
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of α-aminoboronic acids from amines and potassium acyltrifluoroborates (KATs) via trifluoroborate-iminiums (TIMs)†
Tomoya
Shiro‡
,
Anne
Schuhmacher‡
,
Moritz K.
Jackl
and
Jeffrey W.
Bode
 *
*
Laboratorium fur Organische Chemie, Department of Chemistry and Applied Biosciences, ETH Zurich, Zürich, Switzerland 8093. E-mail: bode@org.chem.ethz.ch; Web: http://www.bode.ethz.ch
First published on 16th May 2018
Abstract
We report the facile formation of trifluoroborate-iminiums (TIMs) from potassium acyltrifluoroborates (KATs) and the transformation of TIMs to α-aminotrifluoroborates by reduction or Grignard additions. Conditions for the hydrolysis of α-aminotrifluoroborates to α-aminoboronic acids, which are important biologically active compounds, were established. This new methodology allows access to sterically demanding α-aminoboronic acids that are not easily prepared with currently available methods. This work also introduces TIMs, that can be easily prepared and handled, as a new category of functional groups that serve as precursors to valuable organic compounds.
Introduction
α-Aminoboronic acids are currently of great interest for modern pharmaceutical development and several such compounds including bortezomib and ixazomib have recently been approved as new small molecule drugs. These structures are prized for their ability to inhibit the proteasome and various serine proteases, making them particularly attractive as potential anticancer, antibacterial or antiviral drugs.1 Known synthetic approaches include addition of nitrogen substituted carbon to electrophilic boron,2 addition of nucleophilic boron to activated imines,3 SN2 like displacements of amines to halomethyl boronic acid derivatives,4 decarboxylative deborylations5 or hydroaminations of vinylboronic acid derivatives6 but to date there are few methods suitable for the preparation of molecules derived from tertiary amines and bearing a fully substituted boron containing carbon.In this report, we document the facile formation of stable trifluoroborate-iminiums (TIMs) from KATs and primary or secondary amines and their use for the synthesis of secondary and tertiary α-aminoboronic acids. A few structures similar to TIMs have been reported previously, but little is known about their chemistry and properties.7 Yudin and co-workers observed the formation of imines from MIDA acyl boronates and amines, however they were only able to isolate the reduction products, not the iminium ions.8 In contrast, we have found that TIMs are air, moisture, and chromatographically stable and are easily formed under simple conditions in high yield. They serve as convenient intermediates for the synthesis of α-aminotrifluoroborates – which can be readily hydrolyzed to the boronic acids – by reduction or nucleophilic addition. This route offers a simple, broadly applicable route to α-aminoboronic acids, particularly those that would be difficult to access by established approaches (Fig. 1).
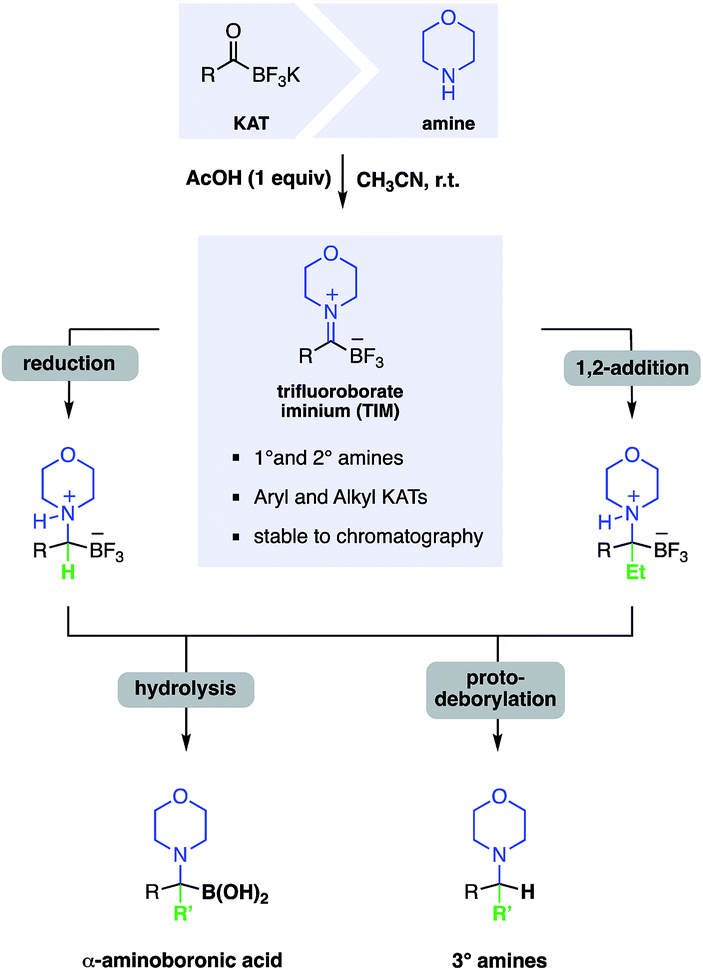 | ||
| Fig. 1 Overview of the chemistry of TIMs including formation from KATs and transformation to α-aminotrifluoroborates and α-aminoboronic acids. | ||
Results and discussion
Experience from our group on amide-forming reactions of KATs with hydroxylamines or N–Cl amines established that KATs show little or no intrinsic reactivity with simple amines under aqueous conditions. For instance, our recent report on protein PEGylation using PEG–KAT reagents was conducted in glycine buffer and we have recently disclosed that amide formation from KATs and amines in the presence of a chlorinating agent does not proceed via an iminium intermediate.9 We reasoned, however, that iminium formation could occur under non-aqueous conditions (Table 1). Although KATs are generally insoluble in most organic solvents, sparing solubility is observed in acetone, DMF, acetonitrile, DMSO and other polar, aprotic solvents. Simply mixing KATs and amines did not lead to substantial iminium formation, however the addition of acid – presumably to form a salt with the liberated potassium ion – led to clean formation of the highly soluble TIMs and the corresponding inorganic salt (entry 2). Based on these observations, we found that zwitterion formation occurs cleanly under a variety of conditions and with numerous acidic additives, including HCl, AcOH, and CF3CO2H. Conveniently, the amine hydrochloride salts can also be employed directly in the reaction.|
|
||||
|---|---|---|---|---|
| Entry | Amine | Acid | Conditions | Conversion |
| a Product detected by LC-MS and TLC and conversion determined by LC-MS. b Isolated yields. | ||||
| 1 | 2.0 M in THF | None | CH3CN (0.2 M), 8 h | <5%a |
| 2 | HCl salt | None | CH3CN (0.2 M), 1 h | 94%b |
| 3 | 2.0 M in THF | AcOH | CH3CN (0.2 M), 15 min | 93%b |
| 4 | 2.0 M in THF | CF3CO2H | CH3CN (0.2 M), 15 min | 90%b |
| 5 | 2.0 M in THF | AcOH | DMF (0.2 M), 1 h | 100%a |
| 6 | 2.0 M in THF | AcOH | CH3CN (0.02 M), 5 h | 98%a |
| 7 | 2.0 M in THF | AcOH | CH3CN (0.002 M), 8 h | 97%a |

The TIMs can be formed from both secondary and primary amines, in which case they are isolated as a chromatographically stable, protonated imine zwitterion (Scheme 1). TIMs are typically white solids stable to air, moisture and standard aqueous workup. They are readily formed chemoselectively in the presence of nearly all common functional groups, including carboxylic acids, esters, nitriles, and aldehydes.
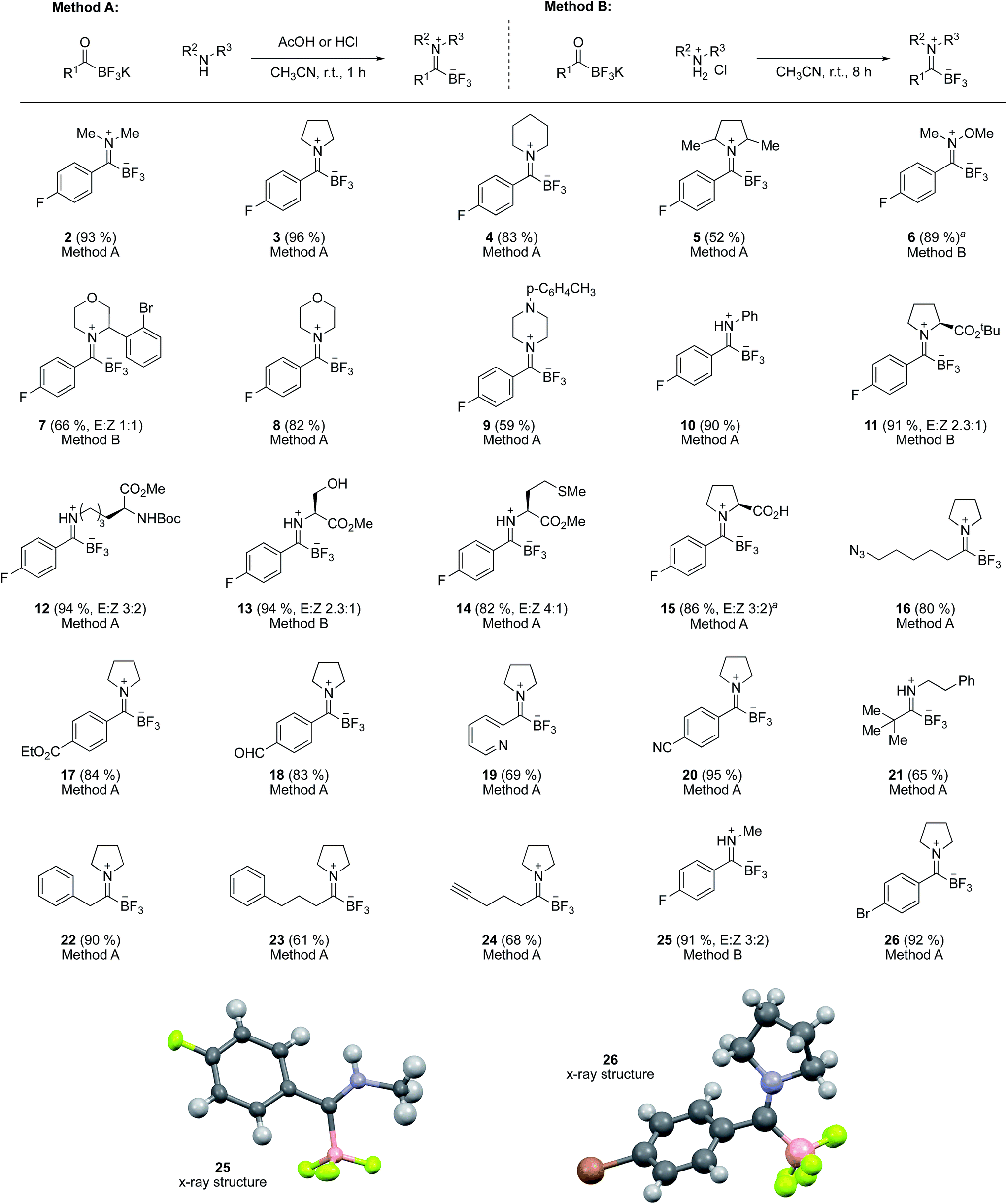 | ||
| Scheme 1 Substrate scope for the formation of TIMs from KATs and amines. aDMF used instead of CH3CN. | ||
Unlike iminium ions derived from aldehydes or ketones, TIMs show no tendency to form enamines and are kinetically inert under aqueous conditions. Studies on exchange reactions with exogenous amines are currently in progress, but in preliminary studies this does not appear to be a major pathway. Intrigued by their unexpectedly high chemical inertness, we performed numerous tests on their stability, as summarized in Table 2. At no point did we observe protodeborylation products. The only products identified from their decomposition were the starting amine and KAT. Under more basic conditions (pH 8.0–9.0), the TIMs eventually broke down to the constituent KATs and amines.
|
|
||||
|---|---|---|---|---|
| Entry | Conditions | Conversiona | ||
| 2 h | 16 h | 24 h | ||
| a Conversion determined using LC-MS. b CH2Cl2 used as solvent instead of CH3CN. Solvolysis of the BF3 group was never observed under aqueous conditions. | ||||
| 1 | Et3N (5.00 equiv.) | 0% | ||
| 2 | Piperidine (20.0 equiv.) | 0% | ||
| 3 | CF3CO2H (0.1 mL)b | 0% | ||
| 4 | aq. HCl (0.1 M, pH 1.2) | 0% | ||
| 5 | aq. citric acid (0.1 M, pH 2.0) | 0% | ||
| 6 | aq. citric acid/K2HPO4 buffer (0.1 M, pH 3.0) | 0% | 2% | |
| 7 | aq. citric acid/K2HPO4 buffer (0.1 M, pH 5.0) | 2% | 5% | |
| 8 | aq. KH2PO4/K2HPO4 buffer (0.1 M, pH 7.0) | 4% | 14% | 19% |
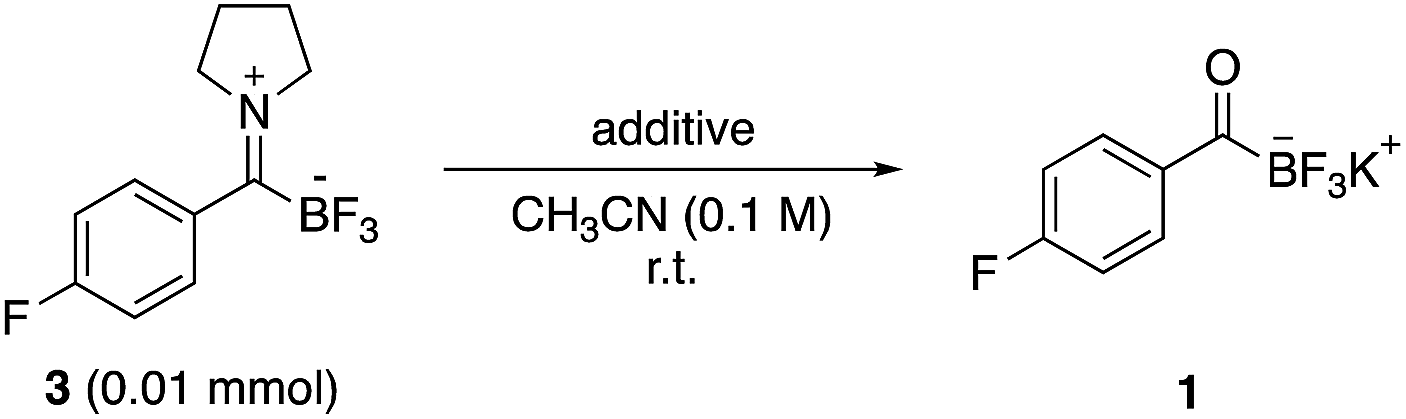
Based on the known chemistry of iminiums as excellent electrophiles in polar addition reactions, we anticipated that TIMs would undergo nucleophilic addition. The formation of stable zwitterionic iminiums offers an opportunity to conduct bond forming reactions on species that would otherwise be difficult to form or prone to enamine formation. We were pleased to find that TIMs, including those derived from secondary amines, underwent clean reduction to give mono-substituted α-aminotrifluoroborates in the presence of KBH4 (Scheme 2).
 | ||
| Scheme 2 Substrate scope for the reduction of TIMs yielding monosubstituted α-aminotrifluoroborates. | ||
A great challenge is nucleophilic addition of carbon nucleophiles to iminiums. A few examples of the addition of organometallic compounds to iminiums are known, but these appear to be limited to aldehyde derived or non-enolizable iminiums.10
We sought to employ the remarkable stability of the TIMs to access the fully substituted α-aminotrifluoroborates, including those containing cyclic tertiary amines. Therefore, we established conditions for the addition of Grignard reagents to TIMs derived from secondary amines. These couplings proceeded well for a broad scope of TIMs and Grignard reagents and the resulting fully substituted α-aminotrifluoroborates were isolated in high yields. Linear alkyl, vinyl and even sterically more demanding branched alkyl and aryl Grignards added smoothly to both aromatic and aliphatic TIMs. At no point did we observe diminished yields due to enamine formation. For the addition of substituted aromatic and heteroaromatic Grignard reagents, Knochel's Turbo-Grignard chemistry11 was successfully employed (Scheme 3).
 | ||
| Scheme 3 Substrate scope for the addition of Grignard reagents to TIMs yielding fully substituted α-aminotrifluoroborates. | ||
The α-aminotrifluoroborates are themselves interesting compounds, isolated as internal salts. A few simpler variants have been prepared and shown to undergo cross-coupling reactions under palladium or photoredox catalysis. Molander demonstrated that aminomethyltrifluoroborates can be coupled to various aryl- and hetaryl halides or mesylates under Suzuki–Miyaura conditions.12 Different amino acid derived Boc-aminomethyltrifluoroborates were coupled to aryl bromides under photoredox conditions by the same group.13 Suginome was able to cross-couple chiral α-(acylamino)benzylboronic esters to aryl bromides with inversion of configuration.14 Unfortunately, all attempts to subject these more substituted substrates to cross coupling conditions were unsuccessful, possibly due to the increased steric demands of the substrates in comparison to the successful examples studied by Molander and Suginome.
In the course of these cross coupling studies, we identified conditions for clean protodeborylation of some substrates. The best results were obtained with Zr(OiPr)4 in toluene (Scheme 4). Unfortunately, these conditions were not successful with all substrates, particularly those lacking an α-aryl substituent on the α-aminotrifluoroborate. Further investigations on the mechanism of this unexpected reaction and the origin of the limitations are in progress, as are continued efforts to effect cross coupling of the α-aminotrifluoroborates.
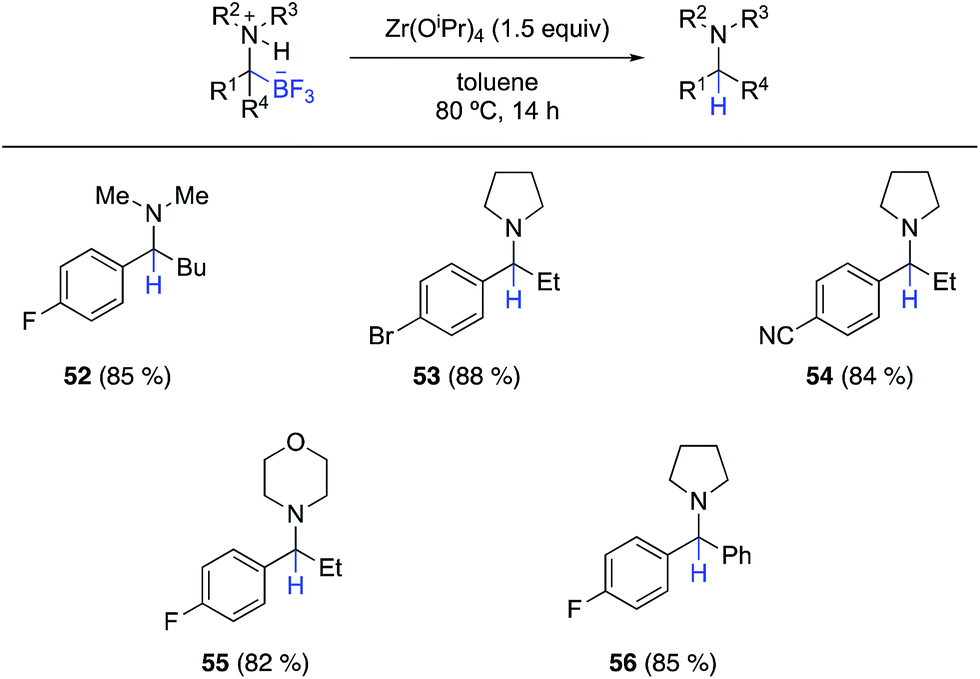 | ||
| Scheme 4 Substrate scope for the protodeborylation of fully substituted α-aminotrifluoroborates using Zr(OiPr)4. | ||
The ultimate goal of this research was the formation of α-aminoboronic acids, as these compounds have emerged as important structures in drug design and lack good, convergent approaches for their synthesis. Several conditions for the formation of boronic acids are known in literature, however these conditions did not yield the desired boronic acids for our substrates.15 SiCl4 is known for defluorination of organotrifluoroborates,16 and we identified SiCl4 in CH3OH as the optimal conditions for the conversion of the α-aminotrifluoroborates to the boronic acids. The boronic acid products are isolated as their HCl salts after treatment with aqueous HCl (Scheme 5).
 | ||
| Scheme 5 Substrate scope for the formation of α-aminoboronic acids from α-aminotrifluoroborates using SiCl4. Compound 58 and 62 were isolated as TFA salts after purification by preparative HPLC. | ||
Conclusions
In summary, we established the facile synthesis of TIMs from amines and KATs, which were found to be stable zwitterionic compounds with properties and stabilities suitable for further development. TIMs can be easily reduced with hydride reagents or participate in C–C bond forming reactions with Grignard reagents to give α-aminotrifluoroborates. The α-aminotrifluoroborates can be hydrolyzed to α-aminoboronic acids. Along with the increasing synthetic and commercial availability of KATs, this chemistry enables the synthesis of fully substituted α-aminoboronic acids, that are difficult to access with methods known to date.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the European Research Council (ERC Starting Grant 306793 – CASAA). Support for T. S. was generously provided by Sumitomo Dainippon. We thank the LOC Mass Spectrometry Service, the LOC NMR Service, and the SmOCC crystallography service for analyses. We want to thank Sizhou M. Liu (ETH Zürich) for preparation of some aliphatic KAT starting materials, Adelaïde Savoy (ETH Zürich) for help with the “Turbo-Grignard” reaction, Alberto Osuna Gálvez (ETH Zürich), and Dino Wu (ETH Zürich) for helpful discussions.Notes and references
- (a) P. C. Trippier and C. McGuigan, Med. Chem. Commun., 2010, 1, 183 RSC; (b) R. Smoum, A. Rubinstein, V. M. Dembitsky and M. Srebnik, Chem. Rev., 2012, 112, 4156 CrossRef PubMed; (c) J. Hiratake and J. Oda, Biosci., Biotechnol., Biochem., 1997, 61, 211 CrossRef; (d) C. Kettner, L. Mersinger and R. Knabb, J. Biol. Chem., 1990, 265, 18289 Search PubMed; (e) N. C. J. Strynadka, R. Martin, S. E. Jensen, M. Gold and J. B. Jones, Nat. Struct. Biol., 1996, 3, 688 CrossRef PubMed; (f) R. M. Dunsdon, J. R. Greening, P. S. Jones, S. Jordan and F. X. Wilson, Bioorg. Med. Chem. Lett., 2000, 10, 1577 CrossRef PubMed; (g) B. A. Teicher, G. Ara, R. Herbst, V. J. Palombella and J. Adams, Clin. Cancer Res., 1999, 5, 2638 Search PubMed.
- E. S. Priestley and C. P. Decicco, Org. Lett., 2000, 2, 3095 CrossRef PubMed.
- (a) M. A. Beenen, C. An and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 6910 CrossRef PubMed; (b) A. W. Buesking, V. Bacauanu, I. Cai and J. A. Ellman, J. Org. Chem., 2014, 79, 3671 CrossRef PubMed; (c) C. Solé, H. Gulyás and E. Fernández, Chem. Commun., 2012, 48, 3769 RSC; (d) K. Hong and J. P. Morken, J. Am. Chem. Soc., 2013, 135, 9252 CrossRef PubMed; (e) D. Wang, P. Cao, B. Wang, T. Jia, Y. Lou, M. Wang and J. Liao, Org. Lett., 2015, 17, 2420 CrossRef PubMed.
- (a) D. S. Matteson and T. C. Cheng, J. Org. Chem., 1968, 33, 3055 CrossRef; (b) D. S. Matteson, K. M. Sadhu and G. E. Lienhard, J. Am. Chem. Soc., 1981, 103, 5241 CrossRef; (c) D. S. Matteson and K. M. Sadhu, Organometallics, 1984, 3, 614 CrossRef; (d) V. Martichonok and J. B. Jones, J. Am. Chem. Soc., 1996, 118, 950 CrossRef; (e) M. B. Kostova, D. M. Rosen, Y. Chen, R. C. Mease and S. R. Denmeade, J. Med. Chem., 2013, 56, 4224 CrossRef PubMed; (f) G. A. Molander and J. Ham, Org. Lett., 2006, 8, 2031 CrossRef PubMed; (g) S. Adachi, A. B. Cognetta III, M. J. Niphakis, Z. He, A. Zajdlik, J. D. St. Denis, C. C. G. Scully, B. F. Cravatt and A. K. Yudin, Chem. Commun., 2015, 51, 3608 RSC.
- (a) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Science, 2017, 356, 1045 Search PubMed; (b) D. Hu, L. Wang and P. Li, Org. Lett., 2017, 19, 2770 CrossRef PubMed; (c) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283 CrossRef PubMed.
- D. Nishikawa, K. Hirano and M. Miura, J. Am. Chem. Soc., 2015, 137, 15620 CrossRef PubMed.
- (a) A. M. Dumas and J. W. Bode, Org. Lett., 2012, 14, 2138 CrossRef PubMed; (b) T. Wang, L. Liu, S. Grimme, C. G. Daniliuc, G. Kehr and G. Erker, Chem.–Asian J., 2016, 11, 1394 CrossRef PubMed; (c) D. J. Brauer, H. Bürger, S. Buchheim-Spiegel and G. Pawelke, Eur. J. Inorg. Chem., 1999, 255 CrossRef.
- D. B. Diaz, C. C. G. Scully, S. K. Liew, S. Adachi, P. Trinchera, J. D. St. Denis and A. K. Yudin, Angew. Chem., Int. Ed., 2016, 55, 12659 CrossRef PubMed.
- (a) A. M. Dumas, G. A. Molander and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 5683 CrossRef PubMed; (b) H. Noda, G. Erős and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 5611 CrossRef PubMed; (c) D. Mazunin, N. Broguiere, M. Zenobi-Wong and J. W. Bode, ACS Biomater. Sci. Eng., 2015, 1, 456 CrossRef; (d) A. O. Gálvez, C. P. Schaack, H. Noda and J. W. Bode, J. Am. Chem. Soc., 2017, 139, 1826 CrossRef PubMed; (e) C. J. White and J. W. Bode, ACS Cent. Sci., 2018, 4, 197 CrossRef PubMed.
- (a) H. Böhme and P. Plappert, Chem. Ber., 1975, 108, 2827 CrossRef; (b) H. Böhme and P. Plappert, Chem. Ber., 1975, 108, 3574 CrossRef; (c) N. Millot, C. Piazza, S. Aviolio and P. Knochel, Synthesis, 2000, 7, 941 CrossRef; (d) T. Murai, Y. Mutoh, Y. Ohta and M. Murakami, J. Am. Chem. Soc., 2004, 126, 5968 CrossRef PubMed; (e) C. Fischer and E. M. Carreira, Org. Lett., 2004, 6, 1497 CrossRef PubMed; (f) M. Suginome, L. Uehlin and M. Murakami, J. Am. Chem. Soc., 2004, 126, 13196 CrossRef PubMed; (g) M. Shimizu, H. Itou and M. Miura, J. Am. Chem. Soc., 2005, 127, 3296 CrossRef PubMed.
- A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333 CrossRef PubMed.
- (a) G. A. Molander and F. Beaumard, Org. Lett., 2011, 13, 1242 CrossRef PubMed; (b) J. Raushel, D. L. Sandrock, K. V. Josyula, D. Pakyz and G. A. Molander, J. Org. Chem., 2011, 76, 2762 CrossRef PubMed.
- M. El Khatib, R. A. M. Serafim and G. A. Molander, Angew. Chem., Int. Ed., 2016, 55, 254 CrossRef PubMed.
- (a) T. Ohmura, T. Awano and M. Suginome, Chem. Lett., 2009, 38, 664 CrossRef; (b) T. Ohmura, T. Awano and M. Suginome, J. Am. Chem. Soc., 2010, 132, 13191 CrossRef PubMed; (c) T. Awano, T. Ohmura and M. Suginome, J. Am. Chem. Soc., 2011, 133, 20738 CrossRef PubMed.
- (a) G. A. Molander, L. N. Cavalcanti, B. Canturk, P. S. Pan and L. E. Kennedy, J. Org. Chem., 2009, 74, 7364 CrossRef PubMed; (b) S. R. Inglis, E. C. Y. Woon, A. L. Thompson and C. J. Schofield, J. Org. Chem., 2010, 75, 468 CrossRef PubMed; (c) D. W. Blevins, M. L. Yao, L. Yong and G. W. Kabalka, Tetrahedron Lett., 2011, 52, 6534 CrossRef; (d) G. W. Kabalka and V. Coltuclu, Tetrahedron Lett., 2009, 50, 6271 CrossRef; (e) A. K. L. Yuen and C. A. Hutton, Tetrahedron Lett., 2005, 46, 7899 CrossRef.
- (a) D. S. Matteson and G. Y. Kim, Org. Lett., 2002, 4, 2153 CrossRef PubMed; (b) B. J. Kim and D. S. Matteson, Angew. Chem., Int. Ed., 2004, 43, 3056 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures, characterisation data, and spectra. CCDC 1833580–1833584. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01486h |
| ‡ T. S. and A. S. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |