
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOncosis-inducing cyclometalated iridium(III) complexes†
Ruilin
Guan
a,
Yu
Chen
a,
Leli
Zeng
 ab,
Thomas W.
Rees
a,
Chengzhi
Jin
a,
Juanjuan
Huang
a,
Zhe-Sheng
Chen
b,
Liangnian
Ji
a and
Hui
Chao
*ac
ab,
Thomas W.
Rees
a,
Chengzhi
Jin
a,
Juanjuan
Huang
a,
Zhe-Sheng
Chen
b,
Liangnian
Ji
a and
Hui
Chao
*ac
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-Sen University, Guangzhou, 510275, P. R. China. E-mail: ceschh@mail.sysu.edu.cn
bCollege of Pharmacy and Health Sciences, St. John's University, New York, NY 11439, USA
cMOE Key Laboratory of Theoretical Organic Chemistry and Functional Molecule, School of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Xiangtan, 400201, P. R. China
First published on 3rd May 2018
Abstract
Oncosis is a non-apoptotic form of programmed cell death (PCD), which differs from apoptosis in both morphological changes and inner pathways, and might hold the key to defeating a major obstacle in cancer therapy – drug-resistance, which is often a result of the intrinsic apoptosis resistance of tumours. However, despite the fact that the term “oncosis” was coined and used much earlier than apoptosis, little effort has been made to discover new drugs which can initiate this form of cell death, in comparison to drugs inducing apoptosis or any other type of PCD. So herein, we present the synthesis of a series of mitochondria-targeting cyclometalated Ir(III) complexes, which activated the oncosis-specific protein porimin and calpain in cisplatin-resistant cell line A549R, and determined their cytotoxicity against a wide range of drug-resistant cancer types. To the best of our knowledge, these complexes are the very first metallo-components to induce oncosis in drug-resistant cancer cells.
Introduction
Ever since cancer was identified as one of the leading causes of human death, scientists and researchers worldwide have been motivated by the urgency to find a cure for this disease. As the first generation of anticancer drugs, platinum complexes such as cisplatin, oxaliplatin, and carboplatin are unfortunately limited by their severe side effects and, most importantly, inherent or acquired resistance by their target cells.1–3 To overcome these limitations, a great deal of research has been done.One strategy has been to change the metal core of the complex. Different coordination modes or valence states of the metal core have been introduced, including Pt(IV) prodrugs, and half-sandwich and cyclometalated complexes.4–9 Lippard et al. developed the idea of Pt(IV) complexes as anticancer agent candidates, and have recently published a review of new platinum drugs and their design.7 Better selectivity towards cancer cells was also achieved by Guo et al., with the introduction of biotin or superparamagnetic iron oxide nanoparticles (MRI agents), in which a boost in cytotoxicity by glutathione was observed.8,9
A variety of heavy metal cores including iridium, ruthenium, osmium, titanium, copper, iron, gold, and other metal compounds have also been developed in this field, including some which have made it as far as clinical studies.10–19 Sadler et al. developed a series of Ru(II) complexes with a novel half-sandwich structure and achieved considerable anticancer activity.16 Using a different approach, Ma et al. designed Ir(III) complexes as inhibitors of bioactivity and bio-targets, such as H-Ras/Raf-1 protein–protein interactions and tumor necrosis factor-α.17–19
Some of the aforementioned groups adopted other approaches, and selective activation of drugs by light is a particularly appealing one.20–24 Sadler et al. reported precisely designed Pt(IV) prodrugs, which can sustain the bioactive reducing agent GSH and be photoactivated to Pt(II) by visible light.20,21 Using a similar strategy, Gasser et al. developed a Ru(II) complex, the irradiation of which caused the activation of a protective cage moiety to release the drug.22 In another example involving photoactivation, photodynamic therapy (PDT) was employed by Gasser et al. to design Ru(II) polypyridyl complexes23 and ruthenium–porphyrin conjugates24 as effective photosensitizers and anticancer agents. Although many clinical agents induce apoptosis in order to cause the death of cancer cells, other modes of cell death have also been employed, including necroptosis, autophagy and paraptosis.25–34 Using NADH as a cofactor, Sadler et al. developed a new strategy for anticancer drug design based on the catalytic properties of Ru(II) compounds, and found the new reductive stress mechanism of cell death.28,29 Lippard et al. synthesized two Re(V) oxo complexes, which induced programmed necrosis, also known as necroptosis, and a Pt(II) complex reported by Guo et al. was proven to induce autophagic cell death in human ovarian carcinoma cells.30,31 Gasser et al. discovered that a Ru(II) polypyridyl complex, a PDT agent they synthesized, can induce either apoptosis or paraptosis depending on the cell cycle phase the cells were in.32 New subcellular locations have also been introduced as new targets for therapeutics. Rhee et al. and Ang et al. presented Ir(III) PDT agents and Ru(II) complexes which were selectively localized in the endoplasmic reticulum (ER), and caused severe damage to cells.33,34
Apoptosis has been thought to be the main death pathway of PCD and it was not until recently that more pathways have been discovered and studied,35 and these modes of cell death include the perhaps underestimated oncosis. Since its discovery over a century ago, this potential strategy for the treatment of drug-resistant cancers has lain relatively forgotten.36,37 Derived from the Greek word “swelling”, oncosis is a unique mode of cell death with its characteristic whole cell swelling, accompanied by severe mitochondrial damage, cytoplasmic vacuolization, plasma membrane blebbing and cytoskeletal collapse.37,38 A surface receptor, porimin, and Ca ion-related protein calpain were reported as key elements in mediating oncotic cell death.39–41
Based on the abundant experience of our group in the design of metal complexes with specific subcellular localization, especially in lysosomes and mitochondria,42–46 and because of the fact that mitochondrial dysfunction plays a critical role at a very early stage of cell death, we decided to synthesize a series of mitochondria-targeting cyclometalated Ir(III) complexes. Benzothiazole has been reported to possess considerable anticancer properties, and much research has been carried out aimed at developing benzothiazole containing chemodrugs with high anticancer activity.47–52 Therefore, we report in this study a series of iridium(III) complexes with benzothiazole substituted ligands: [Ir(ppy)2(bbtb)]+ (OnIr1), [Ir(DFppy)2(bbtb)]+ (OnIr2), [Ir(2pq)2(bbtb)]+ (OnIr3), and [Ir(pbt)2(bbtb)]+ (OnIr4). The crystalline structure of OnIr3 was also obtained (Fig. 1a and S1–S9†). We tested these complexes with more than ten cancer cell lines, ten drug-resistant cell lines and two normal cell lines, in which considerable cytotoxicity was exhibited as well as good selectivity towards cancer cells and normal cells. These complexes displayed mitochondria-targeting properties and further investigation suggested a relatively unusual death mode as oncosis was activated by OnIr1–OnIr4 in drug-resistant cell line A549R.
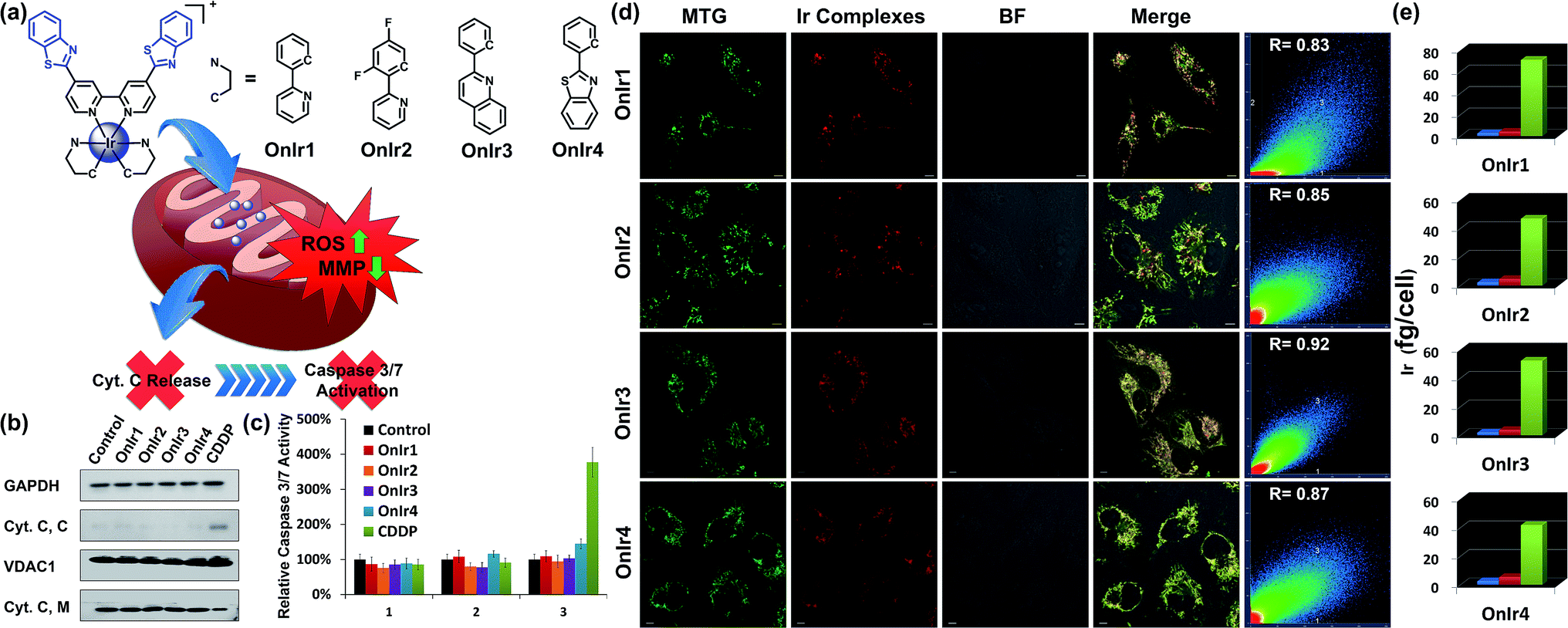 | ||
| Fig. 1 (a) Chemical structures of Ir(III) complexes. (b) Cyt. C content in the cytosol (Cyt. C, C) and mitochondria (Cyt. C, M). A549R cells were incubated with OnIr1 (2 μM), OnIr2 (2 μM), OnIr3 (3 μM), OnIr4 (3 μM), and cisplatin (150 μM) for 24 h and then separated into two cellular portions. (c) Caspase 3/7 activity assay. (d) Confocal images of co-localization assay and (e) ICP-MS results of Ir distribution in the cytosol (blue bar), nucleus (red bar) and mitochondria (green bar). A549R cells were incubated with Ir(III) complexes (2 μM, 8 h) respectively, and then incubated with MTG for 0.5 h after being washed with PBS. Scale bar in (d): 10 μm. | ||
Results and discussion
Subcellular localization
The subcellular localization of drugs determines the initial interactions between cells and drugs, the study of which, fortunately, is facilitated by the good photophysical properties of the Ir(III) complexes (Fig. S10†). We used A549R, a cisplatin-resistant cell line, as a model, to study the behavior of these complexes. The specific cellular target of the complexes was confirmed by a co-localization assay and an ICP-MS assay. As shown in Fig. 1d, the signal of the commercial mitochondrial dye MitoTracker® Green (MTG) correlated well with those of the complexes, giving correlation coefficients between 0.83 and 0.92. In the ICP-MS assay, a majority of the Ir(III) complexes were shown to be localized in the mitochondria (Fig. 1e). Both of the assays confirm that the cellular targets of the Ir(III) complexes are mitochondria. We also confirmed that the mechanism of cellular uptake of OnIr1–OnIr4 was endocytosis, as shown in Fig. S11.† The stability of OnIr1–OnIr4 in FBS and culture media is also shown in Fig. S12 and S13.†Mitochondrial morphological alteration
Subsequently, we focused on the cellular target of our complexes, mitochondria, the abnormal changes of which would cause severe failure in normal physiological activities. Residing in the mitochondrial inner membrane (MIM), ChChd3 is an abundant protein and essential for maintaining the mitochondrial cristae structure. The loss and reduction of ChChd3 reflects an injury in the MIM, which might result in a breakdown in mitochondrial function and cristae architecture.53 A marked decrease in ChChd3 content in a time-dependent manner in the treated cells suggested the occurrence of a traumatic alteration in mitochondria, and all four Ir(III) complexes displayed similar results (Fig. 2c). Transmission electron microscopy (TEM) experiments were than carried out for the observation of the mitochondrial structure. In the TEM images of untreated A549R cells, the double-membrane structure of the mitochondrion was observed and the mitochondrial cristae were obvious and complex. After treatment with OnIr1, the mitochondrial cristae became smoother, and the entire mitochondrion was dilated and swollen (Fig. 2a), indicating more severe damage and a major loss of mitochondrial membrane potential.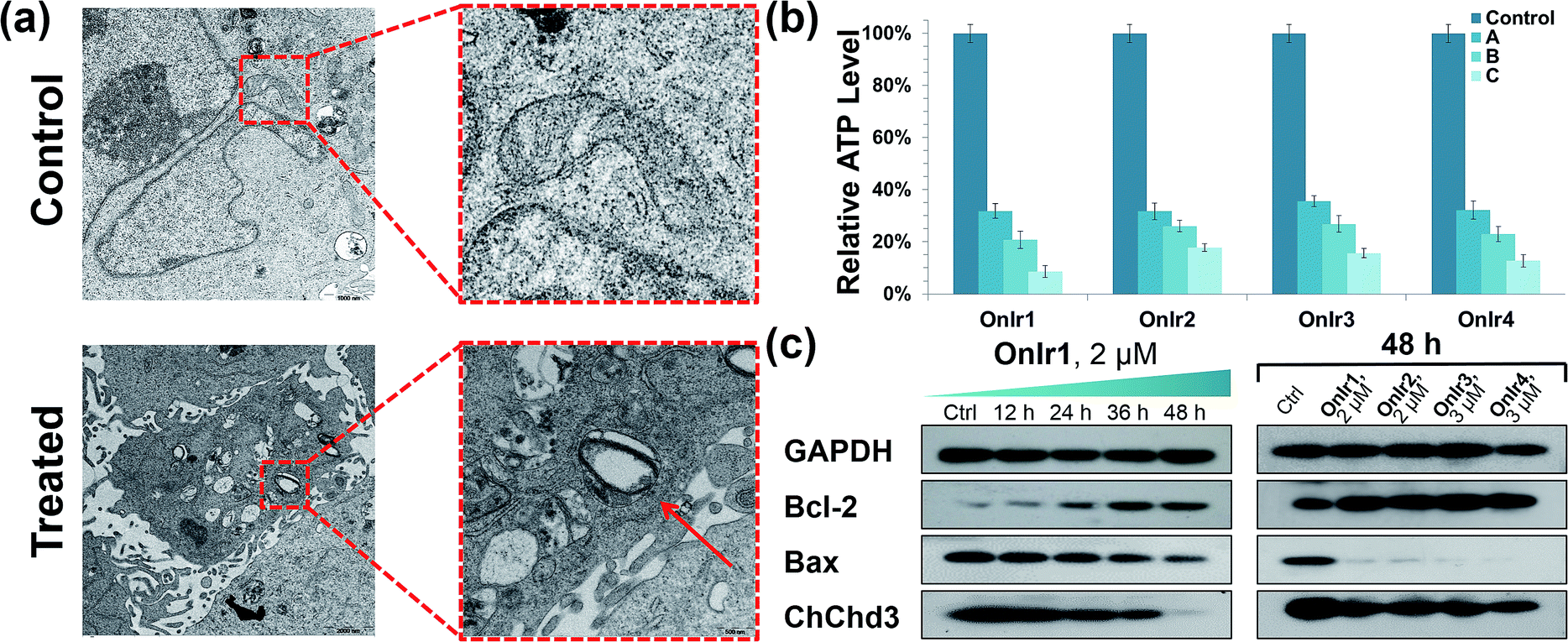 | ||
| Fig. 2 (a) TEM image of swollen mitochondria. The cells were treated with OnIr1 (1 μM, 24 h). (b) ATP deletion assay for A549R cells incubated with OnIr1–OnIr4 for 48 h. Incubation with OnIr1 and OnIr2 was performed at a concentration of (A) 0.5 μM, (B) 1 μM, and (C) 2 μM; incubation with OnIr3 and OnIr4 was performed at a concentration of (A) 1 μM, (B) 2 μM, and (C) 3 μM. (c) Cellular content of bcl-2, bax and ChChd3 in A549R cells. | ||
Mitochondrial membrane potential (MMP) and overload of reactive oxygen species (ROS)
Since one of the main pathways in mitochondria to initiate cell death is the loss of mitochondrial membrane potential (MMP) induced by the overload of reactive oxygen species (ROS),54 flow cytometry with JC-1 and dichlorodihydrofluorescein diacetate (DCFH) was performed. The MMP loss of A549R cells would prevent the aggregation of the MMP-dependent mitochondrial dye JC-1, indicated by a fluorescence change from red to green, which is shown in Fig. 3a in a time-dependent manner, especially the rise in FITC signals. The cause of MMP loss was tested using DCFH, a ROS reactive reagent. In Fig. 3b, a significant increase in the DCFH signal indicated the over-generation of ROS, also in a time-dependent manner, consistent with the JC-1 results.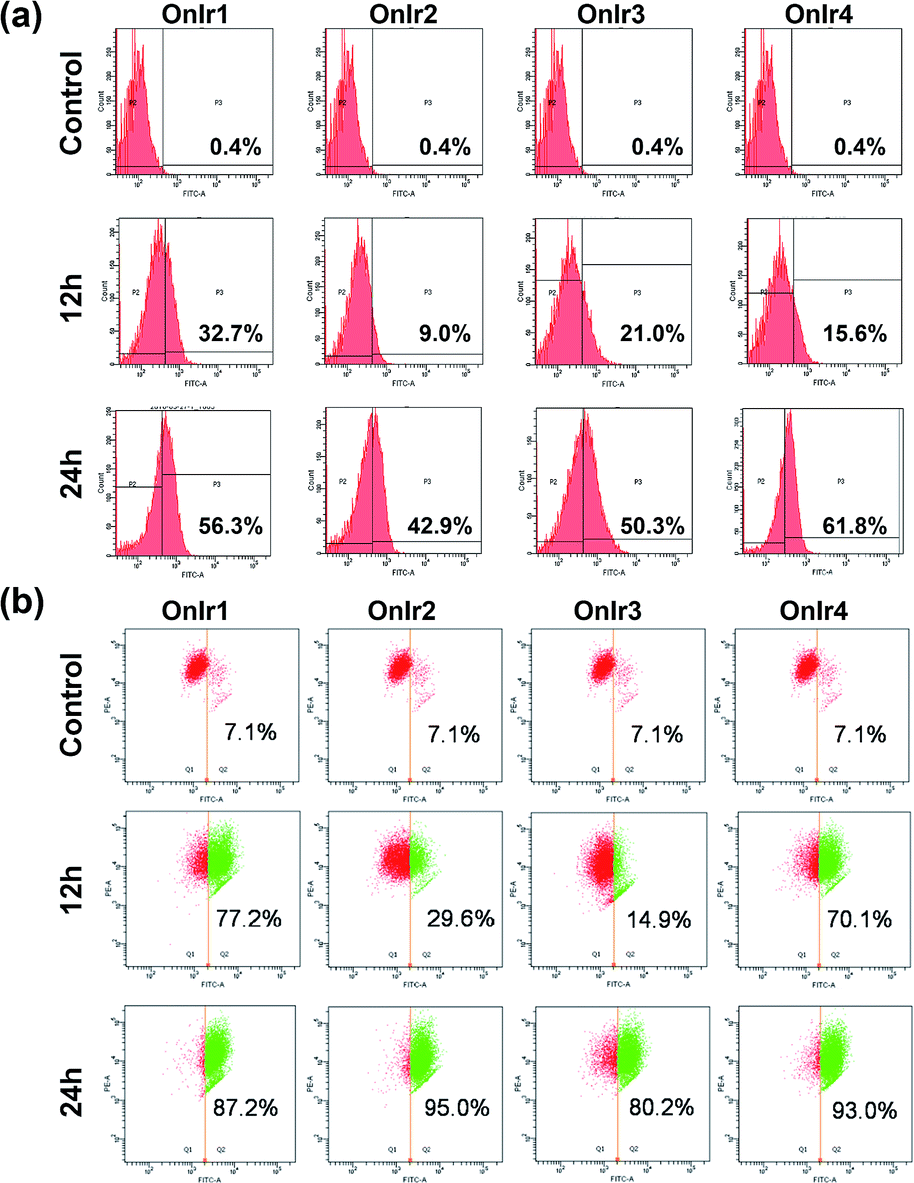 | ||
| Fig. 3 (a) Flow cytometry results of JC-1 assay and (b) ROS generation assay; A549R cells were incubated with OnIr1 and OnIr2 at a concentration of 1 μM, while incubation with OnIr3 and OnIr4 was performed at a concentration of 2 μM. | ||
ATP depletion and increase in the bcl-2/bax ratio
Mitochondria are the power plants of the cell, the dysfunction of which can lead to an issue called ATP depletion.55 As shown in Fig. 2b, the ATP level of cells after treatment with the Ir(III) complexes suffered a rapid decrease to less than 35% of that in the control cells.56 As a pair of death-related proteins, the sites of action of which are in mitochondria, bcl-2 and bax are also influenced by the Ir(III) complexes. Western blotting was applied to detect the expression level of these proteins. The content of bcl-2 increased in a time-dependent manner, while that of bax decreased, as shown in Fig. 2a. The ratio of bcl-2/bax increased; note that this pattern is the exact opposite of typical apoptosis, where the expression of bcl-2 would decrease while that of bax would increase, but consistent with a special death mode called oncosis.57–59 All four Ir(III) complexes exhibited the same tendency (Fig. 2c). This abnormal phenomenon implied a different death mode from apoptosis.Release of cytochrome C and caspase 3/7 activation
After ROS overload and MMP loss we would expect to observe the release of cytochrome C (Cyt. C) from the mitochondria to the cytosol and then the activation of the caspase family, especially caspase 3/7, which would eventually lead to cell death.60 In this study, the cytosol and mitochondria of A549R cells were separated to test their Cyt. C content by western blotting. To our surprise, there was no evidence of Cyt. C release to be observed in the A549R cells (Fig. 1b). In spite of the severe loss of MMP, the content of Cyt. C in the cytosol did not increase and that of the mitochondria did not decrease. The results of the whole-cell caspase 3/7 expression level assay agreed with these results, where no obvious increase in the caspase 3/7 level was observed even after incubation with the four Ir(III) complexes at a concentration of nearly 4 times their IC50 values for one day, while incubation with cisplatin (CDDP) caused an increased signal (Fig. 1c and S14a†). Western blotting was also performed and similar results were obtained (Fig. S16†). Caspase inhibitors Ac-DEVD-CHO and Z-VAD-fmk were co-incubated with the Ir(III) complexes, and no significant changes in cell viabilities were observed (Fig. S14b†). Therefore, combined with the results of the bcl-2/bax ratio, a different death mode from classic caspase-dependent apoptosis was suggested.Morphological alterations of the cells
