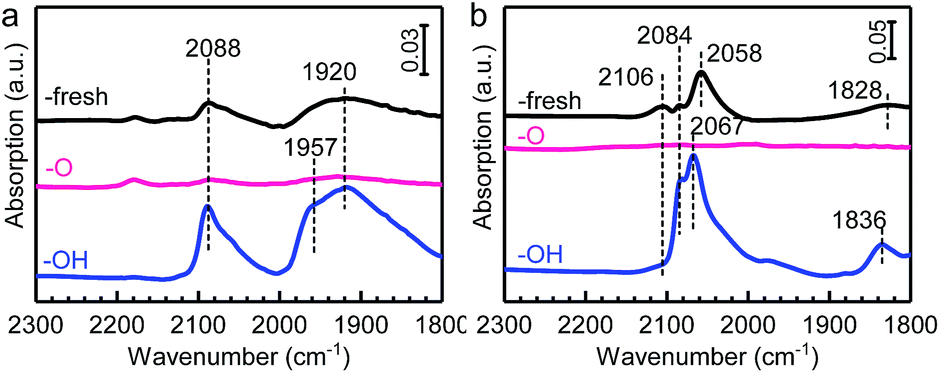 | ||
| Fig. 1 In situ DRIFT spectra of CO adsorption on the (a) Pd/HAP and (b) Pt/HAP series of samples obtained at 20 °C. Spectra were collected by purging gas CO with He after the samples were saturated by CO adsorption. | ||
 | ||
| Fig. 2 HRTEM images of (a) Pd/HAP-RO and (b) Pt/HAP-RO samples which were treated under 10 v% H2/He at 800 °C for 5 h followed by oxidation under 10 v% O2/He at 500 °C for 3 h. | ||
One of the important characteristics of SMSI is that the above phenomena are reversible under the opposite treatment conditions. We therefore further reduced the calcined samples (M/HAP-OH) and measured their adsorption properties. As shown in Fig. 1, the adsorption capacity was restored after reduction: On Pd/HAP-OH both adsorption peaks of linear-CO (2088 cm−1) and bridged-CO (1957 and 1920 cm−1) species were observed. On Pt/HAP-OH only three adsorption peaks at 2084, 2067 and 1836 cm−1 were clearly observed. The disappearance of the 2106 cm−1 peak suggested the reduction of Ptδ+ NPs. The results clearly demonstrated a reversal of the adsorption capacity under redox treatments in our case, as this change is not owing to the dispersion change of the metals (Fig. S2†). The reversibility is exactly the same as that for the OMSI that occurred in the HAP supported Au catalyst.14
The occurrence of SMSI is usually accompanied by electron transfer between metal NPs and the support. The electronic properties of Pd and Pt under redox treatment were therefore examined by X-ray photoelectron spectroscopy (XPS). Unfortunately no effective information was obtained on the Pd/HAP sample due to the signal overlap of Pd 3d and Ca 2p. However, the XPS spectra of Pt suggested the reversible electron transfer clearly. As shown in Fig. S5†, the binding energy (BE) of Pt 2p3/2 in the Pt/HAP-fresh sample is 72.0 eV, suggesting that the Pt species existed in the Pt2+ oxidative state. The BE of Pt 2p3/2 shifted to 72.5 eV after calcination at 500 °C, suggesting that an electron transfer from Pt NPs to the HAP support occurred. After a subsequent 500 °C-reduction treatment, the BE of Pt 2p3/2 shifted to 71.0 eV, the typical value for metallic Pt. This reversibility of the electron transfer under redox treatments in the Pt/HAP sample is exactly the same as that for the OMSI that occurred on the HAP supported Au catalyst.14 It should be noted that the absence of the Pt0 species in the Pt/HAP-fresh sample detected by XPS seems discrepant with the DRIFTS results where a peak of CO–Pt0 at 2058 cm−1 was observed (Fig. 1b). This can be explained by the in situ reduction of the partial surface Pt2+ species during the CO adsorption measurements.20,41
The above results unambiguously demonstrated the occurrence of OMSI between PGMs and HAP, a kind of nonoxide. In our previous work, we have proven that Au can form OMSI with HAP and we proposed that the occurrence of this OMSI is related to the change of the PO43− species (cation didn't affect the occurrence of this SMSI) in an O2 atmosphere (in the inert gas this SMSI didn't occur).14 However, recently, Xiao and co-workers found that on the Mg–Al layered double oxides supported Au catalyst, Au NPs could be encapsulated by the support due to the hydroxide-to-oxide transformation under calcination, although this process is not reversible.17 This report together with the fact that HAP also contains the hydroxyl group (–OH) inspired us to conjecture that the dehydroxylation during the calcination process may also contribute to the occurrence of this OMSI in the HAP system. If this is the case, a hydroxyl compensation may reverse the OMSI. To verify this, we treated the sample of Pd/HAP-O under 3 v% H2O/He at 250 °C for 0.5 h (denoted as Pd/HAP-O-H2O) and examined the CO adsorption by DRIFT. It was clearly observed that the CO adsorption on Pd restored compared with that treated by O2 (Fig. S6†), indicating that the –OH indeed plays an important role in the reverse of OMSI upon the HAP system. Accordingly, we believe that the dehydroxylation/dehydration may also trigger the OMSI. We therefore treated the sample in an inert gas (helium) at 500 °C. It was found that CO adsorption didn't decrease at all, consistent with our previous result of Au/HAP.14 However, when treating the sample at a higher temperature (800 °C, denoted as Pd/HAP-He-800) where the dehydroxylation of HAP starts,42 a complete disappearance of CO adsorption was observed. As most of Pd NPs is in size <5 nm (Fig. S7a and b†), this CO suppression should have been mainly due to the occurrence of OMSI rather than the increased Pd NP size. This was further confirmed by the HRTEM images. As suggested in Fig. S7c and d,† the Pd NPs on the Pd/HAP-He-800 sample were encapsulated by a thin layer. We therefore further treated the Au/HAP sample in He at 800 °C (denoted as Au/HAP-He-800) and examined the CO adsorption by DRIFT. However, as shown in Fig. S8,† a peak at 2103 cm−1, ascribed to linear CO adsorption on metallic Au NPs, was clearly observed, demonstrating that a complete encapsulation of the Au NPs by HAP didn't occur. This is different from the Pd/HAP-He-800 sample which might originate from the different work function and surface energy of Au and PGMs. HRTEM images revealed that the Au NPs were actually partially encapsulated (Fig. S9†), in consistent with the DRIFT results. This set of experiments clearly demonstrated that the dehydroxylation/dehydration during high-temperature treatments contributes, at least partially, to the formation of OMSI. Of course, the contribution of the change of the PO43− species under an O2 atmosphere cannot be excluded. A detailed and systemic study is still needed to clarify the origin and mechanism of the OMSI.
Catalytic performance
The finding of OMSI of PGMs is of great value in practical application. For example, TWCs, consuming over half of annual PGM production, generally work in an oxidative atmosphere at elvated temperatures which requires the catalysts to be stable at HTO conditions. Our sample M/HAP-O, with the encapsulation occurring at HTO conditions, may meet this requirement. To verify this, we tested the durability of Pt/HAP-fresh and Pt/HAP-O (with a Pt loading of ∼0.04 wt% that is in a similar level to the commercial TWCs to avoid activity saturation at higher temperatures) at 400 °C by using CO oxidation in a simulated auto-emission control as the probe reaction.20 As shown in Fig. S11†, for the Pt/HAP-O sample, the conversion kept unchanged during a 40 hour test, showing an excellent stability. However, the Pt/HAP-fresh, as expected, showed a much higher initial CO activity but a much lower CO stability in the identical condition. The abrupt decrease of CO conversion at the 5th hour should be ascribed to the formation of OMSI under the reaction conditions where a net oxidative gas atmosphere was remained after CO being totally converted. For comparison, we also tested a Pt/TiO2-H500 catalyst prepared by the same method with a similar Pt loading. Before the test the sample was pre-reduced at 500 °C under 10 v% H2/He for 1 h which would lead to the formation of classical SMSI. As shown in Fig. 3, although a higher initial CO conversion was obtained which should be due to the lower space velocity, the CO conversion decreased severely within a 23 h test, suggesting a much worse stability. This tendency is more straightforward when the result was presented in a reaction rate vs. reaction time model. As shown in Fig. S12†, the Pt/TiO2-H500 sample had slightly higher initial reaction rate (∼630 molCO gPt−1 h−1) compared with Pt/HAP-O (∼570 molCO gPt−1 h−1) but deactivated quickly. On the contrary, the Pt/HAP-O catalyst is very stable and its reaction rate remained unchanged. The result of stalbity test clearly highlights the advantage of OMSI in this application.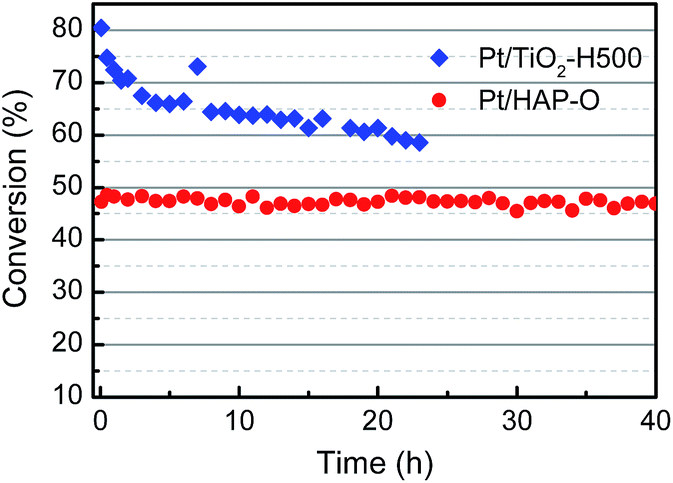 | ||
Fig. 3 CO conversion curves as a function of reaction time on Pt/HAP-O and Pt/TiO2-H500 samples at 400 °C. SV of Pt/HAP-O and Pt/TiO2–H500 are ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 690000 and 1100000 L gPt−1 h−1, respectively. Reaction gas composition: 1.6 v% CO, 1 v% O2, 0.01 v% propene, 0.0087 v% toluene, 10 v% water and balanced with He. 690000 and 1100000 L gPt−1 h−1, respectively. Reaction gas composition: 1.6 v% CO, 1 v% O2, 0.01 v% propene, 0.0087 v% toluene, 10 v% water and balanced with He. | ||
In addition, PGM catalysts are widely used in fine chemical syntheses. They, however, suffer from the deactivation by metal leaching and aggregation during reaction.43,44 Our previous work has demonstrated that the formation of OMSI can effectively inhibit the aggregation and leaching of Au NPs in the liquid phase alcohol oxidation reaction.14 In this work, this effect was further exemplified by using Suzuki cross-coupling, a widely used reaction in the organic synthesis industry catalyzed by Pd-based catalysts.45,46 As shown in Fig. 4, on the Pd/HAP-fresh sample the initial conversion of iodobenzene is 92%. However, it continuously decreased to 50% in the subsequent 4 cycles. The analysis of the used catalyst shows that there is a severe leaching of Pd, with the Pd loading amount decreased from 0.36 wt% to 0.06 wt% (Table S1†). Furthermore, besides a small amount of Pd NPs with a size of ∼2 nm, some big Pd NPs can also be observed in the HAADF-STEM images (Fig. S10a and b†), suggesting the aggregation of the Pd species during the reaction progress. On the contrary, the conversion (∼90%) on the Pd/HAP-O kept almost unchanged in five cycle tests, suggesting an excellent reusability. As expected, after the reaction, the Pd NPs are still small (∼2 nm, Fig. S10c and d†) and the Pd loading is 0.35 wt% (Table S1†), suggesting that the encapsulation layer can effectively prevent the leaching and aggregation of metal NPs, thus significantly enhance the reusability of the catalysts.
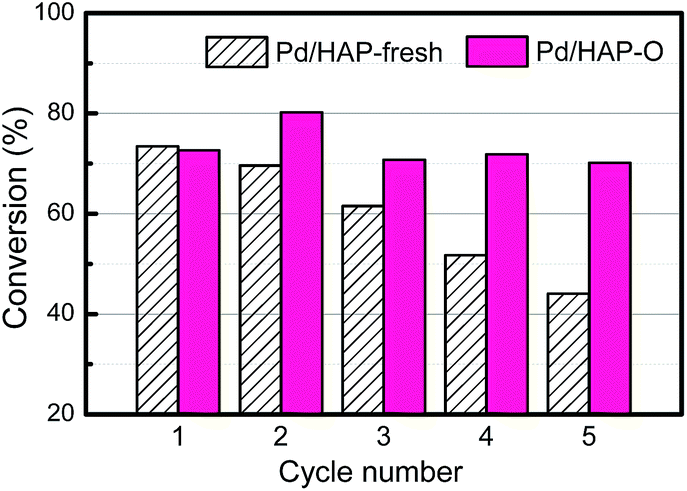 | ||
| Fig. 4 Cycling performance of Pd/HAP-fresh and Pd/HAP-O in the Suzuki cross-coupling reaction. Reaction conditions: 80 °C, 3 h, iodobenzene (4 mmol), phenylboronic acid (4.8 mmol), K2CO3 (8 mmol), H2O (20 mL), and catalyst (50 mg). | ||
OMSI in ZnO supported PGM catalysts
It was also found previously that Au NPs can manifest OMSI with a ZnO-nanorod support.13 We therefore speculated that the OMSI can also occur upon the PGMs/ZnO system. To verify this, we further prepared ZnO-nanorod supported Pd and Pt samples and measured their CO adsorption properties by DRIFTS, and the results are presented in Fig. 5. Similar to that of CO adsorption on HAP supported catalysts, a reversible CO adsorption was detected on both the Pd/ZnO and Pt/ZnO samples, demonstrating the occurrence of OMSI.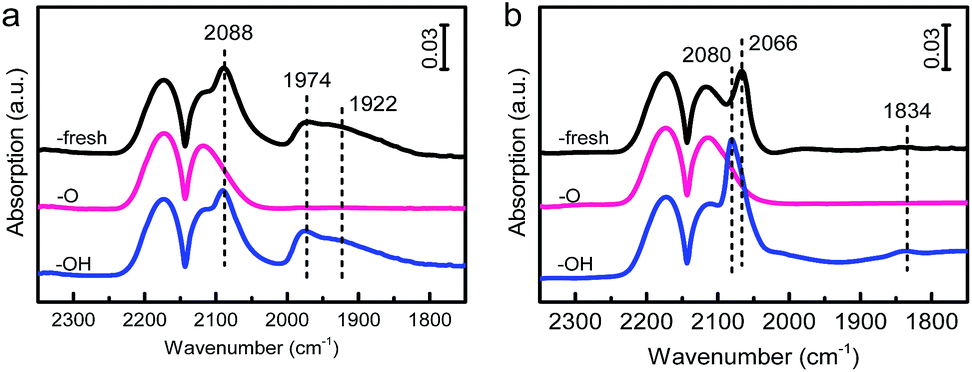 | ||
| Fig. 5 In situ DRIFT spectra of CO adsorption on the (a) Pd/ZnO and (b) Pt/ZnO series samples obtained at 20 °C. | ||
Conclusions
In summary, in this work the OMSI (O2-induced SMSI) between PGM NPs and HAP was discovered for the first time. It takes place with HTO and reversed with HTR. In addition, it also occurs at high-temperature treatment in inert gas atmosphere, and at least partially reversed with water treatment, suggesting that the losing of –OH group at high temperature partially contributes to the formation of OMSI. With this OMSI effect, the PGM NPs were encapsulated by a thin support layer thus their aggregation and leaching were effectively inhibited. As a result, the catalytic stability and the reusability of the M/HAP catalysts were significantly improved. Furthermore, it was found that this OMSI is general and can be extended to ZnO-nanorod supported PGM catalysts. The new findings in this work not only contributes to a deeper understanding of the SMSI effect but also would be beneficial for the development of ultrastable PGM catalysts in various application conditions, thus opening a new window for the design and tailor of PGM catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key R&D Program of China (2016YFA0202804), the Strategic Priority Research Program of the Chinese Academy of Science (XDB17020100), the National Natural Science Foundation of China (21776270, 21476232) and the Department of Science and Technology of Liaoning Province (2015020086-101).Notes and references
- R. J. Seymour and J. I. O'Farrelly, Platinum-Group Metals. Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, 2001 Search PubMed.
- G.-M. Schwab, in Adv Catal, ed. H. P. D. D. Eley and B. W. Paul, Academic Press, 1979, vol. 27, pp. 1–22 Search PubMed.
- J. Liu, ChemCatChem, 2011, 3, 934–948 CrossRef.
- H. Yi, Y. Xia, H. Yan and J. Lu, Chin. J. Catal., 2017, 38, 1581–1587 CrossRef.
- H. Yang, J. Deng, Y. Liu, S. Xie, P. Xu and H. Dai, Chin. J. Catal., 2016, 37, 934–946 CrossRef.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef.
- S. J. Tauster and S. C. Fung, J. Catal., 1978, 55, 29–35 CrossRef.
- R. T. K. Baker, E. B. Prestridge and R. L. Garten, J. Catal., 1979, 56, 390–406 CrossRef.
- S. J. Tauster, S. C. Fung, R. T. K. Baker and J. A. Horsle, Science, 1981, 211, 1120–1125 CrossRef PubMed.
- A. D. Logan, E. J. Braunschweig and A. K. Datye, Langmuir, 1988, 4, 827–830 CrossRef.
- J. P. Belzunegui, J. Sanz and J. M. Rojo, J. Am. Chem. Soc., 1992, 114, 6749–6754 CrossRef.
- X. Liu, M.-H. Liu, Y.-C. Luo, C.-Y. Mou, S. D. Lin, H. Cheng, J.-M. Chen, J.-F. Lee and T.-S. Lin, J. Am. Chem. Soc., 2012, 134, 10251–10258 CrossRef PubMed.
- H. Tang, J. Wei, F. Liu, B. Qiao, X. Pan, L. Li, J. Liu, J. Wang and T. Zhang, J. Am. Chem. Soc., 2016, 138, 56–59 CrossRef PubMed.
- H. Tang, Y. Su, B. Zhang, A. F. Lee, M. A. Isaacs, K. Wilson, L. Li, Y. Ren, J. Huang, M. Haruta, B. Qiao, X. Liu, C. Jin, D. Su, J. Wang and T. Zhang, Sci. Adv., 2017, 3, e1700231 CrossRef PubMed.
- J. C. Matsubu, S. Zhang, L. DeRita, N. S. Marinkovic, J. G. Chen, G. W. Graham, X. Pan and P. Christophe, Nat. Chem., 2017, 9, 120–127 CrossRef PubMed.
- L. Wang, J. Zhang, Y. Zhu, S. Xu, C. Wang, C. Bian, X. Meng and F.-S. Xiao, ACS Catal., 2017, 7, 7461–7465 CrossRef.
- A. Corma, S. Pedro, C. Patricia and C. J. Juan, J. Am. Chem. Soc., 2008, 130, 8748–8753 CrossRef PubMed.
- P. Sonström, D. Arndt, X. Wang, V. Zielasek and M. Bäumer, Angew. Chem., Int. Ed., 2011, 50, 3888–3891 CrossRef PubMed.
- H. Tang, F. Liu, J. Wei, B. Qiao, K. Zhao, Y. Su, C. Jin, L. Li, J. J. Liu, J. Wang and T. Zhang, Angew. Chem., Int. Ed., 2016, 55, 10606–10611 CrossRef PubMed.
- G. L. Haller and D. E. Resasco, Adv. Catal., 1989, 36, 173–235 Search PubMed.
- H. E. Hilliard, U.S. GEOLOGICAL SURVEY MINERALS YEARBOOK, 2002 Search PubMed.
- Y. Zhang, R. W. Cattrall, I. D. McKelvie and S. D. Kolev, Gold Bull., 2011, 44, 145–153 CrossRef.
- H. Shinjoh, Catal. Surv. Asia, 2009, 13, 184–190 CrossRef.
- J.-J. He, C.-X. Wang, T.-T. Zheng and Y.-K. Zhao, Johnson Matthey Technol. Rev., 2016, 60, 196–203 CrossRef.
- E. J. Braunschweig, A. D. Logan, A. K. Datye and D. J. Smith, J. Catal., 1989, 118, 227–237 CrossRef.
- S. Bernal, F. J. Botana, J. J. Calvino, C. López, J. A. Pérez-Omil and J. M. Rodríguez-Izquierdo, J. Chem. Soc., Faraday Trans., 1996, 92, 2799–2809 RSC.
- K. Zhao, B. Qiao, J. Wang, Y. Zhang and T. Zhang, Chem. Commun., 2011, 47, 1779–1781 RSC.
- B. Qiao, J.-X. Liang, A. Wang, C.-Q. Xu, J. Li, T. Zhang and J. J. Liu, Nano Res., 2015, 8, 2913–2924 CrossRef.
- R. Lang, T. Li, D. Matsumura, S. Miao, Y. Ren, Y. T. Cui, Y. Tan, B. Qiao, L. Li, A. Wang, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2016, 55, 16054–16058 CrossRef PubMed.
- C. Lamberti, A. Zecchina, E. Groppo and S. Bordiga, Chem. Soc. Rev., 2010, 39, 4951–5001 RSC.
- A. Vimont, F. Thibault-Starzyk and M. Daturi, Chem. Soc. Rev., 2010, 39, 4928–4950 RSC.
- M. Fernández-García, A. Martínez-Arias, A. Iglesias-Juez, A. B. Hungría, J. A. Anderson, J. C. Conesa and J. Soria, Appl. Catal., B, 2001, 31, 39–50 CrossRef.
- B. Qiao, J. Lin, L. Li, A. Wang, J. Liu and T. Zhang, ChemCatChem, 2014, 6, 547–554 CrossRef.
- K. I. Hadjiivanov and G. N. Vayssilov, Adv. Catal., 2002, 47, 307–511 Search PubMed.
- D. Tessier, A. Rakai and F. Bozon-Verduraz, J. Chem. Soc., Faraday Trans., 1992, 88, 741–749 RSC.
- P. Panagiotopoulou and D. I. Kondarides, J. Catal., 2008, 260, 141–149 CrossRef.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Commun., 2011, 3, 634–641 Search PubMed.
- M. J. Kale and P. Christopher, ACS Catal., 2016, 6, 5599–5609 CrossRef.
- T. Avanesian, S. Dai, M. J. Kale, G. W. Graham, X. Pan and P. Christopher, J. Am. Chem. Soc., 2017, 139, 4551–4558 CrossRef PubMed.
- K. Zhao, H. Tang, B. Qiao, L. Li and J. Wang, ACS Catal., 2015, 5, 3528–3539 CrossRef.
- Y. Liu and Z. Shen, J. Eur. Ceram. Soc., 2012, 32, 2691–2696 CrossRef.
- A. Biffis, M. Zecca and M. Basato, J. Mol. Catal. A: Chem., 2001, 173, 249–274 CrossRef.
- L. D. Pachón and G. Rothenberg, Appl. Organomet. Chem., 2008, 22, 288–299 CrossRef.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef.
- A. Suzuki, J. Organomet. Chem., 1999, 576, 147–168 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental Section, Fig. S1–S12 and Table S1. See DOI: 10.1039/c8sc01392f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |