
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new structure–activity relationship for cyanine dyes to improve photostability and fluorescence properties for live cell imaging†
Christian
Schwechheimer
a,
Franziska
Rönicke
a,
Ute
Schepers
b and
Hans-Achim
Wagenknecht
 *a
*a
aInstitute of Organic Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: Wagenknecht@kit.edu
bInstitute of Toxicology and Genetics, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 3rd July 2018
Abstract
A new set of cyanine-indole dyes was synthesized, characterized by optical and cytotoxic properties and subsequently applied for live cell imaging. Furthermore, these dyes were postsynthetically linked covalently to the 2′-position of uridine anchors in presynthesized oligonucleotides using the copper(I)-catalyzed azide–alkyne cycloaddition in order to evaluate their photostability and imaging properties in living cells. The nucleophilicity at position C-2 of the indole part of the dyes was elucidated as key for a new structure–activity relationship that served as a rational guide to improve the photostability and optical properties of these green-emitting dyes for live cell imaging of nucleic acids. While the photostability rises exponentially with decreasing nucleophilicity, thermal bleaching experiments confirmed an opposite trend supposing that the superoxide radical anion is mainly responsible for the photobleaching of the dyes. Furthermore, the cytotoxicities of the dyes were tested in HeLa cells and moderate to low LD50 values were obtained. This interdisciplinary strategy allowed us to identify one dye with excellent optical properties and even better photostability and decreased cytotoxicity compared to a cyanine-indole dye that bears an additional cyclooctatetraene group as a triplet state quencher.
Introduction
Molecular imaging allows the visualization of biomolecules as they function in their natural cellular environment1 and at a single molecule level2,3 and therefore requires bright and photostable dyes. Covalent labelling is crucial to allow imaging of intracellular transport of DNA and RNA and their processing inside cells. Especially for the fluorescent labeling of DNA and RNA, nucleic-acid sensitive dyes are needed4,5 that should meet the following requirements: (i) high brightness (B), defined as extinction (ε) × quantum yield (ΦF), (ii) no quenching as covalent labels of DNA and RNA, (iii) long-lasting photostability, and (iv) low cytotoxicity. Derivatives of cyanine dyes play an important role in fluorescent DNA staining and imaging since they typically show a drastically enhanced fluorescence by binding to DNA or RNA and are synthetically well accessible.6–8 However, the broadly applied Cy3 and Cy5 dyes have very small Stokes shifts that cause problems in separating the excitation from the emission channel and are the main reason for a self-quenched emission in multiply labelled probes due to homotransfer.9,10 In contrast, the cyanine-indole-quinolinium dye (CyIQ, 1), a member of the cyanine-styryl dyes that originated from a combinatorial library and was initially published as an RNA binder,11 shows promising brightness and improved photostability compared to other dyes which are commonly used for nucleic acid labeling, including fluorescein, BODIPY and thiazole orange (TO).12 Only Cy3 is more photostable but has the disadvantage described above. Using CyIQ as the lead structure, we developed by empirical means a set of differently substituted cyanine-styryl dyes with significantly improved optical properties and photostabilities, which cover the emission colors from blue to green, yellow and red.13 These dyes can be attached by the postsynthetic copper-catalyzed cycloaddition to the 2′-position of ribo- or arabino-configured anchors in oligonucleotides and combined with energy transfer pairs as wavelength-shifting nucleic acid probes (“DNA and RNA traffic lights”).14The answer to the question how photostability may be improved not just by combinatorial15 or empirical efforts13 but by rational and clear structure–reactivity relationships is challenging and rarely found in the literature.16 The major identified photobleaching pathways of fluorophores are initiated by their triplet states that sensitize oxygen and thereby form singlet oxygen. In this regard, basically two different concepts exist to develop photostable dyes.16 Firstly, singlet oxygen can be considered as an electrophile, and thus electron-withdrawing substituents reduce the dye reactivity towards singlet oxygen and thereby improve the photostability.16 When the electron density is reduced by cyano groups at the methine bridge or by fluoro substituents at the aromatic parts of cyanine dyes the photostability of thiazole orange-derived dyes was improved.17 However, the contribution of singlet oxygen to fluorophore bleaching is rather low especially at low fluorophore concentrations that include, for instance, most of the single-molecule experiments.18 Secondly, a more general way to enhance photostability is to attach triplet state quenchers, like cyclooctatetraene (COT) or trolox, in order to depopulate the long-lived triplet state of the dyes19 and to reduce the degrading photochemical pathways that evolve from this state.20 However, these linked triplet state quenchers make the dyes rather large and increase their lipophilicity that may interfere with biological activity. Herein, we present a new approach that focuses on the nucleophilicity of indoles as a structural component in a range of cyanine dyes and thereby propose a new and alternative concept to improve the photostability and optical properties of these dyes for live cell imaging of nucleic acids. Accordingly, a new set of cyanine dyes with indoles was synthesized (Scheme 1), characterized by their optical and cytotoxic properties and subsequently applied for live cell imaging. Furthermore, these dyes were postsynthetically linked covalently to the 2′-position of uridine anchors in presynthesized oligonucleotides using the copper(I)-catalyzed azide–alkyne cycloaddition in order to evaluate their photostability and live cell imaging properties.
 | ||
| Scheme 1 Structure of cyanine-indole dyes 1–12 and postsynthetic attachment to the 2′-position of DNA1–DNA12. | ||
Results and discussion
Synthesis and optical properties of new dye–DNA conjugates
The synthesized dyes 2–12 follow the CyIQ lead 1 and are synthetically accessible by Knoevenagel condensation of the corresponding 2-methylquinolinium salts (prepared by alkylation of quinoline with 3-iodopropanol) and differently substituted indole-3-carboxaldehydes. Cyanine-indole-quinolinium dyes with empirical structural variations at the quinolinium part were published before.21,22 However, the dyes 1–10 differ at the indole part by (i) the methylated vs. non-methylated indole-nitrogen (R1), (ii) the electron-donating (OMe) and electron-withdrawing (CN, Br) substituents R2 at position 5 of the indole, and (iii) by an additional nitrogen in the aromatic indole X. Additionally, we prepared the CyIQ–COT conjugates 11 and 12 by the attachment of cyclooctatetraene (COT) using a short propyl linker to the indole-nitrogen. The COT-conjugated dyes follow the triplet quencher concept as described above19,20 and thus serve as the “best practice” examples to evaluate the properties of the synthesized dyes 1–10. Previous studies have shown that the methoxylation at the 6-position of the quinolinium moiety yields an increase in photostability. For this reason, the COT conjugate 12 with this modification was synthesized to further elucidate the aforementioned differences.The syntheses of all dyes with an azide functionality are described, together with all NMR spectroscopic and mass spectrometric analyses in the ESI.† The phosphoramidite as a DNA building block of 2′-propargylated uridine is commercially available and the postsynthetic modifications of oligonucleotides are also described in the ESI.† All dye-modified oligonucleotides DNA1–DNA12 were strictly purified by HPLC, identified by MALDI-TOF mass spectrometry and quantified by their UV/Vis absorption using ε260 nm (see ESI†).
Of course, all optical properties of the labeled DNA1–DNA12 including quantum yields ΦF and brightnesses B, both for the single (ss) and double strands (ds), were characterized by UV/Vis absorption and fluorescence spectroscopy (Table 1). In general, the fluorescence quantum yields of CyIQ-related dyes are higher as covalent DNA conjugates than non-covalent DNA conjugates (see ESI†).23 A particular focus of this study was set on the different photostabilities of dyes 1–12 as DNA conjugates that were determined by fluorescence bleaching during irradiation with the appropriate LED (468 nm, see ESI†) at 20 °C. All dyes as labels of DNA1–DNA12 absorb light with maxima in a range from 459 nm to 522 nm and emit green-yellow light with maxima between 532 nm and 587 nm. More importantly, they all show strong Stokes' shifts between 43 nm and 85 nm which is better suitable for imaging compared to the rather small Stokes' shifts of only a few nm for standard Cy3 in order to better separate excitation from emission light. Hereafter, DNA1 with the CyIQ dye 1 serves as the starting point for our systematic structure–reactivity investigations. The electron-donating group OMe at R2 reduces the quantum yields of ssDNA4 and ssDNA3 as well as their double strands drastically (ΦF = 1–5%) and permits their use in fluorescence imaging, although their photostabilities have not been significantly altered (t1/2 = 0.5–1.0 h). In contrast, the CN group at R2 improves the quantum yields to ΦF = 19–32% in dsDNA5 and dsDNA6 and the photostabilities to t1/2 = 1.7–4.2 h in their single strands. The latter results support the concept that the electron-withdrawing effect at the aromatic end groups of cyanine dyes enhances the photostability. Br as an alternative electron-withdrawing group at R2 in dsDNA7 and dsDNA8 also improves the quantum yields to ΦF = 30–32% but dramatically lowers the photostabilities to t1/2 = 2–7 min in the single strands, presumably by populating the photochemically reactive triplet state due to the heavy atom effect. This fits well with the above mentioned structure–reactivity relationship concept by an increased (downstream) triplet state reactivity. The introduction of an additional endocyclic N at the indole part is another basically different way to reduce the electron density in this heterocycle that raises the quantum yields of dsDNA9 and dsDNA10 to ΦF = 49–51% and slightly improves the photostabilities to t1/2 = 1.4 h in the single strands. In all regarded pairs of dyes, methylation of the NH group of the indole part improved photostabilities, with a restriction for the ssDNA1–ssDNA2 pair where t1/2 was slightly reduced from 0.8 h to 0.6 h. For instance, methylation of dye 5 resulted in a significantly improved photostability from t1/2 = 1.7 h for ssDNA5 to t1/2 = 4.2 h for ssDNA6, which leads us to assume a reaction with a basic reactive oxygen species (the superoxide radical anion, vide infra). Finally, the COT group as a triplet quencher instead of the methyl group at the indole part in dsDNA11 and dsDNA12 shows quantum yields of ΦF = 17–19% and expectedly good photostability of t1/2 = 3.7 h in their single strands. It is, however, important to note already here that even better optical properties are obtained with ss/dsDNA6 (ΦF = 19% and t1/2 = 4.2 h). In principle, this shows that the COT moiety can be omitted.
| ssDNA | λ exc nm | λ em nm | λ Stokes cm−1 | Φ F | B mM−1 cm−1 | τ F ns | t 1/2 min | dsDNA | λ exc nm | λ em nm | λ Stokes cm−1 | Φ F | B mM−1 cm−1 | τ F ns |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Irradiation by an LED at 468 nm. b Irradiation by an LED at 530 nm: t1/2 = 316 min. c Irradiation by an LED at 530 nm: t1/2 = 8 min. | ||||||||||||||
| 1 | 500 | 572 | 2517 | 0.21 | 8.76 | 2.08 | 50 | 1 | 499 | 571 | 2527 | 0.09 | 3.75 | 1.92 |
| 2 | 507 | 564 | 1993 | 0.35 | 15.0 | 2.99 | 38 | 2 | 502 | 556 | 2935 | 0.20 | 8.55 | 2.47 |
| 3 | 499 | 565 | 2341 | 0.03 | 1.31 | 0.48 | 31 | 3 | 499 | 583 | 2887 | 0.01 | 0.44 | n.d. |
| 4 | 512 | 574 | 2110 | 0.05 | 2.30 | 0.71 | 59 | 4 | 509 | 581 | 2435 | 0.04 | 1.84 | n.d. |
| 5 | 479 | 558 | 2956 | 0.26 | 8.74 | 2.98 | 100b | 5 | 461 | 532 | 2895 | 0.32 | 10.8 | 1.67 |
| 6 | 484 | 556 | 2676 | 0.28 | 8.20 | 5.04 | 253 | 6 | 467 | 541 | 2929 | 0.19 | 5.57 | 3.65 |
| 7 | 490 | 559 | 2519 | 0.22 | 8.69 | 2.17 | 2c | 7 | 475 | 543 | 2636 | 0.32 | 12.6 | 3.68 |
| 8 | 498 | 565 | 2381 | 0.30 | 11.9 | 2.70 | 7 | 8 | 483 | 540 | 2185 | 0.30 | 11.9 | 4.18 |
| 9 | 477 | 546 | 2649 | 0.49 | 18.2 | 3.82 | 44 | 9 | 459 | 541 | 3302 | 0.20 | 7.44 | 2.27 |
| 10 | 479 | 545 | 2528 | 0.51 | 17.0 | 3.17 | 85 | 10 | 466 | 551 | 3310 | 0.22 | 7.35 | 2.16 |
| 11 | 522 | 565 | 1458 | 0.46 | 20.7 | 4.76 | 219 | 11 | 515 | 587 | 2382 | 0.19 | 8.57 | 2.02 |
| 12 | 509 | 567 | 2010 | 0.39 | 17.0 | 3.03 | 224 | 12 | 509 | 580 | 2405 | 0.17 | 7.43 | 2.42 |
Nucleophilicity at C-2 of indole responsible for photobleaching
It becomes clear from the results described above that the electron density at C-2 of the indole plays a significant role in the optical properties and photostabilities since it is regulated by the substituent R2, possibly by more or less effective donor–acceptor interplay in the excited state that leads to a contribution of charge-transfer states. In fact, the C-2-position has also been identified by the major photooxidation product that was formed during irradiation of the CyIQ lead dye 1 in the presence of dsDNA.12 The reactivity of this position towards reactive oxygen species guides towards improved photostability by rational means. In order to obtain a general structure–reactivity relationship between the measured photostabilities and the electron-densities at C-2 of the indole parts of singly labeled ssDNA1–ssDNA12, we applied the nucleophilicity parameter N that was introduced by Mayr et al. to universally compare the nucleophilic reactivity of different compounds on the same scale.24 When the published nucleophilicity parameters N for 5-cyano- (in dye 5), 7-aza- (in dye 9), 5-bromo- (in dye 7), and 5-methoxyindole (in dye 3), unsubstituted indole (in dye 1), and 1-methylindole (in dye 2) were plotted against the extinction coefficient εmax of the appropriate absorption maxima of the dyes 1–3, 5, 7, and 9, a clearly linear relationship was obtained (see ESI, Fig. S2†). This referenced linearity allowed calculating the nucleophilicity parameter Ncalc of the remaining indoles as part of the dyes 4, 6, 8, and 10–12, respectively, according to the linear function Ncalc = a + bεmax (with a and b from the reference fit). When subsequently the experimentally determined fluorescence bleaching half-lifes t1/2 of ssDNA1–ssDNA12 were plotted against the nucleophilicity parameter Ncalc, (see ESI, Fig. S53†), there was an exponential relationship between both values observable at first glance, which, however, needed to be specified by considering the different absorbances of the dyes with respect to the 468 nm LED excitation spectrum. Hence, the spectral overlaps between absorbances and LED emission spectrum were calculated as an integral between 380 nm and 600 nm for each dye and applied to reference the fluorescence bleaching half-life times tref1/2 (see ESI,† Section 10). For these referenced tref1/2 values, a clear monoexponential dependence on the nucleophilicity parameter Ncalc was obtained for the majority of the dye–DNA conjugates (Fig. 1). Only for the 5-bromoindole-based dyes 7 and 8 in ssDNA7 and ssDNA8, respectively, and for the COT-linked dyes 11 and 12 in ssDNA11 and ssDNA12, respectively, our exponential fit is not suitable. For ssDNA7 and ssDNA8 it can be assumed that the Br substituent negatively affects the photostability by a more efficient intersystem crossing (heavy atom effect) and an increased population of the triplet state, whereas in the case of ssDNA11 and ssDNA12, the COT group has an additional positive effect by the triplet quenching pathway that improves photostability to a value that is higher than expected by considering solely the electron density at C-2 of the indole part of the ground state fluorophore. It is important to note here that the latter four dye–DNA conjugates show that the observed exponential structure–reactivity for the other dyes 1–6 and 9–10 obviously follows a photodegradation pathway that differs from the triplet state reactivity. | ||
| Fig. 1 Analysis of the nucleophilicity-dependent photobleaching of ssDNA1–ssDNA12: the fluorescence photobleaching half-lifes referenced to the 468 nm LED excitation spectrum tref1/2 show a monoexponential dependence (dotted line) on Ncalc at C-2 of the indole part of the dyes (for parameters of the fit function see ESI†). | ||
The influence of the nucleophilicity at C-2 of the indole part contradicts a photodegradation pathway by dye-sensitized singlet oxygen that is typically electrophilic (Scheme 2).25 In order to explicitly rule out that singlet oxygen is mainly responsible for bleaching we additionally performed thermal bleaching experiments with dyes 1–10 by treatment with 50 mM H2O2 in the presence of 1 mM Na2MoO4. Under these conditions singlet oxygen is formed without light.26,27 The bleaching was followed by the loss of absorbance at the respective λmax, and half-lifes t1/2 were determined after correction by the background reaction with H2O2 alone. Interestingly, there was also a monoexponential correlation obtained between the calculated nucleophilicity parameter Ncalc and the corresponding half-lifes of the thermal bleaching – but more shallow and with an opposite trend (Fig. 2). Control experiments with Na2MoO4 alone did not give significant bleaching (see ESI†).
 | ||
| Scheme 2 Quenching and proposed photodegradation pathways. | ||
 | ||
| Fig. 2 Analysis of the nucleophilicity-dependent light-independent (thermal) bleaching of the dyes 1–12: the absorption bleaching half-lifes t1/2 show monoexponential dependences (dotted lines) on Ncalc at C-2 of the indole part of the dyes (for parameters of the fit functions see ESI†). Please note that dyes 11 and 12 were not completely soluble and the data was omitted. | ||
The half-lifes tref1/2 of photobleaching showed a strong exponential dependence and were higher for weaker indole nucleophiles (such as the indoles in the dyes of ssDNA5 or ssDNA6), whereas stronger nucleophiles (such as the indoles in the dyes of ssDNA3 or ssDNA4) increased the half-lifes t1/2 for thermal bleaching. This agrees well with a simplified consideration of the participating molecular orbitals. The electron-withdrawing effect of the CN substituent lowers the HOMO energy of dyes 5 and 6, which accelerates the reaction with the low-lying LUMO of singlet oxygen. In fact, Armitage et al. could show for thiazole orange-derived dyes that electron-withdrawing substituents in particular at the benzothiazole core (structural counterpart of the indole in our dyes) lower the energy of the HOMO (and LUMO equally), while those substituents at the quinolinium part leave the HOMO energy rather unaffected (and reduce mainly the LUMO energy).3,28 This can also be assumed for our cyanine-indole-quinolinium dyes because it agrees with the thermal reactivity described above.
Electrophilic singlet oxygen vs. nucleophilic reactive oxygen species
Generally, singlet oxygen sensitization shortens the fluorescence lifetimes τF of the dyes by triplet energy transfer. Especially the dyes in ss/dsDNA2, DNA7, and DNA8 show longer fluorescence lifetimes τF = 2.17–4.18 ns than the lead ss/dsDNA1 with τF = 1.92–2.08 ns although they are less photostable. Contrarily, ss/dsDNA3 and DNA4 show the shortest fluorescence lifetimes of τF = 0.48–0.71 ns, but photostabilities comparable to ss/dsDNA1. Our photostability studies were performed with 2.5 μM dye–DNA conjugates, at which the singlet oxygen contribution to fluorescent dye bleaching is negligible according to Blanchard et al.18 Moreover, singlet oxygen formed by rose bengal sensitization did not yield a significant bleaching of dye 3 (see ESI†). Taken together, this excludes not only the triplet state reactivity (as described above) but also singlet oxygen reactivity as major pathways for photobleaching of our cyanine-indole dyes (Scheme 2). Alternatively, excited singlet states are quenched by photoinduced electron transfer (PET) processes with oxygen according to type I reactions forming the superoxide radical anion and the perhydroxyl radical as important reactive oxygen species.29 In particular, the perhydroxyl radical undergoes electron transfer with electron-rich centers due to its high oxidation potential. According to the strong exponential correlation between the nucleophilicity at C-2 of the indole and the photobleaching half-lifes, we assume that this is the major photodegradation pathway for the dyes described herein. Conclusively, dye 6 has been identified as a top candidate that shows similar optical properties and an even better photostability compared to dye 11 although the covalently linked COT moiety is omitted in 6.Cytotoxicity and live cell imaging
With regard to the live cell imaging approach, we tested the cytotoxicity of these dyes towards HeLa cells in the first instance. The applied cell proliferation assay (see ESI†) is based on the intracellular color change caused by the reduction of a tetrazolium salt (MTT) into a formazan product, which only takes place in metabolic active cells. At a wavelength of 595 nm the absorbance of the formazan product of the lysed cells can be detected after 72 h and used as a direct measure of their viability. For the majority of the dyes, a moderate to low cytotoxicity could be observed with values for the median lethal dose (LD50) between 5–10 μM (Fig. 3). This concludes that these dyes can be used at these concentrations for the following live cell imaging, considering that these experiments require much shorter incubation times of maximum 24 h compared to the 72 h incubation times during the cytotoxicity tests. An exception could be observed for 11, where the toxic effect is probably triggered by the COT moiety, based on comparing it to 1, where the same structure without the COT group (1) showed a much higher cell viability. This is an important result because dye 6 has similar optical properties and a better photostability than the COT-modified dye 11 but is significantly less cell toxic. The other COT-modified dye 12 forms aggregates in aqueous solutions and also inside cells (vide infra) which reduces the apparent cytotoxicity. Furthermore, the bromo-substituted dyes 7 and 8 also show an enhanced cytotoxic effect. | ||
| Fig. 3 Cytotoxicity probed via MTT tests on HeLa cells after 72 h incubation of dye 1–12 with concentrations of 1 μM, 5 μM, 10 μM and 20 μM, respectively. | ||
For live cell imaging, cells were incubated with a final concentration of 10 μM of the respective dye 1–12 for 24 h before confocal fluorescence microscopy imaging. The majority of the dyes were localized in endosomal vesicles (1, 3, 4, 5, 9, 10, and 12), but could also be found in mitochondria (6), in the cytosol (4, 11) and in nucleoli (1, 3, 4) (Fig. 4 and S72–S74 in the ESI†). As expected, the dyes 2, 7 and 8 could not be imaged due to their high cytotoxicity. This effect can probably be avoided by the reduction of the final concentration from 10 μM to 5 μM, ensuring a more than 10 times higher cell viability which is given by the results of the cytotoxicity tests. Moreover, dye 11 shows strong cell penetration properties and an equal distribution in the cytosol. This can be attributed to the lipophilic COT moiety, which likely improves cell penetration and alters the lipid bilayer.30 Interestingly, all methylated dyes (Fig. 3, blue bars) show a kind of plateau, which is affiliated with an intracellular equilibrium concentration. Altogether, dyes 1–6 and 9–10 show good imaging properties inside living cells. Especially, the photostable dye 6 can be used as a mitochondria targeting agent. This localization of dye 6 was checked using MitoTracker Red (see ESI†).
 | ||
| Fig. 4 Live cell images of HeLa cells after treatment with dyes 6 and 10 (for images of the other dyes see ESI, Fig. S70 and S71†), left column: excitation at 488 nm, emission detected at 500–515 nm, middle column: brightfield image, right column: overlay of both images. The images were taken after 24 h treatment of the dyes, because this time is needed for the efficient transport of DNA1–DNA12 (Fig. 5). | ||
Transfection experiments were representatively done for ssDNA6 and ssDNA11, which bear the dyes with the best optical properties and photostabilities. Therefore, HeLa cells were transfected with a final concentration of 75 nM of the respective dye-modified DNA for 24 h at 37 °C and 5% CO2 with the help of ScreenFect A®, used according to the manufacturer's protocol (see ESI,† Section 14). 24 h was needed for efficient cellular uptake. To determine the intracellular locations, cells were washed to get rid of the non-transfected DNA and subsequently imaged via live cell confocal fluorescence microscopy. The corresponding images (Fig. 5) reveal a high degree of similarity between the two dye–DNA conjugates as both of them can be mainly found in endosomes and in the cytosol by their bright fluorescence.
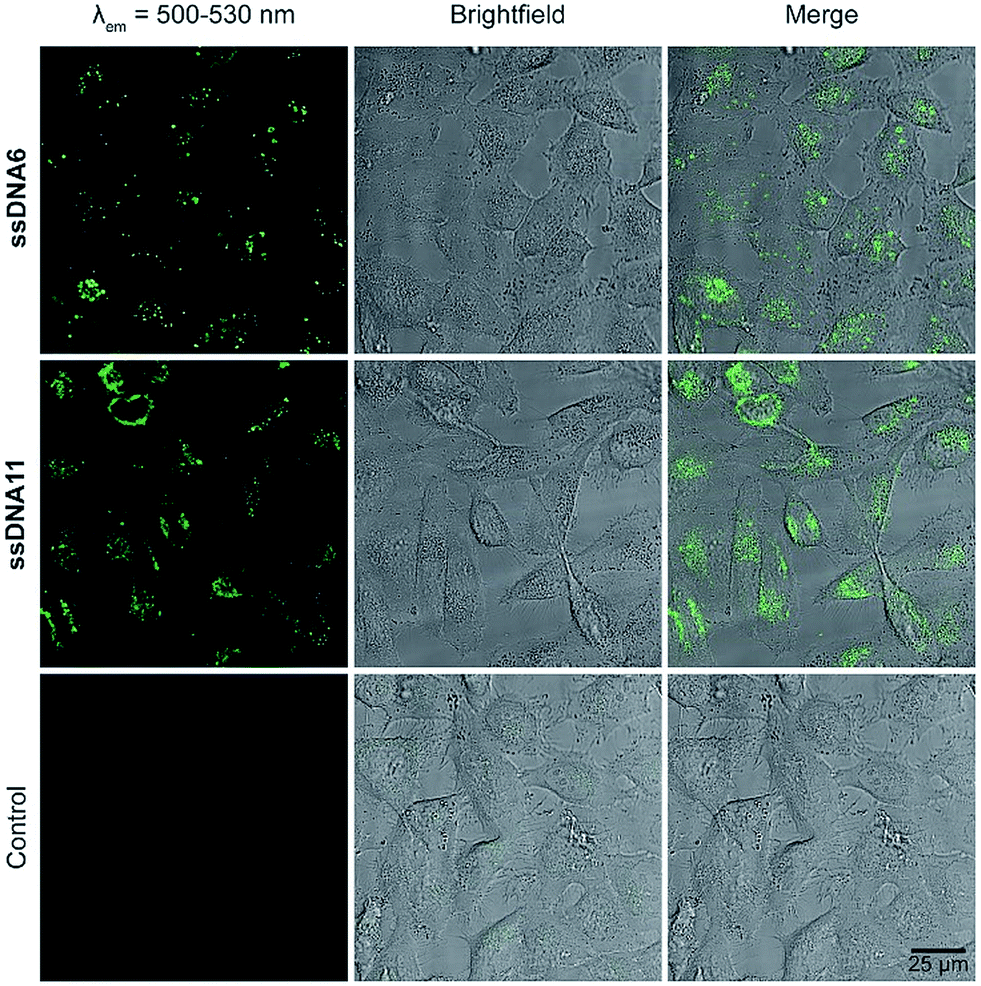 | ||
| Fig. 5 Live cell images after transfection with ssDNA6 (top), ssDNA11 (middle) and without DNA (control, bottom), left column: excitation at 488 nm, emission detected at 500–530 nm, middle column: brightfield image, right column: overlay of both images. | ||
Conclusions
We synthesized a new set of green-emitting cyanine-indole dyes based on the lead structure 1 and focused on the electronic character of indoles as the structural component of these dyes. Furthermore, these dyes were postsynthetically linked covalently to the 2′-position of uridine anchors in presynthesized DNA by the copper(I)-catalyzed azide–alkyne cycloaddition in order to evaluate their photostabilities and live cell imaging properties as DNA conjugates. We elucidated the nucleophilicity at position C-2 of the indoles of dyes 1–10 as key for a new structure–activity relationship that yielded a rational guide to improve the photostability and optical properties of these green-emitting dyes for live cell imaging of nucleic acids. Concretely, the nucleophilicity parameters of the indoles were referenced to the extinction coefficient of the synthesized dyes and subsequently correlated with the fluorescence bleaching half-lifes. Accordingly, the photobleaching half-lifes by LED irradiation were higher for weaker indole nucleophiles and lower for stronger indole nucleophiles. Taken together with thermal bleaching experiments, which excluded a significant contribution of singlet oxygen to photobleaching, type I reactions with oxygen were evidenced as primary photobleaching pathways, especially by the highly reactive perhydroxyl radicals. This strategy allowed us to identify dye 6 as the best candidate with comparable optical properties and even better photostability than dye 11 that bears an additional COT group as a triplet quencher. The cell viability of dye 11 in comparison with the structurally related dye 1 lacking the COT group revealed a strong cytotoxic effect of the COT group, which limits the applicability of its DNA conjugates (like ssDNA11) for live cell imaging. In contrast, dye 6 did not show such a strongly enhanced cell toxicity. We conclude that the additional COT substituent can be omitted to obtain smaller, but more photostable and brighter fluorescent labels for nucleic acids.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft (grant Wa 1386/17–1), the Graduiertenkolleg GRK 2039 (also funded by DFG), and KIT is gratefully acknowledged.Notes and references
- P. V. Chang and C. R. Bertozzi, Chem. Commun., 2012, 48, 8864–8879 RSC
.
- E. M. S. Stennett, M. A. Ciuba and M. Levitus, Chem. Soc. Rev., 2014, 43, 1057–1075 RSC
- B. A. Armitage, Top. Curr. Chem., 2005, 253, 55–76 CrossRef
- R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579–2619 CrossRef PubMed
- F. Hövelmann and O. Seitz, Acc. Chem. Res., 2016, 49, 714–723 CrossRef PubMed
- H. Ihmels and D. Otto, Top. Curr. Chem., 2005, 258, 161–204 CrossRef
- A. Mishra, R. K. Behera, B. K. Mishra and G. B. Behera, Chem. Rev., 2000, 100, 1973–2011 CrossRef PubMed
- M. Levitus and S. Ranjit, Q. Rev. Biophys., 2011, 44, 123–151 CrossRef PubMed
- I. D. Johnson, H. C. Kang and R. P. Haugland, Anal. Biochem., 1991, 198, 228–237 CrossRef PubMed
- J. E. Berlier, A. Rothe, G. Buller, J. Bradford, D. R. Gray, B. J. Filanoski, W. G. Telford, S. Yue, J. Liu, C.-Y. Cheung, W. Chang, J. D. Hirsch, J. M. B. R. P. Haugland and R. P. Haugland, J. Histochem. Cytochem., 2003, 51, 1699–1712 CrossRef PubMed
- Q. Li, Y. Kim, J. Namm, A. Kulkarni, G. R. Rosania, Y.-H. Ahn and Y.-T. Chang, Chem. Biol., 2006, 13, 615–623 CrossRef PubMed
- P. Bohländer and H.-A. Wagenknecht, Org. Biomol. Chem., 2013, 11, 7458–7462 RSC
- P. R. Bohländer and H.-A. Wagenknecht, Methods Appl. Fluoresc., 2015, 3, 044003 CrossRef PubMed
- P. R. Bohländer and H.-A. Wagenknecht, Eur. J. Org. Chem., 2014, 7547–7551 CrossRef
- G. R. Rosania, J. W. Lee, L. Ding, H.-S. Yoon and Y.-T. Chang, J. Am. Chem. Soc., 2003, 125, 1130–1131 CrossRef PubMed
- Q. Zheng and L. D. Lavis, Curr. Opin. Chem. Biol., 2017, 39, 32–38 CrossRef PubMed
- N. I. Shank, K. J. Zanotti, F. Lanni, P. B. Berget and B. A. Armitage, J. Am. Chem. Soc., 2009, 131, 12960–12969 CrossRef PubMed
- Q. Zheng, S. Jockusch, Z. Zhou and S. C. Blanchard, Photochem. Photobiol., 2014, 90, 448–454 CrossRef PubMed
- Q. Zheng, S. Jockusch, G. G. Rodriguez-Calero, Z. Zhou, H. Zhou, R. B. Altman, H. D. Abruna and S. C. Blanchard, Photochem. Photobiol. Sci., 2016, 15, 196–203 RSC
- Q. Zheng, M. F. Juette, S. Jockusch, M. R. Wasserman, Z. Zhou, R. B. Altman and S. C. Blanchard, Chem. Soc. Rev., 2014, 43, 1044–1056 RSC
- X. H. Zhang, Q. Liu, H. J. Shi, L. Y. Wang, Y. L. Fu, X. C. Wei and L. F. Yang, Dyes Pigm., 2014, 100, 232–240 CrossRef
- X.-H. Zhang, L.-Y. Wang, Z.-X. Nan, S.-H. Tan and Z.-X. Zhang, Dyes Pigm., 2008, 79, 205–209 CrossRef
-
C. Schwechheimer, M. Merkel, P. R. Bohländer and H.-A. Wagenknecht, in Modified Nucleic Acids, ed. Y. T. Kazuhiko Nakatani, Springer, 2016, ch. 4, pp. 83–100 Search PubMed
- S. Lakhdar, M. Westermaier, F. Terrier, R. Goumont, T. Boubaker, A. R. Ofial and H. Mayr, J. Org. Chem., 2006, 71, 9088–9095 CrossRef PubMed
- R. R. Nani, J. A. Kelley, J. Ivanic and M. J. Schnermann, Chem. Sci., 2015, 6, 6556–6563 RSC
- J. M. Aubry and B. Cazin, Inorg. Chem., 1988, 27, 2013–2014 CrossRef
- K. Böhme and H.-D. Brauer, Inorg. Chem., 1992, 31, 3468–3471 CrossRef
- E. E. Rastede, M. Tanha, D. Yaron, S. C. Watkins, A. S. Waggoner and B. A. Armitage, Photochem. Photobiol. Sci., 2015, 14, 1703–1712 RSC
-
K. Krumova and G. Cosa, in Singlet Oxygen: Applications in Biosciences and Nanosciences, eds. S. Nonell and C. Flors, Royal Society of Chemistry, Cambridge, 2016, pp. 3–21 Search PubMed
- J. L. Alejo, S. C. Blanchard and O. S. Andersen, Biophys. J., 2013, 104, 2410–2418 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, experimental procedures, DNA preparation, optical measurements, cell images and complete analytical data. See DOI: 10.1039/c8sc01574k |
| This journal is © The Royal Society of Chemistry 2018 |