
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadical difluoromethylthiolation of aromatics enabled by visible light†
Jianbin
Li‡
 ,
Dianhu
Zhu‡
,
Leiyang
Lv
and
Chao-Jun
Li
*
,
Dianhu
Zhu‡
,
Leiyang
Lv
and
Chao-Jun
Li
*
Department of Chemistry, McGill University, 801 Sherbrooke Street West, Montreal, QC H3A 0B8, Canada. E-mail: cj.li@mcgill.ca
First published on 1st June 2018
Abstract
Direct introduction of a difluoromethylthio group (–SCF2H) to arenes represents an efficient route to access a valuable catalogue of organofluorines; however, to realize this transformation under metal-free and mild conditions still remains challenging and rarely reported. Herein, a metal-catalyst-free and redox-neutral innate difluoromethylthiolation method with a shelf-stable and readily available reagent, PhSO2SCF2H, under visible light irradiation is described. This light-mediated protocol successfully converts a broad spectrum of arenes and heteroarenes to difluoromethylthioethers in the absence of noble metals and stoichiometric amounts of additives.
The difluoromethylthio group, as a member of the fluoroalkyl family, has been receiving growing attention from both academia and industry.1 This is not only because it incorporates two instrumental elements, sulfur and fluorine, into one functionality, but also due to its unique properties (Fig. 1b): (1) –SCF2H is intermediately lipophilic (Hansch lipophilicity parameter, πR = 0.68 for –SCF2H, 0.56 for –CH3 and 1.44 for –SCF3),2 providing flexibility to medicinal chemists in the rational design of drug candidates; (2) –SCF2H features a slightly acidic proton, rendering it a weak hydrogen bond donor (pKa = 35.2; hydrogen bond acidity parameter A = 0.098) to tune the molecule's binding ability;1b,3 (3) the electron-withdrawing nature of –SCF2H could promote the metabolic stability of target compounds; and (4) difluoromethylsulfides can participate in some late-stage modification events, which could diversify this functionality and may regulate the bio-activity of host molecules. Pyriprole, patented in 2008 as a novel pest control agent, showed advantageous performance (Fig. 1a).4 Its invention detailed that the C-4 position bearing –SCF2H was identified as the most preferable structure. Furthermore, the important role of –SCF2H in pharmaceuticals and agrochemicals is evidenced by its frequent enrolment in other bioactive compounds, e.g., herbicide SSH-108,5 nifedipine analogue,6 and thymol analogue7 (Fig. 1a).
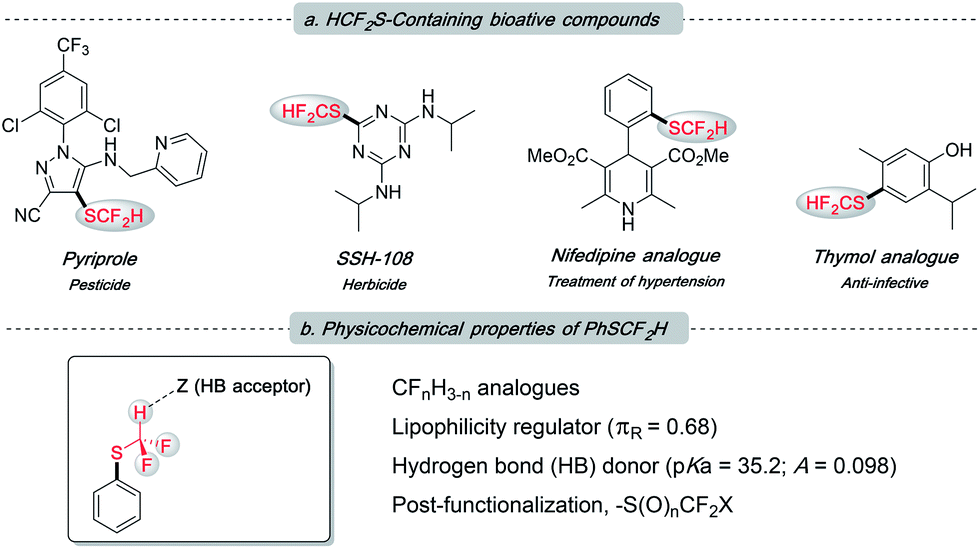 | ||
| Fig. 1 (a) Frequent appearance of the –SCF2H residue in bioactive molecules; (b) overview of the uniqueness of –SCF2H. | ||
Despite the intriguing pharmaceutical potential exhibited by difluoromethylthioethers, their widespread application remains limited possibly owing to a lack of efficient preparative methods.8 Classical and commonly used approaches to synthesize difluoromethylthioethers employ the nucleophilic attack of an appropriate thiolate (RS−) to some “CF2” species,9 typically a difluoromethyl carbene (:CF2),10 (Scheme 1a, left). A complementary but less common approach to assemble –SCF2H is difluoromethylation of disulfides using nucleophilic difluoromethyl sources (e.g., activated TMSCF2H).11 A major step forward to expand the substrate scope was made by the Gooβen group who described a stepwise synthetic route involving pre-formed thiocyanates and the subsequent copper-mediated Langlois type nucleophilic substitution by TMSCF2H (Scheme 1a, right).3a,12 Nevertheless, these indirect methods still suffer from a limited substrate scope. In addition, they usually necessitate strong bases, harsh thermal conditions and environmentally unfriendly reagents to generate reactive thiolates and “CF2” species.
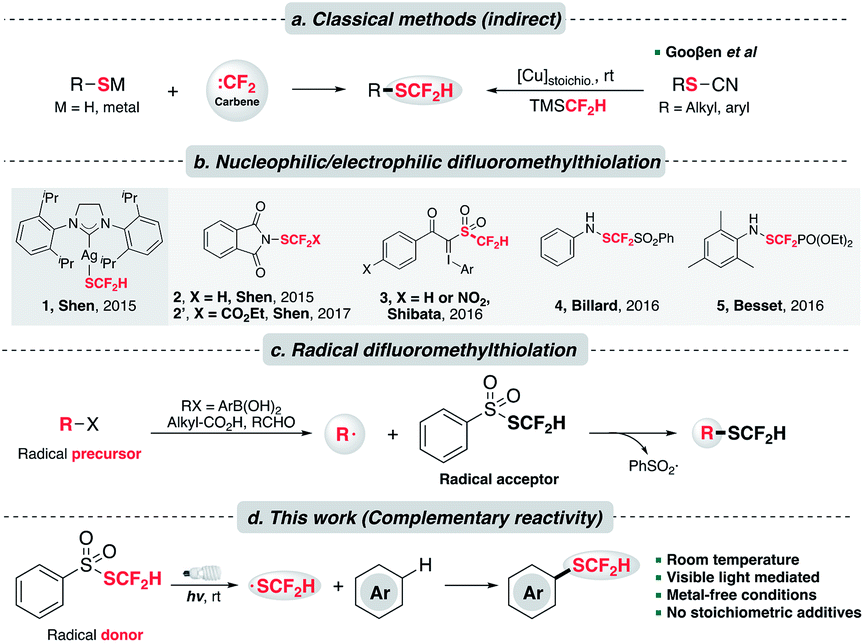 | ||
| Scheme 1 Different recipes to install –SCF2H on arenes. (a) Classical methods to prepare difluoromethylthioethers; (b) direct difluoromethylthiolating reagents (1: nucleophilic; 2–5: electrophilic); (c) radical difluoromethylthiolation wherein PhSO2SCF2H acts as a radical acceptor; (d) this work: catalyst-free and redox-neutral innate difluoromethylthiolation with PhSO2SCF2H as the radical source under visible light. | ||
To address these issues, a key contribution was made by Shen and his co-workers who delineated the first nucleophilic difluoromethylthiolating reagent 1, [(SIPr)Ag(SCF2H)] (Scheme 1b).13 In the presence of transition metals (M = Pd, Cu), this complex could couple with diverse aryl and heteroaryl halides, triflates and diazonium compounds to prepare difluoromethylthioethers. Complementarily, the same group debuted an electrophilic difluoromethylthiolating reagent 2,14 which could undergo a Friedel–Craft type Csp2–H difluoromethylthiolation on various N-heteroarenes. Shortly after, Shibata et al. uncovered a hypervalent difluoromethanesulfonyliodonium ylide reagent 3, which efficiently difluoromethylthiolated N-heterocycles under copper catalysis.15 Moreover, the Billard,16 Besset17 and Shen18 groups independently synthesized three electrophilic –SCF2FG group transfer reagents (FG = PhSO2, PO(OEt)2 and CO2Et respectively), although a separate reductive workup was necessary to give difluoromethylthioethers. Inspired by these elegant examples, other difluoromethylthiolating systems were unveiled.7,19
Alternatively, difluoromethylthiolation could be operated in a radical pathway. S-(Difluoromethyl)benzenesulfonothioate, PhSO2SCF2H, invented in 2016 by Shen et al., was revealed as an effective difluoromethylthiolating reagent (Scheme 1c).20 Mechanistically, it was proposed to execute as a radical acceptor and combine with the alkyl or aryl radicals generated from the Ag/persulfate-involved oxidation event. This reactivity was further elaborated in two recent studies on the ring-opening difluoromethylthiolation of cycloalkanols21 and the preparation of difluoromethylthioesters by using aldehydes as acyl radical precursors.22
Promising though these strategies are, the main drawbacks lie in their requirement of relatively high thermal energy (ranging from 50 to 120 °C), or involvement of precious metal catalysts and stoichiometric amounts of additives. In the case of –SCF2FG group transfer, additional steps were required to furnish the final thioether products. Clearly, a more sustainable synthetic protocol that features mild conditions and step economy is highly desired.
In 2017, our group disclosed a radical trifluoromethylation reaction on arenes with a novel sulfone reagent 6 (Scheme 2a).23 With light irradiation, 6 engenders two twin radicals via Norrish type I cleavage as the sulfinyl radical fragments further to produce the CF3 radical and implements trifluoromethylation. The success of this chemistry lies in the judicious design of the reagent structure. In the so-called “dummy group” strategy, the undesirable radical produced by homolysis of 6 is diminished in reactivity by the “captodative effect”24 as well as sterics.
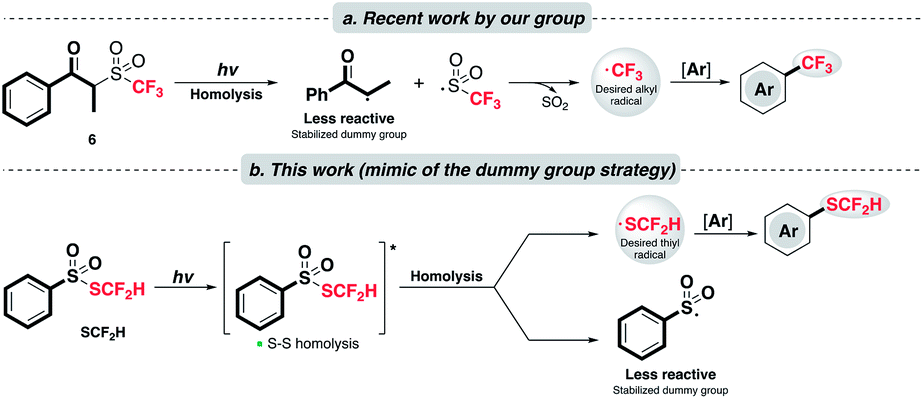 | ||
| Scheme 2 (a) Controlled radical generation via the “dummy fragment” concept; (b) this work: catalyst-free and redox-neutral innate difluoromethylthiolation enabled by light. | ||
Inspired by this work, we would like to apply a similar strategy and achieve a rarely known direct radical difluoromethylthiolation (Scheme 2b). A key component of this project is to explore and identify a suitable SCF2H radical precursor. After a careful examination of the literature, we consider that the thiosulfonate reagent, PhSO2SCF2H, might be applicable, although it was documented to be a radical acceptor as its principle utility.20,22 We proposed that the homolytic fragmentation of PhSO2SCF2H with a suitable light source is feasible due to the well-studied propensity of homolysis of the S–S bond25 and the stabilization of the resulting sulfonyl radical exerted by the resonance effect of the phenyl group.26 The stabilization of the dummy radical is beneficial in two ways: (1) it facilitates homolytic bond scission and allows the accumulation of SCF2H radicals; (2) it is expected to cause less competition toward the targeted reaction pathway. Radical addition of the desired SCF2H radical will furnish the target product.
As a part of our interest in photo-induced fluorine chemistry, we wish to document a photo-induced metal-free aromatic difluoromethylthiolation protocol with PhSO2SCF2H, wherein PhSO2SCF2H was utilized as a difluoromethylthiyl radical source. To the best of our knowledge, direct aromatic difluoromethylthiolation realized by radical attack of SCF2H radical on arenes remained unexplored.
To commence the study, N-methylindole 1a was chosen as a model substrate and treated with difluoromethylthiolating sources in CH3CN under an inert atmosphere based on some literature parallels on difluoromethylthiolation (see ESI† for full details). Although no desired product was detected when using BnSCF2H, the reaction with PhSO2SCF2H proceeded as expected and upon 16 hours of UV irradiation, 20% yield of the desired product was obtained (Table 1, entries 1 and 2). Using compact fluorescence lamps (CFL) as a light source gave 64% yield of the target compound (entry 3). Fortunately, prolonging the reaction time enabled the consumption of unreacted substrates and increased the yield to 80% (entry 4). The essential role played by light was illustrated by the control experiment as the dark condition disabled the reaction completely (entry 5 and see ESI† for details on control experiments). Recent work by our group revealed the unique properties of NaI, e.g., high reducing ability along with low nucleophilicity,27 which were helpful in radical generation. Therefore, we expected the SCF2H radical generation would be accelerated by a complementary reductive pathway. Gratifyingly, catalytic incubation of iodide gave similarly good yield in a shortened reaction time (entries 6–8). Among the tested iodides, tetrabutylammonium iodide (TBAI) offered the highest yield (entry 8). Further increment of TBAI loading could promote the reaction to be quantitative (entry 9). Conforming to other electrophilic difluoromethylthiolation studies, 1a as a limiting reagent would be more profitable as an excess of difluoromethylthiolating reagent is crucial to maintain a decent level of active difluoromethylthiolating species (entries 10 and 11).
| a Abbreviations: CFL, compact fluorescent lamp; rt, room temperature; TBAI, tetrabutylammonium iodide; NR, no reaction. b All reactions were conducted with 0.10 mmol 1a, 0.20 mmol PhSO2SCF2H, 0.020 mmol TBAI in 1.0 mL CH3CN under argon with irradiation of two 40 W CFL unless otherwise noted. c The yield was determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as the internal standard. d BnSCF2H as the difluoromethylthiolating source. e Six 254 nm 2.5 W UV lamps (photo-box). f In the dark. |
|---|
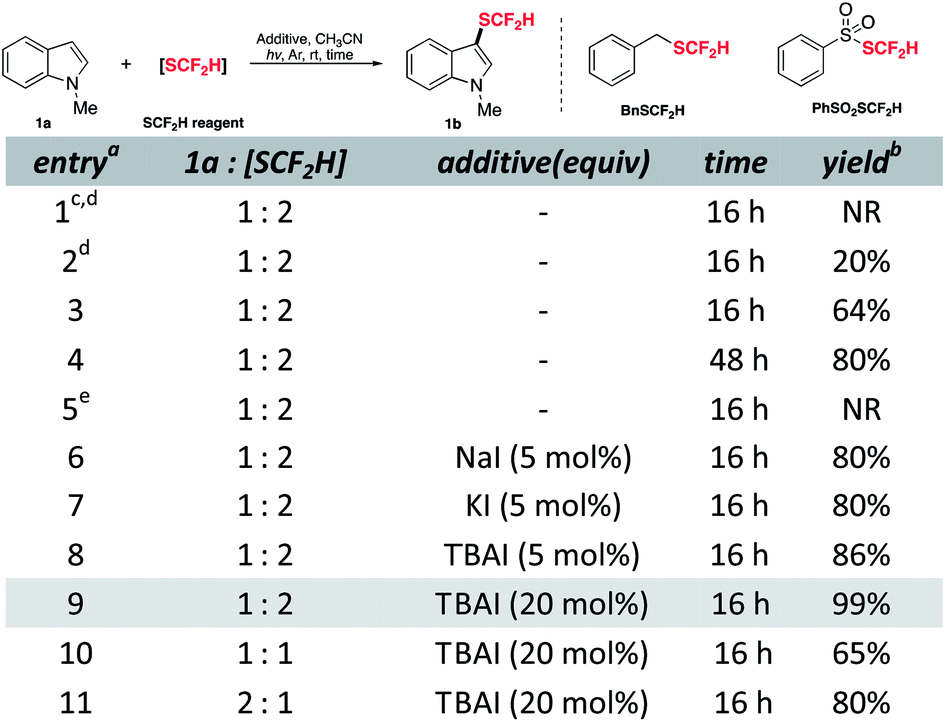
|
With the optimal conditions identified, the generality of this method was examined (Scheme 3). Initially, the functional group tolerance of different indoles was investigated. In general, indoles bearing substituents with different electronic and steric properties at various sites are compatible with the optimal conditions. Reaction rates of substrates with electron-donating groups were higher than those with electron-withdrawing groups. Satisfactorily, quantitative yield was obtained for non-substituted indole (2b). Product formation was not affected by the methyl group at the C-5 or C-7 position (3b and 4b). Even in the presence of a phenyl group at the C-2 position, a decent yield can also be achieved (5b). Notably, the reaction proceeded smoothly in indole tethering a phenolic proton (6b). Methoxylated indoles could result in 98% and 78% of desired products respectively (7b and 8b). Indole derivatives bearing the chloro, bromo and boryl groups resulted in good to excellent yields of products, which allow further cross coupling to achieve more complex settings (9b to 11b). Unexpectedly, regiomers were isolated for substrates with an ester group (12b and 12b′). Then we moved the focus to a class of closely related N-heterocycles, pyrroles. Pyrroles with phenyl, methyl, benzyl, dimethylamino and carbonato groups all gave around 70% of target difluoromethylthioethers (13b to 17b). Of note is the substrate bearing a double bond, which is known to be reactive toward PhSO2SCF2H. Gratifyingly, the Csp2–H difluoromethylthiolation product was selectively obtained (18b).
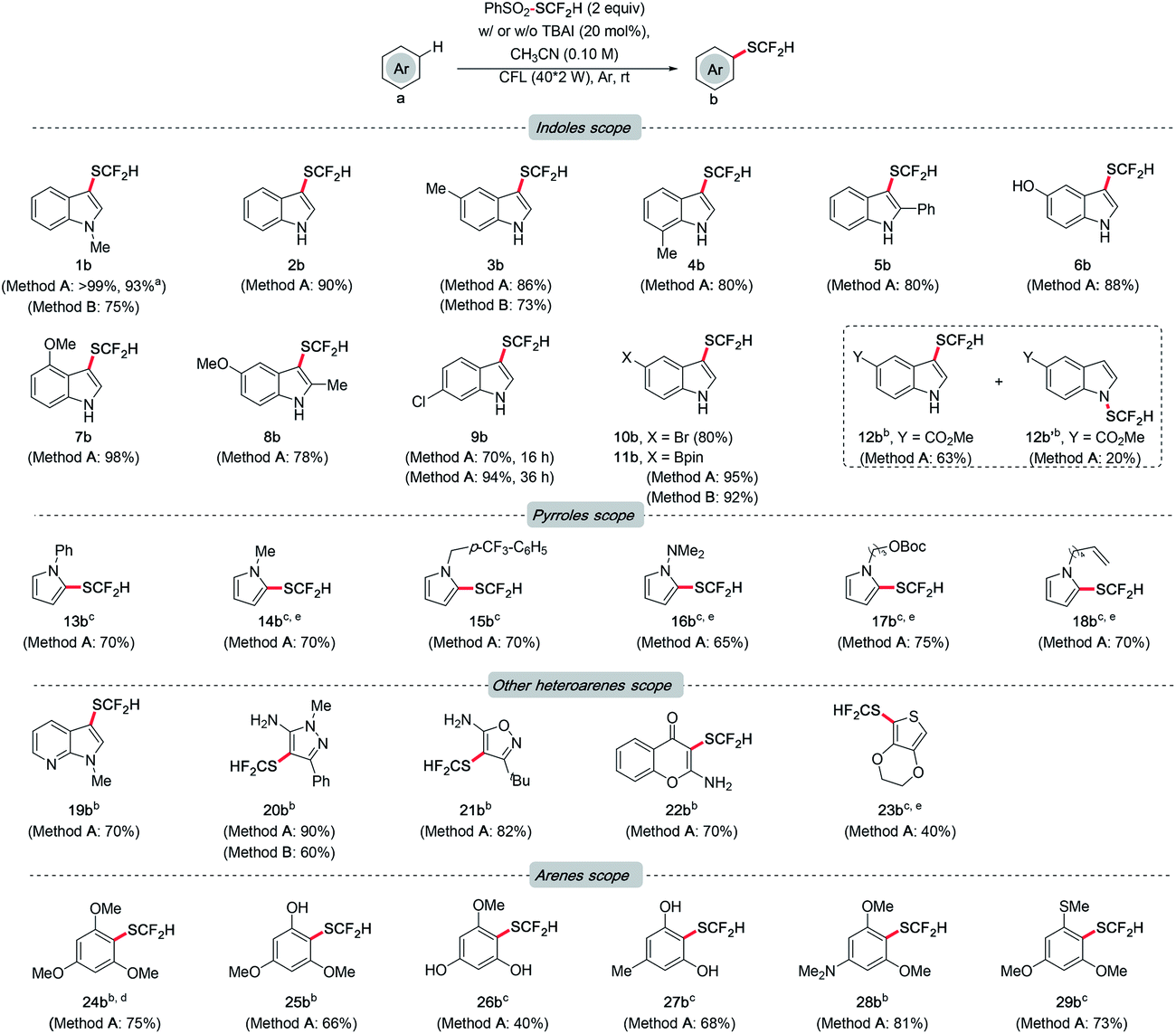 | ||
| Scheme 3 Scope of arenes. Method A: arene (0.10 mmol), PhSO2SCF2H (0.20 mmol), TBAI (0.020 mmol) in 1.0 mL CH3CN under argon for CFL irradiation at rt for 16 h. Method B: arene (0.10 mmol), PhSO2SCF2H (0.20 mmol) in 1.0 mL CH3CN under argon for CFL irradiation at rt for 48 h. The yields in the parentheses refer to the isolated ones unless otherwise specified. Volatility resulted in the low isolated yield of 17b and 18b. aReaction performed on the 0.40 mmol scale. bThe reactions were performed for 24 h. cThe reactions were performed for 48 h. d4 equiv. PhSO2SCF2H were used. eYields are quantified by GC-MS due to the volatility of target compounds. | ||
Other heteroarenes, e.g., azaindole, pyrazole, isoxazole, and chromone, which are pharmaceutically important scaffolds, were also effective (19b to 22b). Notably, this protocol was viable for thiophene, which was generally unreactive in other difluoromethylthiolating recipes (23b).10m,20 Finally, the versatility of this method on some arenes was explored. 1,3,5-Trimethoxybenzene was difluoromethylthiolated successfully (24b). As this method was proved adaptable to phenolic protons, installing –SCF2H on phenol (25b) and resorcinol derivatives (26b and 27b) was efficient. Aniline and sulfide underwent difluoromethylthiolation smoothly as well, resulting in good yields (28b and 29b).
Generally, high regioselectivity was observed for this difluoromethylthiolation reaction, which was a combined effect of electronics and sterics. In most cases, the site of the highest electron density was difluoromethylthiolated, e.g., C-2 in indoles and C-1 in pyrroles. When multiple sites of similar electron richness were available, the reaction occurred at the sterically less hindered Csp2–H (25b and 29b). Although an excess amount of PhSO2SCF2H was used, the difunctionalization product was rarely detected during our scope exploration (see ESI† for details of mono-/difunctionalization and regioselectivity issues).
Knowing that the difluoromethylsulfoxides and difluoromethylsulfones28 are valuable entities in medicinal chemistry, we were encouraged to alter the oxidation state of sulfur in –SCF2H. As expected, the oxidation of difluoromethylthioethers could be accomplished in a controlled manner under different oxidizing conditions (Scheme 4a). Besides, the versatility of this photochemical strategy was extended to produce other synthetically useful thiyl radicals. Under the optimal conditions, phenylthioether (30b) and naphthylthioether (31b) were furnished smoothly. However, alkylthiolation (32b) was undermined by the homocoupling event of alkylthiyl radicals. Moreover, 33a was subjected to a gram-scale experiment to demonstrate the practicality of this method (Scheme 4c). We were pleased to see that the reaction proceeded smoothly and a good yield of the desired product was obtained (33b).
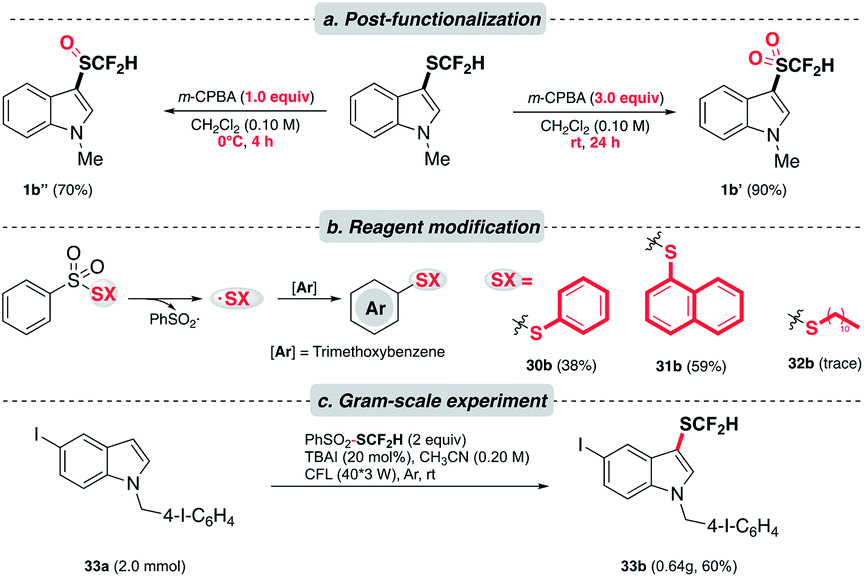 | ||
| Scheme 4 Application. (a) Controlled oxidation of thioethers to corresponding sulfoxides and sulfones; (b) modified reagents to afford arylthiolation products; (c) scaled-up experiments. | ||
Although the underlying mechanism remained obscure at this stage, our preliminary study hints at the radical nature of this reaction as designed. The presence of various radical quenchers significantly impacted the desired reactivity (Scheme 5, eqn (1) and (2)). When diallylmalonate (34a) was employed as the radical clock-type trapper, a cyclization adduct (34b) with PhSO2SCF2H was observed irrespective of the presence of 1a (eqn (2) and (3)). This result, to a degree, supports our postulation of the radical mechanism.
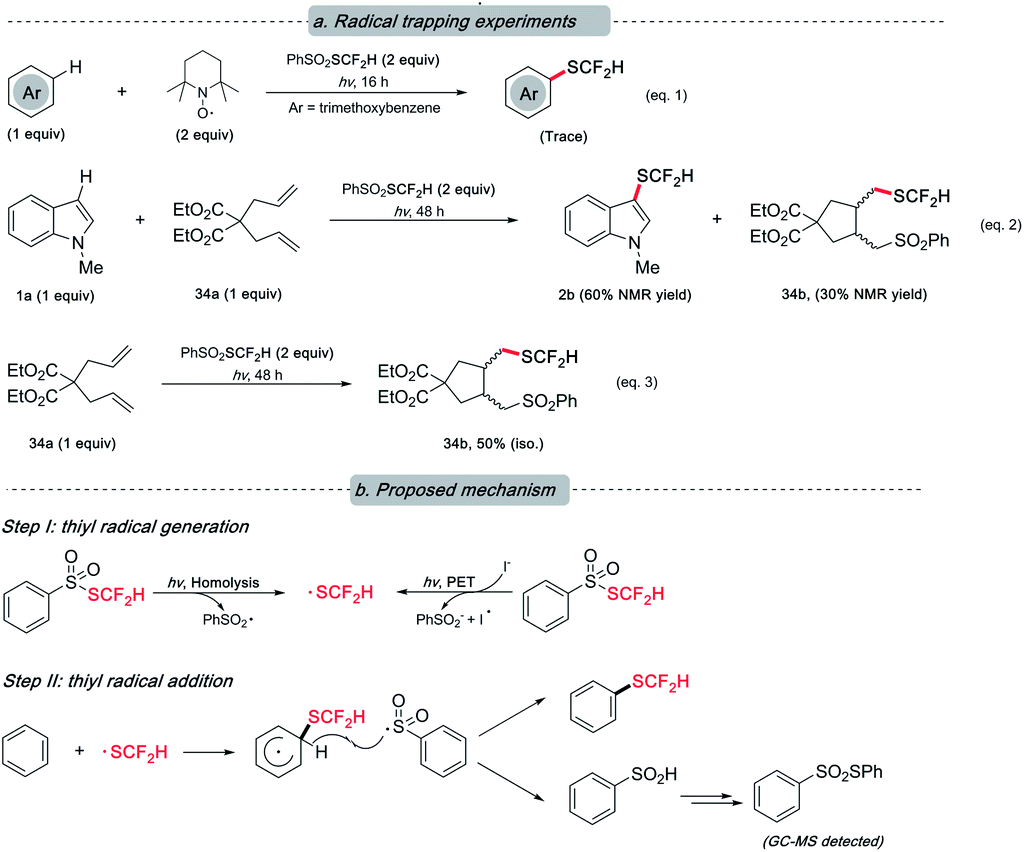 | ||
| Scheme 5 Mechanistic study. (a) Termination of the desired reactivity in the presence of radical trappers; (b) plausible mechanism. | ||
Based on the previous literature20,22a and the abovementioned experiments (see ESI† for details that rationalized the intermediacy of the proposed radical and the by-product formation), we envisioned that a radical-involved mechanism was operative. Under light irradiation, a difluoromethylthiyl radical resulted from either the homolysis of PhSO2SCF2H or the photo-induced electron transfer (PET) event between PhSO2SCF2H and iodide. Subsequently, the action of SCF2H radical on arenes was followed by hydrogen atom abstraction by phenylsulfonium (radical) to furnish the target compound. The resulting phenylsulfinic acid was unstable and further transformed into thiosulfonate.29
Conclusions
In summary, we have developed a metal-catalyst-free aromatic difluoromethylthiolation reaction at room temperature enabled by visible light. This operationally simple strategy features the synthesis of a series of difluoromethylthioethers under mild conditions, which are a class of compounds with high medicinal value.1,2,3b These difluoromethylthioethers could be readily diversified into corresponding sulfones and sulfoxides. Moreover, this “dummy group” strategy holds great potential for achieving other types of radical thiolations by simply switching the functionalities tethered on thiosulfonate reagents. Details of mechanistic insight remain to be explored and we are dedicated to introducing fluorine-containing functional groups on arenes with similar strategies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Canada Research Chair Foundation (for supporting C.-J. L.), the Canada Foundation for Innovation, the FQRNT Center in Green Chemistry and Catalysis, the Natural Sciences and Engineering Research Council of Canada, and McGill University for supporting our research. The authors also thank the Shanghai Institute of Organic Chemistry (SIOC) for supporting D. Zhu with a postdoctoral fellowship and supporting L. L with a National Postdoctoral Program for Innovative Talents fellowship (BX201700110). We are also grateful to Prof. Dr Dmitrii Perepichka (McGill University) for our access to the fluorescence facilities and Dr Robin Stein for her patience and helpful guidance on spectrometry.Notes and references
- (a) F. Leroux, P. Jeschke and M. Schlosser, Chem. Rev., 2005, 105, 827–856 CrossRef PubMed; (b) B. Manteau, S. Pazenok, J.-P. Vors and F. R. Leroux, J. Fluorine Chem., 2010, 131, 140–158 CrossRef; (c) C. Ni, M. Hu and J. Hu, Chem. Rev., 2014, 115, 765–825 CrossRef PubMed; (d) X.-H. Xu, K. Matsuzaki and N. Shibata, Chem. Rev., 2014, 115, 731–764 CrossRef PubMed.
- (a) T. Fujita, J. Iwasa and C. Hansch, J. Am. Chem. Soc., 1964, 86, 5175–5180 CrossRef; (b) I. Rico and C. Wakselhan, Tetrahedron Lett., 1981, 22, 323–326 CrossRef.
- (a) B. Bayarmagnai, C. Matheis, K. Jouvin and L. J. Goossen, Angew. Chem., Int. Ed., 2015, 54, 5753–5756 CrossRef PubMed; (b) Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797–804 CrossRef PubMed.
- S. Okui, N. Kyomura, T. Fukuchi, K. Okano, L. He and A. Miyauchi, US Pat., 7 371 768, 2008.
- K. Morita, K. Ide, Y. Hayase, T. Takahashi and Y. Hayashi, Agric. Biol. Chem., 1987, 51, 1339–1343 Search PubMed.
- L. Yagupolskii, I. Maletina, K. Petko, D. Fedyuk, R. Handrock, S. Shavaran, B. Klebanov and S. Herzig, J. Fluorine Chem., 2001, 109, 87–94 CrossRef.
- Z. Huang, O. Matsubara, S. Jia, E. Tokunaga and N. Shibata, Org. Lett., 2017, 19, 934–937 CrossRef PubMed.
- H.-Y. Xiong, X. Pannecoucke and T. Besset, Chem.–Eur. J., 2016, 22, 16734–16749 CrossRef PubMed.
- Selected examples for difluoromethylthiolation by reacting thiolates with “CF2” species other than CF2 difluorocarbene: (a) W. Zhang, J. Zhu and J. Hu, Tetrahedron Lett., 2008, 49, 5006–5008 CrossRef; (b) G. S. Prakash, Z. Zhang, F. Wang, C. Ni and G. A. Olah, J. Fluorine Chem., 2011, 132, 792–798 CrossRef; (c) Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494–1497 CrossRef PubMed.
- Selected examples for difluoromethylthiolation by reacting thiolates with :CF2 difluorocarbene: (a) B. R. Langlois, J. Fluorine Chem., 1988, 41, 247–261 CrossRef; (b) Q.-Y. Chen and S.-W. Wu, J. Fluorine Chem., 1989, 44, 433–440 CrossRef; (c) P. Deprez and J.-P. Vevert, J. Fluorine Chem., 1996, 80, 159–162 CrossRef; (d) W. Zhang, F. Wang and J. Hu, Org. Lett., 2009, 11, 2109–2112 CrossRef PubMed; (e) Y. Zafrani, G. Sod-Moriah and Y. Segall, Tetrahedron, 2009, 65, 5278–5283 CrossRef; (f) F. Wang, W. Huang and J. Hu, Chin. J. Chem., 2011, 29, 2717–2721 CrossRef; (g) L. Li, F. Wang, C. Ni and J. Hu, Angew. Chem., 2013, 125, 12616–12620 CrossRef; (h) P. S. Fier and J. F. Hartwig, Angew. Chem., 2013, 125, 2146–2149 CrossRef; (i) C. S. Thomoson and W. R. Dolbier Jr, J. Org. Chem., 2013, 78, 8904–8908 CrossRef PubMed; (j) K. Fuchibe, M. Bando, R. Takayama and J. Ichikawa, J. Fluorine Chem., 2015, 171, 133–138 CrossRef; (k) J. Yu, J.-H. Lin and J.-C. Xiao, Angew. Chem., Int. Ed., 2017, 56, 16669–16673 CrossRef PubMed; (l) V. P. Mehta and M. F. Greaney, Org. Lett., 2013, 15, 5036–5039 CrossRef PubMed; (m) T. Ding, L. Jiang and W. Yi, Org. Lett., 2017, 20, 170–173 CrossRef PubMed.
- Selected examples for difluoromethylthiolation by reacting disulfides with nucleophilic “CF2H” sources: (a) J. L. Howard, C. Schotten, S. T. Alston and D. L. Browne, Chem. Commun., 2016, 52, 8448–8451 RSC; (b) J.-B. Han, H.-L. Qin, S.-H. Ye, L. Zhu and C.-P. Zhang, J. Org. Chem., 2016, 81, 2506–2512 CrossRef PubMed; (c) Y.-m. Lin, W.-b. Yi, W.-z. Shen and G.-p. Lu, Org. Lett., 2016, 18, 592–595 CrossRef PubMed.
- K. Jouvin, C. Matheis and L. J. Goossen, Chem.–Eur. J., 2015, 21, 14324–14327 CrossRef PubMed.
- (a) J. Wu, Y. Gu, X. Leng and Q. Shen, Angew. Chem., Int. Ed., 2015, 54, 7648–7652 CrossRef PubMed; (b) J. Wu, Y. Liu, C. Lu and Q. Shen, Chem. Sci., 2016, 7, 3757–3762 RSC.
- (a) D. Zhu, Y. Gu, L. Lu and Q. Shen, J. Am. Chem. Soc., 2015, 137, 10547–10553 CrossRef PubMed; (b) D. Zhu, X. Hong, D. Li, L. Lu and Q. Shen, Org. Process Res. Dev., 2017, 21, 1383–1387 CrossRef.
- S. Arimori, O. Matsubara, M. Takada, N. Shibata and M. Shiro, R. Soc. Open Sci., 2016, 3, 160102–160110 CrossRef PubMed.
- E. Ismalaj, D. Le Bars and T. Billard, Angew. Chem., Int. Ed., 2016, 55, 4790–4793 CrossRef PubMed.
- H.-Y. Xiong, A. Bayle, X. Pannecoucke and T. Besset, Angew. Chem., Int. Ed., 2016, 55, 13490–13494 CrossRef PubMed.
- F. Shen, P. Zhang, L. Lu and Q. Shen, Org. Lett., 2017, 19, 1032–1035 CrossRef PubMed.
- (a) X. Zhao, A. Wei, T. Li, Z. Su, J. Chen and K. Lu, Org. Chem. Front., 2017, 4, 232–235 RSC; (b) Q. Yan, L. Q. Jiang, W. B. Yi, Q. R. Liu and W. Zhang, Adv. Synth. Catal., 2017, 359, 2471–2480 CrossRef.
- D. Zhu, X. Shao, X. Hong, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2016, 55, 15807–15811 CrossRef PubMed.
- B. Xu, D. Wang, Y. Hu and Q. Shen, Org. Chem. Front., 2018, 5, 1462–1465 RSC.
- (a) S.-H. Guo, X.-L. Zhang, G.-F. Pan, X.-Q. Zhu, Y.-R. Gao and Y.-Q. Wang, Angew. Chem., Int. Ed., 2017, 57, 1663–1667 CrossRef PubMed; (b) S. H. Guo, M. Y. Wang, G. F. Pan, X. Q. Zhu, Y. R. Gao and Y.-Q. Wang, Adv. Synth. Catal., 2018, 360, 1861–1869 CrossRef; (c) B. Xu, D. Li, L. Lu, D.-C. Wang, Y. Hu and Q. Shen, Org. Chem. Front., 2018 10.1039/c8qo00327k.
- P. Liu, W. Liu and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 14315–14321 CrossRef PubMed.
- H. G. Viehe, Z. Janousek, R. Merenyi and L. Stella, Acc. Chem. Res., 1985, 18, 148–154 CrossRef.
- (a) W. E. Lyons, Nature, 1948, 162, 1004 CrossRef PubMed; (b) O. Curcuruto, J. Winders, D. Franchi and M. Hamdan, Rapid Commun. Mass Spectrom., 1993, 7, 670–672 CrossRef.
- F. Freeman and C. N. Angeletakis, J. Am. Chem. Soc., 1983, 105, 4039–4049 CrossRef.
- (a) L. Li, W. Liu, H. Zeng, X. Mu, G. Cosa, Z. Mi and C. J. Li, J. Am. Chem. Soc., 2015, 137, 8328–8331 CrossRef PubMed; (b) L. Li, W. Liu, X. Mu, Z. Mi and C. J. Li, Nat. Protoc., 2016, 11, 1948–1954 CrossRef PubMed; (c) W. Liu, X. Yang, Y. Gao and C.-J. Li, J. Am. Chem. Soc., 2017, 139, 8621–8627 CrossRef PubMed.
- (a) C. Rosinger, S. Shirakura, E. Hacker, Y. Sato, S. Heibges and S. Nakamura, Julius-Kühn-Archiv, 2012, 2, 544–548 Search PubMed; (b) T. Yoshimura, T. Ikeuchi, S. Ohno, S. Asakura and Y. Hamada, J. Pestic. Sci., 2013, 38, 171–172 CrossRef.
- C. J. M. Stirling, in The chemistry of sulphinic acids, esters and their derivatives, ed. S. Patai, John Wiley & Sons, Chichester, New York, Brisbane, Toronto, Singapore, 1990, ch. 1, pp. 1–7 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01669k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |