
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA soft phosphorus atom to “harden” an erbium(III) single-ion magnet†
Shi-Ming
Chen
 a,
Jin
Xiong
a,
Yi-Quan
Zhang
b,
Qiong
Yuan
a,
Bing-Wu
Wang
*a and
Song
Gao
*a
a,
Jin
Xiong
a,
Yi-Quan
Zhang
b,
Qiong
Yuan
a,
Bing-Wu
Wang
*a and
Song
Gao
*a
aBeijing National Laboratory of Molecular Science, State Key Laboratory of Rare Earth Materials Chemistry and Applications, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China. E-mail: wangbw@pku.edu.cn; gaosong@pku.edu.cn
bJiangsu Key Laboratory for NSLSCS, School of Physical Science and Technology, Nanjing Normal University, Nanjing 210023, P. R. China
First published on 4th August 2018
Abstract
Beyond the lanthanide organometallic single-ion magnet (SIM) (Cp*)Er(COT)1 ([Cp*]− = pentamethylcyclopentadienide; COT2− = cyclooctatetraenide) that has a good performance, we managed to replace one coordinated carbon atom on the [Cp*]− ring by a soft phosphorus atom and obtained (Dsp)Er(COT) (CCDC No. 1835955; Dsp− = 3,4-dimethyl-2,5-bis(trimethylsilyl)phospholyl) whose sandwich structure is reported here for the first time. This substitution results in a remarkable change of magnetic dynamics. It exhibits slow magnetic relaxation under a zero applied direct current (DC) magnetic field with an energy barrier (Δ/kB) of 358 K and magnetic hysteresis up to 9 K, both of which are higher than those of (Cp*)Er(COT). With the descended local symmetry of (Dsp)Er(COT), the energy barrier and blocking temperature (TB) both improve unexpectedly and are among the highest ones in Er(III)-based single-molecule magnets (SMMs).
Magnetism is one of many basic physical properties of molecules which we chemists could explore and understand by studying the geometrical and electronic structures.2 This area has flourished as a lively crossroad for physics, chemistry and materials science. Single-molecule magnets (SMMs) are one of the most cutting-edge fields in molecular magnetism thriving from the inception of the noted {Mn12} in 1993.3 SMMs belong to a class of molecules which can exhibit slow magnetic relaxation purely of a molecular origin rather than the collective behavior facilitated by magnetic exchange coupling in ensembles through a 3D network.4 If there's only a single spin center, they're also noted as single-ion magnets (SIMs).4c The famed [Pc2Tb]− ([Pc]2− = dianion of phthalocyanine) opened the prosperous stage to lanthanide SIMs (Ln-SIMs) because of their unparalleled single-ion anisotropy due to unquenched orbital angular momentum especially for heavy lanthanide ions.5,6 The success of [Pc2Tb]− offers an intriguing possibility for molecular spintronics.7 In 2011, an organometallic SIM (Cp*)Er(COT)1a was reported by our group. It shows two magnetic relaxation processes under a zero applied DC magnetic field arising from two statically disordered conformers with energy barriers of 197 and 323 K, respectively, which were several times higher than those of cluster-based SMMs at that time. From then on concerted efforts have been made toward Ln-SIMs with a high energy barrier (Δ/kB) for the reversal of magnetic quantum states and magnetic blocking temperature (TB).8
In order to construct a good SIM, hard bases containing O or N coordinating atoms are often introduced to enhance the uniaxial ligand field and hence the zero field splitting which is the main source of Ising-type anisotropy for Ln-SIMs.8,9 But in this work we do exactly the opposite. Based on (Cp*)Er(COT), using a soft phosphorus analogue of the Cp− ligand – a phosphacyclopentadienyl (phospholyl) ligand – can improve the SIM performance unexpectedly.
Phospholyl ligands have already become well-established alternatives to cyclopentadienyl (Cp−) groups10 in organometallic chemistry for polymerization catalysis11 and have captivated synthetic chemists in coordination chemistry.12 Thanks to the isolobal analogy13 between –C(H/Me) and phosphorus, phospholyl ligands can retain aromaticity to some extent relative to Cp− or [Cp*]−.14,15 And they still tend to keep their η5 mode like Cp− in coordinating with Ln ions in most cases.16 However, according to the theory of hard and soft acids and bases,17 the substitution of C by a larger and floppy P makes them softer bases and poor π-donors.18,19 This substitution will undoubtedly lead to the lowered affinity of phospholyl ligands with some high-valent metal ions Ln(III) for instance which act as hard acids. Furthermore, in Dsp− two electron-withdrawing groups –SiMe3 further reduce the nucleophilicity. Meanwhile, they are less inclined to be in η1 coordination mode using the lone-pair electrons of P atoms for the lower affinity of P with Ln ions with only three exceptions.20 So we can safely replace the Cp− ring by a phospholyl group and keep the same coordination mode, and make a rational structural comparison between the two. For Ln(II), the lower oxidation state, larger ionic radius and hence softer acidity make it more easily bind to phospholyl ligands. Phospholyl Ln(II) complexes can be synthesized either by salt metathesis19 or by a redox reaction between metal Ln and 1,1′-biphospholyls.21 However, especially for heavy Ln(III), it's a different scenario. Comparatively very few phospholyl heavy Ln(III) complexes have been successfully synthesized and structurally characterized so far, such as, (Dtp)2TmI (Dtp− = 3,4-dimethyl-2,5-di-tert-butyl phospholyl),22 {(Htp)2TmI}2 (Htp− = 2,5-di-tert-butyl phospholyl),22, (Dtp)2DyI,23 {(Dsp)2DyI}2 (ref. 23) and {(Dtp)2Tm(μ-S)}2.24 Moreover, the single crystal structure of sandwiched (phospolyl)Ln(COT) has not been reported yet, and only two close examples of early Ln(III) compounds (Dsp)Nd(COT)(THF)25 and (Tmp)Nd(COT)[OP(NMe2)3]26 (Tmp− = tetramethyl phospholyl) were found but not in a double-decker structure in the presence of solvent THF and other ancillary ligands OP(NMe2)3. In this work, we obtained a phospholyl heavy Ln(III) complex (Dsp)Er(COT) with a sandwich structure.
With the change of coordination capability described above when phospholyl ligands replace [Cp*]−, ligand field and consequently the electronic structure of the whole complexes will be different accordingly, which may bring about significant variation of optical and magnetic properties. Alternating current (AC) magnetic susceptibility measurement of (Dsp)Er(COT) demonstrated a significant change of magnetic dynamics compared with its parent analogue (Cp*)Er(COT) (vide infra).
Following the work by François Nief and Louis Ricard25,27 with some modifications, we first get phospholyl potassium K(Dsp), which then reacts with ErI(COT) (THF)x (x = 2–3, see the ESI†) in toluene under reflux to produce (Dsp)Er(COT) with insoluble KI (Scheme 1). Trying to precipitate KCl by reacting with {(THF)Er(COT)(μ-Cl)}2 is unsuccessful, although K(Dsp) can eliminate Cl− in {(THF)2Nd(COT)(μ-Cl)}2 (ref. 25) which is probably due to different acidity between light and heavy Ln(III) ions (vide supra). Orange single crystals suitable for X-ray diffraction analysis can be obtained by cooling the concentrated toluene solution of (Dsp)Er(COT) at −30 °C for several days (anal. calc.: C, 45.59%; H, 6.12%; found: C, 45.73%; H, 6.12%.).
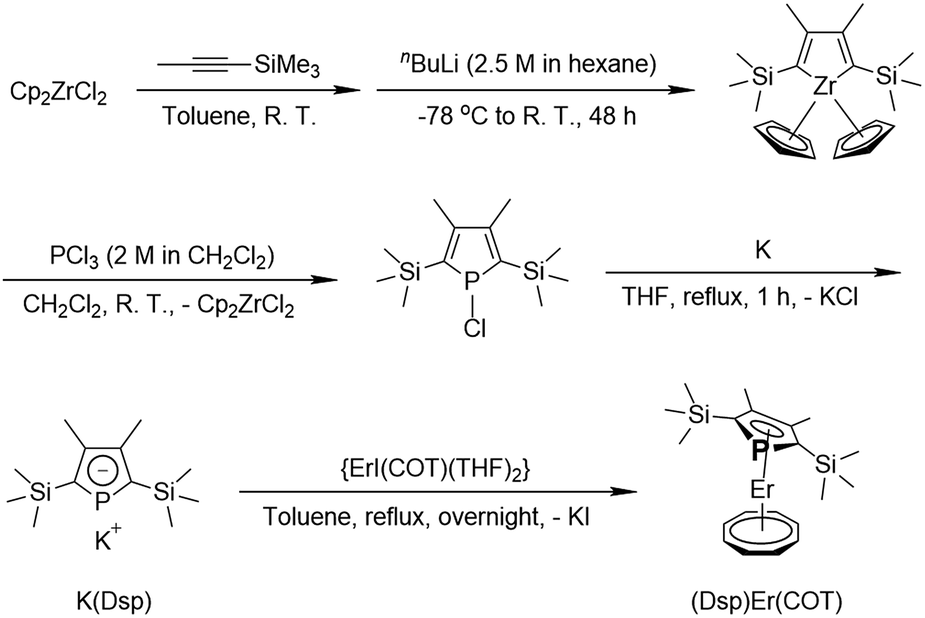 | ||
| Scheme 1 Synthesis of (Dsp)Er(COT). | ||
As depicted in Fig. 1, the single crystal structure of (Dsp)Er(COT) at 180 K (structure refined according to ref. 28) reveals itself as a double-decker structure, reminiscent of its Cp* parent.29 It crystallizes in the P21/c space group. The nearest Er⋯Er distance is 7.832 Å. It's noteworthy that the COT2− ring is in static disorder as well with the ratios 0.56 and 0.44 (Fig. S4 in the ESI†). The tilting angle between Dsp− and COT2− planes is 10.5°. The distance from Er(III) to the five-membered ring is 2.321 Å, larger than that in (Cp*)Er(COT) (2.268 Å), which crystallographically shows a weaker coordination between the two, yet Er(III) is much closer to COT2− (1.686 Å in (Dsp)Er(COT), 1.727 Å in the Cp* version and 1.875 Å in [Er(COT)2]−).30a The Er−C(Dsp−) bond lengths range from 2.635 to 2.645 Å, while Er–C(COT2−) bond lengths are from 2.471 to 2.491 Å. The Er–P bond length is 2.793 Å, being close to the reported Er–P bond length.31 To the best of our knowledge, this is the first sandwich structure reported with a phospholyl ligand and COT2− coordinating simultaneously to a heavy Ln(III).
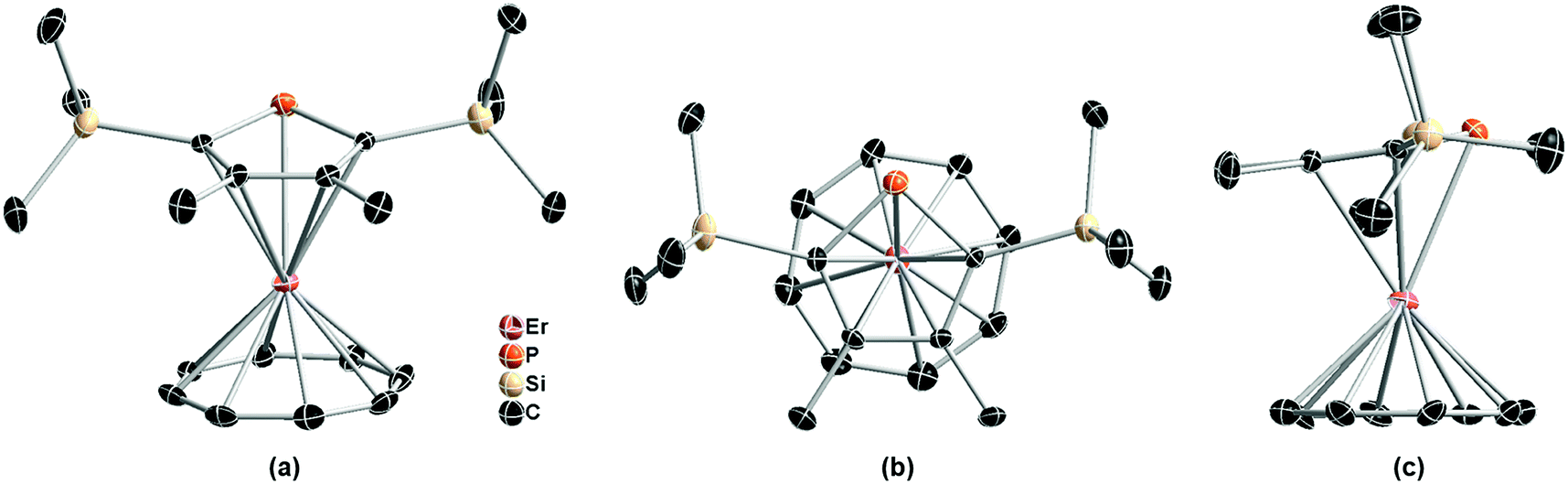 | ||
| Fig. 1 Front view (a), top view (b) and side view (c) of the single crystal structure of (Dsp)Er(COT) with pink, orange, tan, and black ellipsoids (30% possibility) representing Er, P, Si, and C, respectively (another disorder fragment of COT2− is shown in Fig. S4†). Hydrogen atoms and another disordered part of COT2− (Fig. S4†) have been omitted for clarity. Selected bond lengths: Er–P = 2.793 Å; Er–C(Dsp−) = 2.635–2.645 Å; Er–C(COT2−) = 2.471–2.510 Å. | ||
We wonder if a slight change of only one coordinated atom could trigger a pronounced difference of magnetic properties. We resort to AC susceptometry which is often applied to probe magnetic dynamics.4 The out-of-phase component  of molar AC susceptibility for (Dsp)Er(COT) exhibits noticeable frequency-dependence over 1 to 1000 Hz from 2 to 28 K under a zero applied DC field (Fig. 2). As with its prototype, this slow magnetic relaxation of (Dsp)Er(COT) unveils itself as a SIM. There is a single peak in the plot of
of molar AC susceptibility for (Dsp)Er(COT) exhibits noticeable frequency-dependence over 1 to 1000 Hz from 2 to 28 K under a zero applied DC field (Fig. 2). As with its prototype, this slow magnetic relaxation of (Dsp)Er(COT) unveils itself as a SIM. There is a single peak in the plot of  versus T at a given frequency while there are two for (Cp*)Er(COT). The narrow distribution of relaxation time τ (α = 0.01–0.2) from the fitting of the Argand plot32 (Fig. S10†) indicates almost a single relaxation despite the existence of two static disorder conformers which instead gives rise to two relaxation processes corresponding to two peaks of
versus T at a given frequency while there are two for (Cp*)Er(COT). The narrow distribution of relaxation time τ (α = 0.01–0.2) from the fitting of the Argand plot32 (Fig. S10†) indicates almost a single relaxation despite the existence of two static disorder conformers which instead gives rise to two relaxation processes corresponding to two peaks of 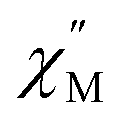 in the measured temperature range for (Cp*)Er(COT)1.
in the measured temperature range for (Cp*)Er(COT)1.
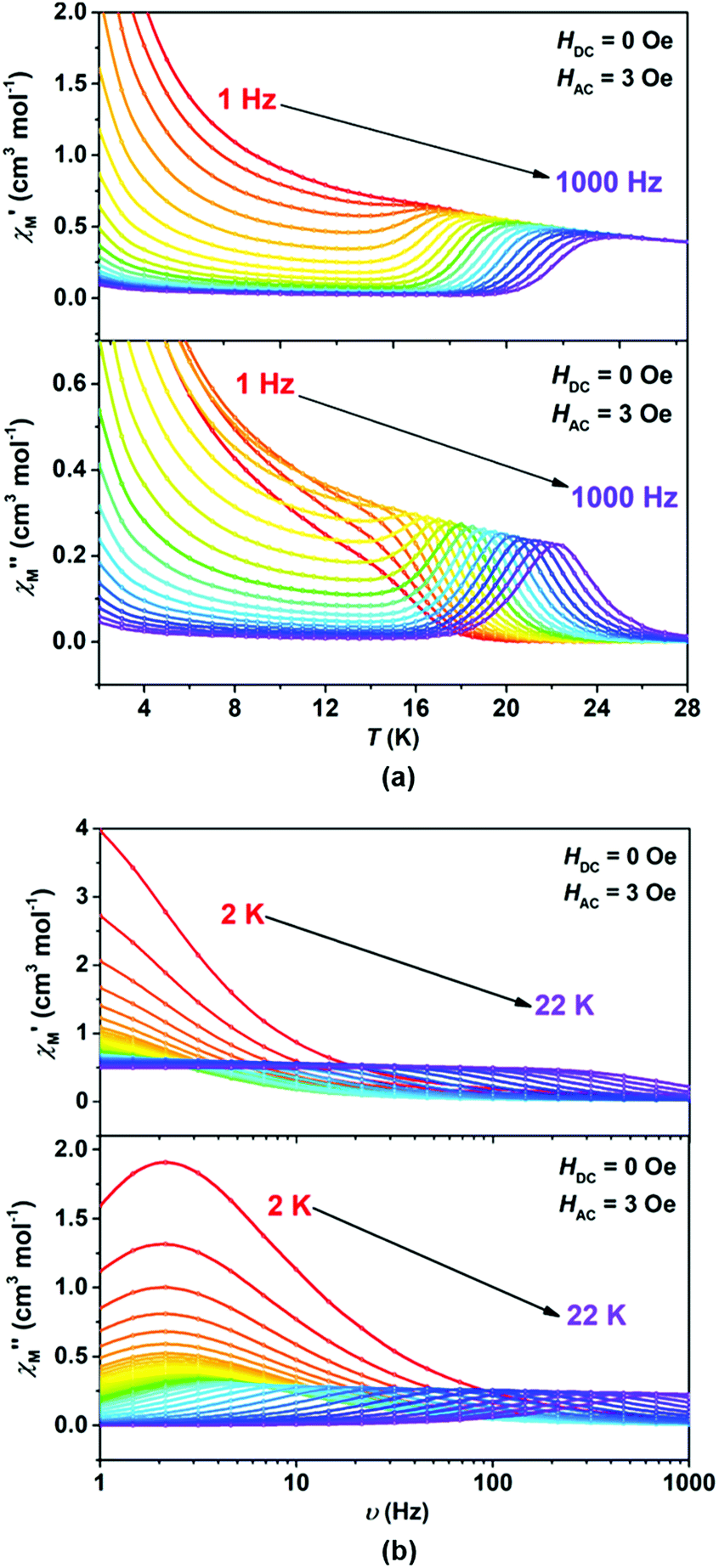 | ||
Fig. 2 Temperature (a) and frequency-dependence (b) of molar AC susceptibility 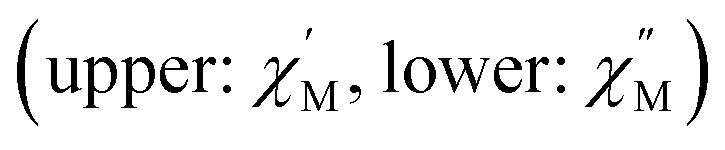 for (Dsp)Er(COT) under a zero applied DC field over the temperature range of 2–28 K and the frequency range of 1–1000 Hz. for (Dsp)Er(COT) under a zero applied DC field over the temperature range of 2–28 K and the frequency range of 1–1000 Hz. | ||
A combination of an Orbach process and quantum tunnelling of magnetization (QTM) (τ−1 = τ0−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[−Δ/(kBT)] + τQTM−1, τ is the relaxation time) can fit the data of lnτ versus T−1 well (Fig. 3). It demonstrates that the thermally activated Orbach process is dominant above 12 K and QTM considerably masks other possible relaxations below 10 K. The fitting returns a relaxation barrier Δ/kB = 358(3) K which is much higher than that of (Cp*)Er(COT) (197, 323 K)1a and [Er(COT)2]− (206 K), and among the highest ones in Er(III)-based SIMs.30,33 Magnetic dynamics measurements were also performed under a 1000 Oe DC field to effectively suppress the QTM (Fig. S12†). A similar workup to that described above gives Δ/kB = 367(1) K (Fig. S13†).
exp[−Δ/(kBT)] + τQTM−1, τ is the relaxation time) can fit the data of lnτ versus T−1 well (Fig. 3). It demonstrates that the thermally activated Orbach process is dominant above 12 K and QTM considerably masks other possible relaxations below 10 K. The fitting returns a relaxation barrier Δ/kB = 358(3) K which is much higher than that of (Cp*)Er(COT) (197, 323 K)1a and [Er(COT)2]− (206 K), and among the highest ones in Er(III)-based SIMs.30,33 Magnetic dynamics measurements were also performed under a 1000 Oe DC field to effectively suppress the QTM (Fig. S12†). A similar workup to that described above gives Δ/kB = 367(1) K (Fig. S13†).
 | ||
| Fig. 3 Plot of natural log of relaxation time versus inverse temperature under a zero applied DC field. The purple and red circles represent the data from MPMS and PPMS, respectively. The black solid line represents the best fitting using a combination of Orbach and QTM processes. The fitting gives Δ/kB = 358(3) K and τ0 = 1.6(3) × 10−11 s. | ||
The subsequent measurement of zero field cooled and field cooled magnetization (ZFC-FC) presents a divergence at about 8.5 K between the two curves (Fig. S7†), which reminds us of a probable magnetic hysteresis. As expected, typical butterfly-shaped magnetic hysteresis loops in mesoscopic SMMs3,4 can be observed below 9 K with a field-sweep rate of 200 Oe s−1 (Fig. 4 and S8†). At 2 K, there is still a magnetic remnant of 0.3 Nβ at a zero field and the value of the coercive field Hc is 55 Oe. The magnetic blocking temperature (TB) of about 9 K (Fig. S8†) is much higher than that of (Cp*)Er(COT) (5 K, 550 Oe min−1) and almost rivals that of [Er(COT)2]− (10 K, 0.78 mT s−1).30a This is a robust confirmation that (Dsp)Er(COT) is a magnet which can reserve its magnetization even under a zero field like a block magnet. The above results explicitly unveil (Dsp)Er(COT) as a SIM with a good performance.
 | ||
| Fig. 4 Variable-field magnetization for (Dsp)Er(COT) with a field-sweeping rate of 200 Oe s−1. (Inset) Expanded view of the variable-field magnetization near the zero field at 9 K and 10 K. | ||
Why does the seemingly weaker ligand field of Dsp− produce stronger anisotropy than [Cp*]−? For one thing, we think that the phospholyl ligand and its parents Cp− and [Cp*]− are more like axial ligands favorable for the oblate electron distribution of Dy(III) to stabilize its Ising-type MJ ground state34,35 as exemplified by plenty of excellent Cp-based Dy(III) SMMs.36 But they're unsuitable for prolate Er(III) under Ising-limit conditions. This sort of difference between Dy(III) and Er(III) originates from the opposite signs of axial crystal field parameters (B02 and B04)30b for the two lanthanide ions. The moving away of the Dsp− ligand from the axial electron cloud of Er(III) could reduce the electron repulsion. On the other hand, a large equatorial ligand such as the COT2− ring with high rotational symmetry is suitable for prolate Er(III) to stabilize its MJ ground state as corroborated by (Cp*)Er(COT) and [Er(COT)2]−. The closer the Er(III) is to the COT2− ring, the stronger the ligand field the −2 charged COT2− exerts on Er(III), and hence the larger the anisotropic splitting of the ground J state. Although there are two COT2− rings in [Er(COT)2]−,30a the distance between Er(III) and COT2− (dEr−COT) becomes larger (1.875 Å) due probably to the repulsion between the two COT2− rings which results in a weakened ligand field instead and its anisotropic energy barrier is lower than that of (Cp*)Er(COT). In this regard, the closer the Er(III) is to the COT2− ring within a certain range, the lower and less mixed the ground state doublets are, making them closer to the Ising limit at least for the lowest Kramers doublets. Compared with (Cp*)Er(COT), the weak adhesion of Er(III) in (Dsp)Er(COT) with the phospholyl ligand leads to a shorter distance between Er(III) and COT2− (vide supra) in order to maintain a balanced and conservative electrostatic potential of Er(III). So SIM performance improves as elaborated above in spite of lower symmetry (Table 1). We believe that dEr−COT plays an essential role in causing different magnetic dynamics between (Dsp)Er(COT) and (Cp*)Er(COT).
| (Cp*)Er(COT) | (Dsp)Er(COT) | |
|---|---|---|
| Er-centroid (COT2−)/Å | 1.7267(3) | 1.6855(3) |
| Er-centroid (five-membered ring)/Å | 2.2679(3) | 2.3220(3) |
| Tilting angle between the two rings/° | 7.3 | 10.5 |
| Δ/K | 197, 323 | 358 |
As a statistical induction, we have found the COT-based Er(III) SIMs reported to-date and plotted each scattered point representing the data of Δ/kBversus dEr−COT (Fig. 5a). In a panoramic view, when the COT2− ring moves gradually away from Er(III), the barrier goes through a precipitous fall below 1.7 Å, and then changes less with increasing dEr−COT. In order to theoretically understand the trend, we calculated the energy barrier of the model molecular fragment [Er(COT)]+ with a different dEr−COT using the ab initio method37 (Table S5; Fig. 5b and S24†). Previous computation30b involved two COT2− rings based on the single crystal structure of [Er(COT)2]−. However, we think it's not a single factor story in which high negative charge repulsion between COT2− rings may result in an unstable ground state within a certain distance (vide supra). So the result may not be very convincing if we only want to examine the relationship between Δ/kB and dEr−COT. In our model with only one COT2− ring, as expected, when dEr−COT becomes larger than 1.6 Å, the energy barrier goes down approximately in a linear way. The downtrend of the energy barrier with increasing dEr−COT in Fig. 5 shows that a short dEr−COT is conducive to a high energy barrier, and the chemically adjustable soft P atom in the five-membered ring is the pushing hand.
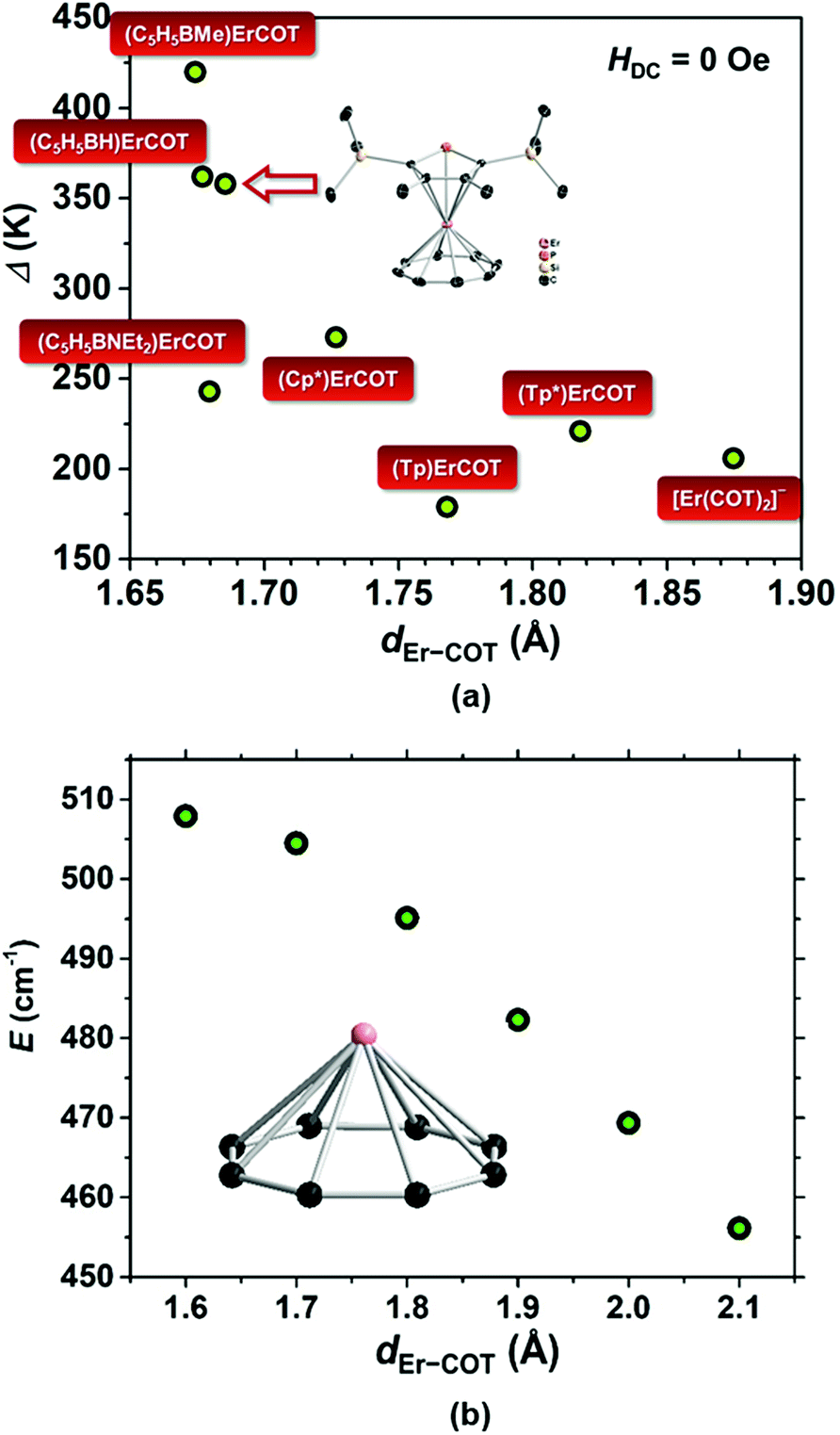 | ||
| Fig. 5 (a) Scattered points representing the data of Δ/kBvs. dEr−COT from reported COT-based Er(III) SIMs.1,30a,33,38 The data of (Cp*)Er(COT) are from the calculation based on the ligand field for easy comparison. ([Tp]− = tris(1-pyrazolyl)borate and [Tp*]− = tris(3,5-dimethyl-1-pyrazolyl)borate); (b) plot of energy barrier vs. series of dEr−COT for the hypothetical fragment [Er(COT)]+ calculated based on ab initio; inset: [Er(COT)]+. | ||
From the above elaboration, we can see the main line clearly whereby the introduction of a soft P atom leads to a larger distance from Er(III) to the Cp analogue which reflects a weaker affinity between the two, and hence a shorter dEr−COT which enhances the ligand field and anisotropic splitting. So a higher energy barrier is possibly reached.
In this series, we have also examined (Dsp)Ln(COT) (Ln = Tb (CCDC No. 1835954), Dy (CCDC No. 1835957), and Tm (CCDC No. 1835958)) among which (Dsp)Dy(COT) exhibits slow magnetic relaxation under a 2000 Oe applied DC field with Δ/kB = 57 K (Fig. S15†). (Dsp)Tm(COT) is also a field-induced SIM under a 2500 Oe applied DC field with an energy barrier of 109 K (Fig. S18†), which is almost the highest in Tm(III)-based SIMs38 so far.
Conclusions
We have successfully synthesized (Dsp)Er(COT) whose sandwiched single crystal structure is reported here for the first time. Compared with its parent compound (Cp*)Er(COT), the replacement of only one coordinated carbon atom on the cyclopentadienyl ligand by a softer phosphorus atom results in a considerable change of magnetic dynamics which returns a relaxation energy barrier of 358 K under a zero applied DC field and blocking temperature of 9 K. Both manifest it as a better SIM than (Cp*)Er(COT). The essential reason mainly resides in the short distance between Er(III) and COT2− caused by the weak affinity of Er(III) with Dsp−. Through the introduction of a softer coordinating atom, we have obtained a better Er(III) SIM. Inspired by this work, we will continue to focus on introducing more soft P atoms and expect better Er(III)-based SIMs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21621061, 21290171, 21571008 and 91422302), National Key R&D Program of China (2017YFA0206301, 2017YFA0204903) and High-performance Computing Platform at Peking University. We sincerely thank Dr Ling Xu for helpful discussion on synthesis, Dr Yin-Shan Meng for discussion on magnetic measurements, and Mrs Zhi-Xian Wang, Mrs Xiu Zhang and Mr Ming-Xing Chen at the Analytical Instrumental Center of Peking University.Notes and references
- (a) S.-D. Jiang, B.-W. Wang, H.-L. Sun, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2011, 133, 4730 CrossRef PubMed; (b) S.-D. Jiang, S.-S. Liu, L.-N. Zhou, B.-W. Wang, Z.-M. Wang and S. Gao, Inorg. Chem., 2012, 51, 3079 CrossRef PubMed.
- (a) R. L. Carlin, Magnetochemistry, Springer-Verlag, Berlin, 1986 CrossRef; (b) O. Kahn, Molecular Magnetism, VCH, Weinheim, 1993 Search PubMed; (c) C. Benelli and D. Gatteschi, Introduction to Molecular Magnetism: From Transition Metals to Lanthanides, Wiley-VCH, Weinheim, 2015 Search PubMed.
- (a) R. Sessoli, H.-L. Tsai, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804 CrossRef; (b) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef.
- (a) D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, 2006 CrossRef; (b) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110 CrossRef PubMed; (c) J. M. Frost, K. L. M. Harriman and M. Murugesu, Chem. Sci., 2016, 7, 2470 RSC.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694 CrossRef PubMed.
- S.-D. Jiang, B.-W. Wang and S. Gao, Struct. Bonding, 2015, 164, 111 CrossRef.
- M. Urdampilleta, S. Klyatskaya, J.-P. Cleuziou, M. Ruben and W. Wernsdorfer, Nat. Mater., 2011, 10, 502 CrossRef PubMed.
- (a) Y.-S. Ding, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, Angew. Chem., Int. Ed., 2016, 55, 16071 CrossRef PubMed; (b) Y.-C. Chen, J.-L. Liu, L. Ungur, J. Liu, Q.-W. Li, L.-F. Wang, Z.-P. Ni, L. F. Chibotaru, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 2829 CrossRef PubMed; (c) J. Liu, Y.-C. Chen, J.-L. Liu, V. Vieru, L. Ungur, J.-H. Jia, L. F. Chibotaru, Y.-H. Lan, W. Wernsdorfer, S. Gao, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 5441 CrossRef PubMed.
- (a) N. Ishikawa, M. Sugita, T. Okubo, N. Tanaka, T. Iino and Y. Kaizu, Inorg. Chem., 2003, 42, 2440 CrossRef PubMed; (b) M. A. AlDamen, J. M. Clemente-Juan, E. Coronado, C. Martí-Gastaldo and A. Gaita-Ariño, J. Am. Chem. Soc., 2008, 130, 8847 CrossRef PubMed; (c) S.-D. Jiang, B.-W. Wang, G. Su, Z.-M. Wang and S. Gao, Angew. Chem., Int. Ed., 2010, 49, 7448 CrossRef PubMed; (d) Lanthanides and Actinides in Molecular Magnetism, ed. R. A. Layfield and M. Murugesu, Wiley-VCH, Weinheim, 2015 Search PubMed.
- (a) F. Nief, Eur. J. Inorg. Chem., 2001, 891 CrossRef; (b) F. Nief, P. Riant, L. Ricard, P. Desmurs and D. Baudry-Barbier, Eur. J. Inorg. Chem., 1999, 1041 CrossRef; (c) P. Gradoz, D. Baudry, M. Ephritikhine, M. Lance, M. Nierlich and J. Vigner, J. Organomet. Chem., 1994, 466, 107 CrossRef; (d) P. Gradoz, C. Boisson, D. Baudry, M. Lance, M. Nierlich, J. Vigner and M. Ephritikhine, J. Chem. Soc., Chem. Commun., 1992, 1720 RSC.
- (a) E. J. M. de Boer, I. J. Gilmore, F. M. Korndorfer, A. D. Horton, A. van der Linden, B. W. Royan, B. J. Ruisch, L. Schoon and R. W. Shaw, J. Mol. Catal. A: Chem., 1998, 128, 155 CrossRef; (b) S. J. Brown, X. Gao, D. G. Harrison, L. Koch, R. E. v. H. Spence and G. P. A. Yap, Organometallics, 1998, 17, 5445 CrossRef; (c) D. Barbier-Baudry, S. Heiner, M. M. Kubicki, E. Vigier and M. Visseaux, Organometallics, 2001, 20, 4207 CrossRef.
- (a) P. Desmurs, M. Visseaux, D. Baudry, A. Dormond, F. Nief and L. Ricard, Organometallics, 1996, 15, 4178 CrossRef; (b) F. Nief and L. Ricard, Organometallics, 2001, 20, 3884 CrossRef.
- R. Hoffmann, Angew. Chem., Int. Ed., 1982, 21, 711 CrossRef.
- F. Mathey, J. F. Nixon and K. Dillon, Phosphorus: the Carbon Copy, Wiley, New York, 1998 Search PubMed.
- Phosphorous – Carbon Heterocyclic Chemistry. The Rise of a New Domain, ed. F. Mathey, Pergamon, Oxford, 2001 Search PubMed.
- (a) E. Le Roux, F. Nief, F. Jaroschik, K. W. Törnroos and R. Anwander, Dalton Trans., 2007, 4866 RSC; (b) S. M. Cendrowski-Guillaume, G. Le Gland, M. Nierlich and M. Ephritikhine, Z. Kristallogr. NCS., 2002, 217, 35 Search PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef.
- F. Jaroschik, T. Shima, X. Li, K. Mori, L. Ricard, X.-F. Le Goff, F. Nief and Z.-M. Hou, Organometallics, 2007, 26, 5654 CrossRef.
- (a) F. Nief, D. Turcitu and L. Ricard, Chem. Commun., 2002, 1646 RSC; (b) D. Turcitu, F. Nief and L. Ricard, Chem.–Eur. J., 2003, 9, 4916 CrossRef PubMed; (c) F. Nief, B. T. de Borms, L. Ricard and D. Carmichael, Eur. J. Inorg. Chem., 2005, 637 CrossRef.
- (a) F. Nief and L. Ricard, Organometallics, 2001, 20, 3884 CrossRef; (b) F. Nief and L. Ricard, J. Organomet. Chem., 1994, 417, 149 CrossRef.
- (a) F. Nief, L. Ricard and F. Mathey, Polyhedron, 1993, 12, 19 CrossRef; (b) F. Nief and L. Ricard, J. Chem. Soc., Chem. Commun., 1994, 2723 RSC.
- F. Jaroschik, F. Nief, X.-F. Le Goff and L. Ricard, Organometallics, 2007, 26, 3552 CrossRef.
- F. Jaroschik, F. Nief and X.-F. Le Goff, Polyhedron, 2009, 28, 2744 CrossRef.
- D. Turcitu, F. Nief and L. Ricard, Chem.–Eur. J., 2003, 9, 4916 CrossRef PubMed.
- M. Visseaux, F. Nief and L. Ricard, J. Organomet. Chem., 2002, 647, 139 CrossRef.
- S. M. Cendrowski-Guillaume, G. Le Gland, M. Nierlich and M. Ephritikhine, Eur. J. Inorg. Chem., 2003, 1388 CrossRef.
- M. Westerhausen, M. H. Digeser, C. Gückel, H. Nöth, J. Knizek and W. Ponikwar, Organometallics, 1999, 18, 2491 CrossRef.
- (a) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef; (b) L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef; (c) G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532 CrossRef.
- W. J. Evans, M. A. Johnston, R. D. Clarka and J. W. Ziller, J. Chem. Soc., Dalton Trans., 2000, 1609 RSC.
- (a) K. R. Meihaus and J. R. Long, J. Am. Chem. Soc., 2013, 135, 17952 CrossRef PubMed; (b) L. Ungur, J. J. Le Roy, I. Korobkov, M. Murugesu and L. F. Chibotaru, Angew. Chem., Int. Ed., 2014, 53, 4413 CrossRef PubMed.
- (a) P. W. Roesky, M. T. Gamer, M. Puchner and A. Greiner, Chem.–Eur. J., 2002, 8, 5265 CrossRef PubMed; (b) P. W. Roesky, M. T. Gamer and N. Marinos, Chem.–Eur. J., 2004, 10, 3537 CrossRef PubMed; (c) M. T. Gamer and P. W. Roesky, Inorg. Chem., 2005, 44, 5963 CrossRef PubMed; (d) T. K. Panda, M. T. Gamer and P. W. Roesky, Inorg. Chem., 2006, 45, 910 CrossRef PubMed.
- (a) K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341 CrossRef; (b) C. Dekker, A. F. M. Arts, H. W. de Wijn, A. J. van Duyneveldt and J. A. Mydosh, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 11243 CrossRef.
- Y.-S. Meng, C.-H. Wang, Y.-Q. Zhang, X.-B. Leng, B.-W. Wang, Y.-F. Chen and S. Gao, Inorg. Chem. Front., 2016, 3, 828 RSC.
- J. Sievers, Z. Phys. B: Condens. Matter, 1982, 45, 289 CrossRef.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078 RSC.
- (a) F. Tuna, C. A. Smith, M. Bodensteiner, L. Ungur, L. F. Chibotaru, E. J. L. McInnes, R. E. P. Winpenny, D. Collison and R. A. Layfield, Angew. Chem., Int. Ed., 2012, 51, 6976 CrossRef PubMed; (b) S. Demir, J. M. Zadrozny, M. Nippe and J. R. Long, J. Am. Chem. Soc., 2012, 134, 18546 CrossRef PubMed; (c) T. Pugh, F. Tuna, L. Ungur, D. Collison, E. J. L. McInnes, L. F. Chibotaru and R. A. Layfield, Nat. Commun., 2015, 6, 7492 CrossRef PubMed; (d) T. Pugh, A. Kerridge and R. A. Layfield, Angew. Chem., Int. Ed., 2015, 54, 4255 CrossRef PubMed; (e) T. Pugh, V. Vieru, L. F. Chibotaru and R. A. Layfield, Chem. Sci., 2016, 7, 2128 RSC; (f) T. Pugh, N. F. Chilton and R. A. Layfield, Angew. Chem., Int. Ed., 2016, 128, 11248 CrossRef; (g) F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445 CrossRef PubMed; (h) C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439 CrossRef PubMed.
- (a) G. Karlström, R. Lindh, P. Å. Malmqvist, B. O. Roos, U. Ryde, V. Veryazov, P. O. Widmark, M. Cossi, B. Schimmelpfennig, P. Neogrady and L. Seijo, Comput. Mater. Sci., 2003, 28, 222 CrossRef; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 Search PubMed; (c) L. F. Chibotaru and L. Ungur, J. Chem. Phys., 2012, 137, 064112 CrossRef PubMed; (d) L. F. Chibotaru, L. Ungur, C. Aronica, H. Elmoll, G. Pilet and D. Luneau, J. Am. Chem. Soc., 2008, 130, 12445 CrossRef PubMed; (e) F. Aquilante, et al. , J. Comput. Chem., 2016, 37, 506 CrossRef PubMed.
- Y.-S. Meng, Y.-S. Qiao, Y.-Q. Zhang, S.-D. Jiang, Z.-S. Meng, B.-W. Wang, Z.-M. Wang and S. Gao, Chem.–Eur. J., 2016, 22, 4704 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1835954–1835958. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01626g |
| This journal is © The Royal Society of Chemistry 2018 |