To clarify which death mode occurred in the process, confocal laser scanning microscopy (CLSM) and TEM were applied to observe the alterations in the A549R cells. The most significant morphological alteration was the vacuolization in cytoplasm, which none of the known forms of apoptosis would cause. During the process, vacuoles generated from lysosomes filled up the entire cytoplasm (Fig. S15†),37 taking up most of the space (Fig. 4a), and showing the absence of organelles, as shown in Fig. 4b. As the incubation time increased, the whole cells were swollen and rounded, sometimes detaching from the substrate, and started to bleb (Fig. 4c). This helped us to rule out paraptosis as the mode of cell death,25 as neither swelling nor blebbing occurs in paraptosis. Notably, the bubbles around the cells were absolutely clear on the inside, suggesting that there was no process of budding, a typical action of apoptosis, taking place. Another thing worth mentioning is that there was also no sign of interruptions in plasmalemmal continuity in both CLSM and TEM micrographs, a typical feature of necrosis and necroptosis.61 The results of co-incubation of autophagy inhibitor 3-methyladenine, paraptosis inhibitor cycloheximide, lysosomal protease-mediated cell death inhibitor leupeptin and necrosis inhibitor necrostatin-1, and the Ir(III) complexes showed no increase in cell viability (Fig. S14c†).62,63 To have a deeper look into it, western blotting was introduced to check the level of LC3, activated during the process of autophagy,64,65 RIP3, activated during necrosis and necroptosis,66,67 and ERK/p-ERK, a marker of paraptosis.68 Again, no significant changes after treatment with OnIr1–OnIr4 were observed (Fig. S16†). All of the evidence thus far pointed toward oncosis, and so we decided to investigate oncosis more comprehensively.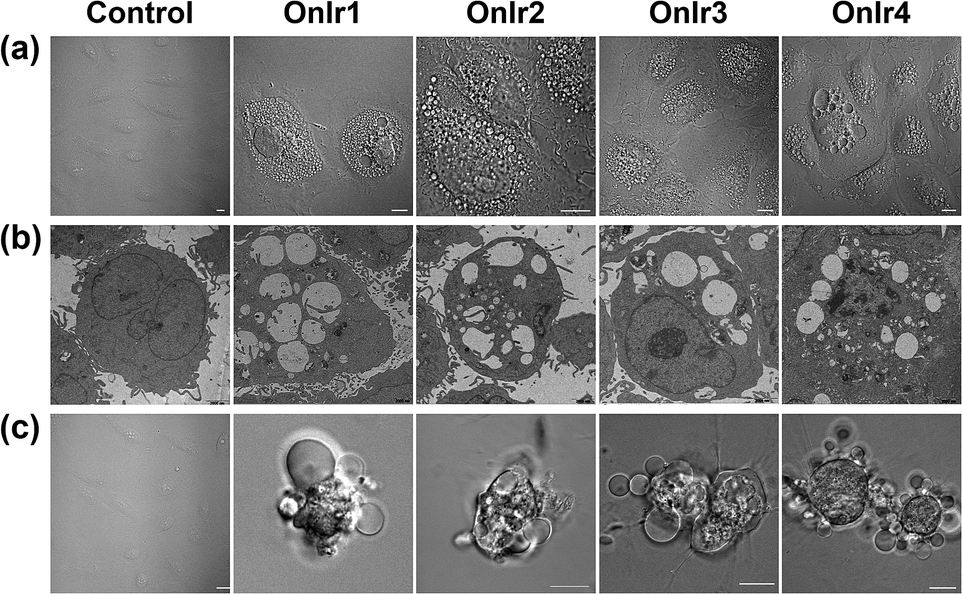 | ||
| Fig. 4 (a) Confocal images and (b) TEM images of cytoplasmic vacuolization. A549R cells were incubated with OnIr1 (1 μM), OnIr2 (1 μM), OnIr3 (2 μM) and OnIr4 (2 μM) for 24 h. (c) Confocal images of plasma membrane blebbing. A549R cells were incubated with OnIr1 (2 μM), OnIr2 (2 μM), OnIr3 (3 μM) and OnIr4 (3 μM) for 24 h. Scale bar in confocal images: 10 μm. | ||
Cytoskeleton and calpain 1 activation
The change of the shape and volume of the whole cell also implied a profound impact on the cytoskeleton. Both actin and tubulin were altered during this phase.61,69–71 By the use of western blotting, a marked decrease of the basic elements of the cytoskeleton, β-actin and α-tubulin, appeared in a time-dependent manner,72 suggesting a possible breakdown and collapse of the cytoskeleton (Fig. 5c). Further research by CLSM and staining with commercial dyes Actin-Tracker Green and Tubulin-Tracker Red allowed the actual visualization of the alterations of actin and tubulin. As shown in Fig. 5a and S17,† control cells have prominent thick actin cables across the whole cells.69 After incubation with 0.5 μM OnIr1, a punctate pattern of actin appeared, and fewer actin cables were observed in many of the cells. Upon increasing the concentration of OnIr1 to 1 μM, a lower density of actin fibers was observed as well as fragmentation of the actin filament. We could also see the interruption of actin fibers by the vacuoles (red arrowhead). After incubation with up to 2 μM OnIr1, the pattern of actin cables effectively disappeared, leaving behind the punctate actin, sometimes only some fragments (Fig. S18†), and somehow those fragments managed to slip out of the cell membrane, probably through the bursting of the bubbles,70 around which a band of fluorescent stain was observed.69 The stain of Actin-Tracker Green faded along with the increase of incubation concentration. Meanwhile, a network of microtubules radiated from the perinuclear centers in control cells,71 and the cells underwent mitosis metaphase as shown in Fig. 5a and S19† with Tubulin-Tracker Red staining. After incubation with 0.5 μM OnIr1, the pattern of the microtubules was less rigid. Upon increasing the concentration of OnIr1 to 1 μM, the loss in length and number of microtubules began and the pattern was vague. A significant interruption of microtubules by the vacuoles (yellow arrowhead) appeared. After further increasing the incubation concentration of OnIr1 up to 2 μM, the shape of the microtubules became completely loose, and only the carcasses remained both in the cells and the big blebs. Similar to that of Actin-Tracker Green, the stain of Tubulin-Tracker Red decreased with an increase in the incubation concentration of OnIr1, and was completely lost in some of the cells (Fig. S20†). These images showed the collapse of the cytoskeleton vividly, and that the breakdown of it could be induced by these Ir(III) complexes in a dose-dependent manner.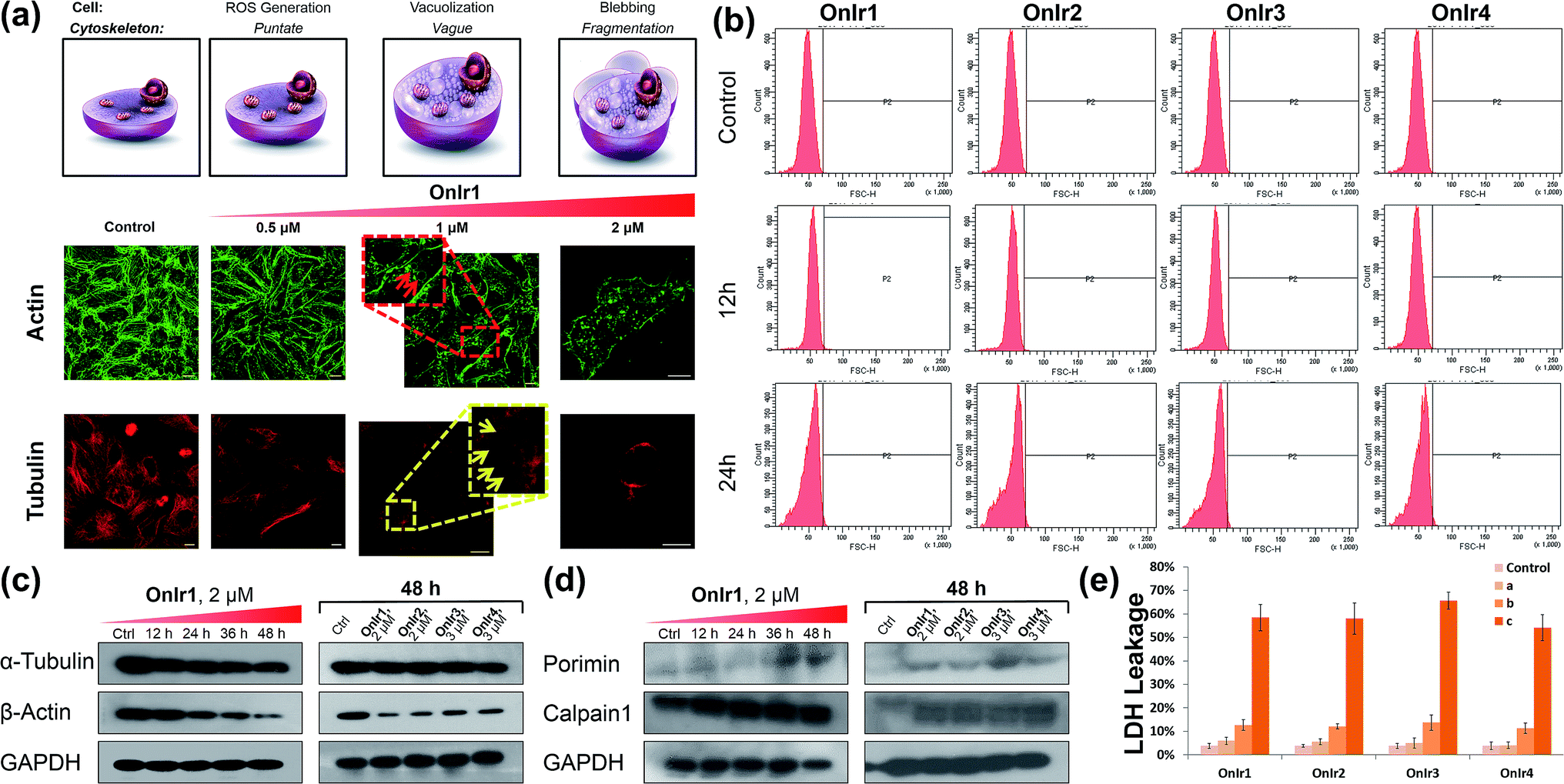 | ||
| Fig. 5 (a) Actin-Tracker Green and (b) Tubulin-Tracker Red staining of fixed A549R cells pre-treated with OnIr1 for 24 h in a dose-dependent manner. Confocal images of A549R cells treated with OnIr1 for 24 h. (b) Flow cytometry results of cell volume changes. A549R cells were incubated with OnIr1 (1 μM), OnIr2 (1 μM), OnIr3 (2 μM) and OnIr4 (2 μM). Cellular content of (c) α-tubulin and β-actin and (d) porimin and calpain 1. (e) LDH leakage assay for A549R cells incubated with OnIr1–OnIr4 for 24 h. Incubation with OnIr1 and OnIr2 was performed at a concentration of (A) 0.5 μM, (B) 1 μM, and (C) 2 μM; incubation with OnIr3 and OnIr4 was performed at a concentration of (A) 1 μM, (B) 2 μM, and (C) 3 μM. Scale bar in (a): 10 μM. | ||
Growing evidence indicates that calpains, a calcium-activated protease family, are involved in processes from mitochondrial dysfunction to cytoskeletal collapse. These neutral cysteine proteases were considered to be among the iconic proteins in the procedure of oncotic cell death, and their substrates include cytoskeletal proteins, the activation of which would cause cytoskeletal breakdown.41,72 Although the members of the calpain family were considered as cytoplasmic proteins, one of the calpains, calpain 1, was found to be not only rich in, but also navigated by its own structure towards, mitochondria.73 The results of western blotting in calpain 1 displayed its activation by the appearance of a new band below the other one after treatment with Ir(III) complexes, and specifically the treatment with OnIr1 gave rise to the new band in a time-dependent manner (Fig. 5d). The impact of calpain activation is profound in oncosis. There are numerous pieces of evidence suggesting that the activation of calpain leads to mitochondrial dysfunction and the breakdown of not only the cytoskeleton but also the plasma membrane.41
Plasma membrane permeability and porimin expression
Since the symbolic phenomenon of oncosis is cell swelling, flow cytometry was employed to measure the enlargement of cell volumes. Apparently, limited by the plasma membrane, a cell cannot swell infinitely, so the peak should not shift entirely to a new volume section. As shown in Fig. 5b, instead of a shift, the shape of the peak representing cell volumes of A549R was changed and tended to a certain extremum, which is the maximum of the control cells, showing an increase in average cell volume. To elucidate how the cells were swollen, we measured the membrane permeability by lactate dehydrogenase (LDH) leakage assay. A dramatic increase of signals was observed (Fig. 5d), indicating a large leakage of LDH, and therefore an increase in membrane permeability in a majority of cells. The increase of cell membrane permeability is ascribed to the increase of cell volume.A surface receptor, porimin (pro-oncosis receptor inducing membrane injury), is supposed to be responsible for the abnormal membrane permeability, which directly causes the iconic whole cell swelling in the process of oncosis, and the activation of porimin is also a marker of oncotic cell death.39,40,74 In Fig. 5d, the increase in expression of porimin upon incubation with OnIr2–OnIr4, as well as OnIr1, is demonstrated in a time-dependent manner.
Cytotoxicity against drug-resistant cancer cells
Now that we had confirmed that the Ir(III) complexes induced a unique death mode, oncosis, which is quite the opposite of apoptosis in many ways, we tried to apply these complexes to fight against cancer cells, especially drug-resistant cancer cells. 11 cancer cell lines from various body parts including lungs, colon, kidneys and liver were tested, along with 10 drug-resistant cell lines and 2 normal cell lines (HL-7702, normal liver cells; HEK 293, normal kidney cells). All four Ir(III) complexes exhibited considerable cytotoxicity in normal cancer cell lines, drug-resistant cell lines and their parental cell lines (IC50 values down to ∼500 nM, Table S3†). The drugs causing resistance included cisplatin, another metal complex like our complexes; mitoxantrone, a type II topoisomerase inhibitor; doxorubicin, a multi-target drug and As2O3, a classic inorganic nonmetal drug. None of the resistances of these different types of drugs were sufficient for the cancer cells to prevent their death on exposure to the Ir(III) complexes. They also exhibited moderate cytotoxicity towards normal cell lines, but their selectivity towards cancer cells and normal cells can be as significant as ∼30 times the IC50 value (OnIr1, HL-7702/A549R).Discussion
From subcellular localization to cell membrane injury, we look into some major alterations the treatment with these Ir(III) complexes led to in the cells. The verification of the death mode caused by certain compounds is often delicate, especially for PCD modes like oncosis, because like any other death modes and pathways, certain phenomena or syndromes of oncosis might be shared with other modes and PCD modes are in a much less acknowledged situation compared with apoptosis. For example, blebbing in cellular membranes could occur both in necrosis and in oncosis; cytoplasmic vacuolization is also a significant feature in paraptosis, etc. Moreover, there are crosstalks between two death modes and that leads to concepts like necroptosis, oncotic apoptosis and even oncotic necrosis, to name a few. In other words, since the word “oncosis” originally means “swelling”, every cell death accompanied by whole cell swelling ought to be oncosis, perhaps a crosstalk such as “apoptotic oncosis”. When it comes to drug-resistant cancer cells, the situation gets even more complicated. As a matter of fact, after treatment with cisplatin, a classic apoptosis-inducing agent, A549R cells exhibited a sign of autophagy and necrosis with a slight increase in LC3II and RIP3, as shown in Fig. S16.† This situation was somewhat expected, since it was reported that high-dose cisplatin can induce autophagy and necrosis.75,76 However, when the pieces of evidence were put together, the fog began to clear, and some of the evidence has been mentioned in previous paragraphs, e.g. the morphological changes of the cells and the bcl-2/bax ratio. Although there is caspase-independent apoptosis which makes the whole caspase 3/7 detection and caspase inhibitor experiments seem less convincing for ruling out this common death mode,77–79 apoptosis per se often exhibits other features that are in no way related to what we observed in this paper. While cell shrinkage is a well-known consequence of apoptosis, the exact opposite cell swelling was observed, and cytoplasmic vacuolization is rare in the process of apoptosis. We also carefully checked other PCD modes either by their specific inhibitors or the detection of their characteristic protein expression and activation. Moreover, oncosis matched every single phenomenon perfectly, and most importantly, we demonstrated the activation of calpain 1, which is highly related to oncotic cell death in many aspects, and the expression of porimin, which is supposed to be the marker of oncosis.The process of oncosis induced by these complexes can be briefly outlined as follows: (1) the complexes entered the cells and localized in the mitochondria; (2) this caused the over-generation of ROS and (3) the loss of MMP, (4) followed by ATP depletion and an increased ratio of bcl-2/bax, leading to (5) the failure of the ionic pumps resulting in a high cellular membrane permeability and vacuolization from the lysosomes,37 (6) rounding and swelling of the cells and blebbing in the plasma membrane, during which (7) a collapse of the cytoskeleton occurred, and finally (8) the bubbles burst and the remainder would experience phagocytosis or inflammation, the very end of an oncotic cell.70
Conclusion
To conclude, we synthesized a series of Ir(III) complexes, OnIr1, OnIr2, OnIr3, and OnIr4, and found that they can target mitochondria and eventually proved that they can induce oncosis in drug-resistant cancer cell line A549R. Finally, we screened the anticancer activity of these oncosis-inducing complexes. They showed IC50 values indicating considerable cytotoxicity against various cancer cell lines and drug-resistant cell lines, with selectivity towards normal cells lines. These results suggest that the unique oncosis-inducing cyclometalated Ir(III) complexes can be potent candidates to fight against drug-resistant cancers.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21525105, 21471164, 21501201 and 21778079), the 973 Program (No. 2015CB856301), and the Fundamental Research Funds for the Central Universities.Notes and references
- V. Brabec and J. Kasparkova, Drug Resist. Updates, 2005, 8, 131–146 CrossRef PubMed.
- Z. H. Siddik, Oncogene, 2003, 22, 7265–7279 CrossRef PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef PubMed.
- T. Zou, J. Liu, C. T. Lum, C. Ma, R. C. Chan, C. N. Lok, W. M. Kwok and C. M. Che, Angew. Chem., Int. Ed., 2014, 53, 10119–10123 CrossRef PubMed.
- N. P. E. Barry and P. J. Sadler, Chem. Soc. Rev., 2012, 41, 3264–3279 RSC.
- Z. T. Cao, Z. Y. Chen, C. Y. Sun, H. J. Li, H. X. Wang, Q. Q. Cheng, Z. Q. Zuo, J. L. Wang, Y. Z. Liu, Y. C. Wang and J. Wang, Biomaterials, 2016, 94, 9–19 CrossRef PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef PubMed.
- Z. Z. Zhu, Z. H. Wang, Y. G. Hao, C. C. Zhu, Y. Jiao, H. C. Chen, Y. M. Wang, J. Yan, Z. J. Guo and X. Y. Wang, Chem. Sci., 2016, 7, 2864–2869 RSC.
- N. Muhammad, N. Sadia, C. Zhu, C. Luo, Z. Guo and X. Wang, Chem. Commun., 2017, 53, 9971–9974 RSC.
- D. L. Ma, H. Z. He, K. H. Leung, D. S. Chan and C. H. Leung, Angew. Chem., Int. Ed., 2013, 52, 7666–7682 CrossRef PubMed.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z. S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- R. J. Needham, C. Sanchez-Cano, X. Zhang, I. Romero-Canelon, A. Habtemariam, M. S. Cooper, L. Meszaros, G. J. Clarkson, P. J. Blower and P. J. Sadler, Angew. Chem., Int. Ed., 2017, 56, 1017–1020 CrossRef PubMed.
- Z. G. Yue, W. Wei, Z. X. You, Q. Z. Yang, H. Yue, Z. G. Su and G. H. Ma, Adv. Funct. Mater., 2011, 21, 3446–3453 CrossRef.
- R. W. Y. Sun and C. M. Che, Coord. Chem. Rev., 2009, 253, 1682–1691 CrossRef.
- Z. J. Guo and P. J. Sadler, Angew. Chem., Int. Ed., 1999, 38, 1512–1531 CrossRef PubMed.
- T. Sriskandakumar, H. Petzold, P. C. Bruijnincx, A. Habtemariam, P. J. Sadler and P. Kennepohl, J. Am. Chem. Soc., 2009, 131, 13355–13361 CrossRef PubMed.
- L. J. Liu, W. H. Wang, S. Y. Huang, Y. J. Hong, G. D. Li, S. Lin, J. L. Tian, Z. W. Cai, H. M. D. Wang, D. L. Ma and C. H. Leung, Chem. Sci., 2017, 8, 4756–4763 RSC.
- C. H. Leung, H. J. Zhong, H. Yang, Z. Cheng, D. S. Chan, V. P. Ma, R. Abagyan, C. Y. Wong and D. L. Ma, Angew. Chem., Int. Ed., 2012, 51, 9010–9014 CrossRef PubMed.
- T. S. Kang, Z. Mao, C. T. Ng, M. Wang, W. Wang, C. Wang, S. M. Lee, Y. Wang, C. H. Leung and D. L. Ma, J. Med. Chem., 2016, 59, 4026–4031 CrossRef PubMed.
- N. J. Farrer, J. A. Woods, L. Salassa, Y. Zhao, K. S. Robinson, G. Clarkson, F. S. Mackay and P. J. Sadler, Angew. Chem., Int. Ed., 2010, 49, 8905–8908 CrossRef PubMed.
- Y. Zhao, J. A. Woods, N. J. Farrer, K. S. Robinson, J. Pracharova, J. Kasparkova, O. Novakova, H. Li, L. Salassa, A. M. Pizarro, G. J. Clarkson, L. Song, V. Brabec and P. J. Sadler, Chem.–Eur. J., 2013, 19, 9578–9591 CrossRef PubMed.
- T. Joshi, V. Pierroz, C. Mari, L. Gemperle, S. Ferrari and G. Gasser, Angew. Chem., Int. Ed., 2014, 53, 2960–2963 CrossRef PubMed.
- A. Frei, R. Rubbiani, S. Tubafard, O. Blacque, P. Anstaett, A. Felgentrager, T. Maisch, L. Spiccia and G. Gasser, J. Med. Chem., 2014, 57, 7280–7292 CrossRef PubMed.
- C. Mari, V. Pierroz, R. Rubbiani, M. Patra, J. Hess, B. Spingler, L. Oehninger, J. Schur, I. Ott, L. Salassa, S. Ferrari and G. Gasser, Chem.–Eur. J., 2014, 20, 14421–14436 CrossRef PubMed.
- S. Sperandio, I. de Belle and D. E. Bredesen, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 14376–14381 CrossRef PubMed.
- R. R. Ye, C. P. Tan, Y. N. Lin, L. N. Ji and Z. W. Mao, Chem. Commun., 2015, 51, 8353–8356 RSC.
- K. N. Dalby, I. Tekedereli, G. Lopez-Berestein and B. Ozpolat, Autophagy, 2010, 6, 322–329 CrossRef PubMed.
- S. Betanzos-Lara, Z. Liu, A. Habtemariam, A. M. Pizarro, B. Qamar and P. J. Sadler, Angew. Chem., Int. Ed., 2012, 51, 3897–3900 CrossRef PubMed.
- J. J. Soldevila-Barreda, I. Romero-Canelon, A. Habtemariam and P. J. Sadler, Nat. Commun., 2015, 6, 6582 CrossRef PubMed.
- W. J. Guo, Y. M. Zhang, L. Zhang, B. Huang, F. F. Tao, W. Chen, Z. J. Guo, Q. Xu and Y. Sun, Autophagy, 2013, 9, 996–1008 CrossRef PubMed.
- K. Suntharalingam, S. G. Awuah, P. M. Bruno, T. C. Johnstone, F. Wang, W. Lin, Y. R. Zheng, J. E. Page, M. T. Hemann and S. J. Lippard, J. Am. Chem. Soc., 2015, 137, 2967–2974 CrossRef PubMed.
- V. Pierroz, R. Rubbiani, C. Gentili, M. Patra, C. Mari, G. Gasser and S. Ferrari, Chem. Sci., 2016, 7, 6115–6124 RSC.
- J. S. Nam, M. G. Kang, J. Kang, S. Y. Park, S. J. Lee, H. T. Kim, J. K. Seo, O. H. Kwon, M. H. Lim, H. W. Rhee and T. H. Kwon, J. Am. Chem. Soc., 2016, 138, 10968–10977 CrossRef PubMed.
- M. J. Chow, C. Licona, G. Pastorin, G. Mellitzer, W. H. Ang and C. Gaiddon, Chem. Sci., 2016, 7, 4117–4124 RSC.
- L. E. Broker, F. A. Kruyt and G. Giaccone, Clin. Cancer Res., 2005, 11, 3155–3162 CrossRef PubMed.
- P. Weerasinghe and L. M. Buja, Exp. Mol. Pathol., 2012, 93, 302–308 CrossRef PubMed.
- P. Weerasinghe, S. Hallock, R. E. Brown, D. S. Loose and L. M. Buja, Exp. Mol. Pathol., 2013, 94, 289–300 CrossRef PubMed.
- A. A. Peters, S. Y. N. Jamaludin, K. Yapa, S. Chalmers, A. P. Wiegmans, H. F. Lim, M. J. G. Milevskiy, I. Azimi, F. M. Davis, K. S. Northwood, E. Pera, D. L. Marcial, E. Dray, N. J. Waterhouse, P. J. Cabot, T. J. Gonda, P. A. Kenny, M. A. Brown, K. K. Khanna, S. J. Roberts-Thomson and G. R. Monteith, Oncogene, 2017, 36, 6490–6500 CrossRef PubMed.
- C. Zhang, Y. Xu, J. Gu and S. F. Schlossman, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6290–6295 CrossRef.
- F. R. Ma, C. H. Zhang, K. V. S. Prasad, G. J. Freeman and S. F. Schlossman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9778–9783 CrossRef PubMed.
- X. Liu, T. Van Vleet and R. G. Schnellmann, Annu. Rev. Pharmacol. Toxicol., 2004, 44, 349–370 CrossRef PubMed.
- K. Q. Qiu, H. Y. Huang, B. Y. Liu, Y. K. Liu, Z. Y. Huang, Y. Chen, L. N. A. Ji and H. Chao, ACS Appl. Mater. Interfaces, 2016, 8, 12702–12710 Search PubMed.
- H. Huang, B. Yu, P. Zhang, J. Huang, Y. Chen, G. Gasser, L. Ji and H. Chao, Angew. Chem., Int. Ed., 2015, 54, 14049–14052 CrossRef PubMed.
- G. Y. Li, Y. Chen, J. Q. Wang, Q. Lin, J. Zhao, L. N. Ji and H. Chao, Chem. Sci., 2013, 4, 4426–4433 RSC.
- J. Liu, Y. Chen, G. Li, P. Zhang, C. Jin, L. Zeng, L. Ji and H. Chao, Biomaterials, 2015, 56, 140–153 CrossRef PubMed.
- J. Liu, C. Jin, B. Yuan, Y. Chen, X. Liu, L. Ji and H. Chao, Chem. Commun., 2017, 53, 9878–9881 RSC.
- E. Brantley, V. Trapani, M. C. Alley, C. D. Hose, T. D. Bradshaw, M. F. Stevens, E. A. Sausville and S. F. Stinson, Drug Metab. Dispos., 2004, 32, 1392–1401 CrossRef PubMed.
- T. D. Bradshaw and A. D. Westwell, Curr. Med. Chem., 2004, 11, 1009–1021 CrossRef PubMed.
- H. K. Kim, M. K. Kang, K. H. Jung, S. H. Kang, Y. H. Kim, J. C. Jung, G. H. Lee, Y. Chang and T. J. Kim, J. Med. Chem., 2013, 56, 8104–8111 CrossRef PubMed.
- E. Kashiyama, I. Hutchinson, M. S. Chua, S. F. Stinson, L. R. Phillips, G. Kaur, E. A. Sausville, T. D. Bradshaw, A. D. Westwell and M. F. Stevens, J. Med. Chem., 1999, 42, 4172–4184 CrossRef PubMed.
- M. S. Chua, E. Kashiyama, T. D. Bradshaw, S. F. Stinson, E. Brantley, E. A. Sausville and M. F. Stevens, Cancer Res., 2000, 60, 5196–5203 Search PubMed.
- S. Yu, X. Gong and W. Chan, Macromolecules, 1998, 31, 5639–5646 CrossRef.
- M. Darshi, V. L. Mendiola, M. R. Mackey, A. N. Murphy, A. Koller, G. A. Perkins, M. H. Ellisman and S. S. Taylor, J. Biol. Chem., 2011, 286, 2918–2932 CrossRef PubMed.
- N. Zamzami, P. Marchetti, M. Castedo, D. Decaudin, A. Macho, T. Hirsch, S. A. Susin, P. X. Petit, B. Mignotte and G. Kroemer, J. Exp. Med., 1995, 182, 367–377 CrossRef PubMed.
- E. M. Mills, D. Xu, M. M. Fergusson, C. A. Combs, Y. Xu and T. Finkel, J. Biol. Chem., 2002, 277, 27385–27392 CrossRef PubMed.
- Q. Huang, R. Zhang, L. y. Zou, X. Cao and X. Chu, PLoS One, 2013, 8, e61345 Search PubMed.
- E. H. Y. Cheng, T. V. Sheiko, J. K. Fisher, W. J. Craigen and S. J. Korsmeyer, Science, 2003, 301, 513–517 CrossRef PubMed.
- S. Cory and J. M. Adams, Nat. Rev. Cancer, 2002, 2, 647–656 CrossRef PubMed.
- P. Weerasinghe, S. Hallock and A. Liepins, Exp. Mol. Pathol., 2001, 71, 89–98 CrossRef PubMed.
- L. M. Dejean, S. Martinez-Caballero and K. W. Kinnally, Cell Death Differ., 2006, 13, 1387–1395 CrossRef PubMed.
- B. F. Trump, I. K. Berezesky, S. H. Chang and P. C. Phelps, Toxicol. Pathol., 1997, 25, 82–88 CrossRef PubMed.
- B. Li, J. Zhao, C. Z. Wang, J. Searle, T. C. He, C. S. Yuan and W. Du, Cancer Lett., 2011, 301, 185–192 CrossRef PubMed.
- M. E. Guicciardi, M. Leist and G. J. Gores, Oncogene, 2004, 23, 2881–2890 CrossRef PubMed.
- N. Mizushima, T. Yoshimori and B. Levine, Cell, 2010, 140, 313–326 CrossRef PubMed.
- D. J. Klionsky, F. C. Abdalla, H. Abeliovich, R. T. Abraham, A. Acevedo-Arozena, K. Adeli, L. Agholme, M. Agnello, P. Agostinis, J. Zschocke and B. Zuckerbraun, et. al. , Autophagy, 2012, 8, 445–544 CrossRef PubMed.
- D. W. Zhang, J. Shao, J. Lin, N. Zhang, B. J. Lu, S. C. Lin, M. Q. Dong and J. Han, Science, 2009, 325, 332–336 CrossRef PubMed.
- P. Vandenabeele, L. Galluzzi, T. Vanden Berghe and G. Kroemer, Nat. Rev. Mol. Cell Biol., 2010, 11, 700–714 CrossRef PubMed.
- W. B. Wang, L. X. Feng, Q. X. Yue, W. Y. Wu, S. H. Guan, B. H. Jiang, M. Yang, X. Liu and D. A. Guo, J. Cell. Physiol., 2012, 227, 2196–2206 CrossRef PubMed.
- K. A. Elliget, P. C. Phelps and B. F. Trump, Cell Biol. Toxicol., 1991, 7, 263–280 Search PubMed.
- G. Majno and I. Joris, Am. J. Pathol., 1995, 146, 3–15 Search PubMed.
- P. C. Phelps, K. A. Elliget and B. F. Trump, FASEB J., 1996, 10, A1425 Search PubMed.
- G. Zhang, C. Jiang, Z. Wang, W. Chen, W. Gu and Y. Ding, BioMed Res. Int., 2014, 2014, 682197 Search PubMed.
- R. Badugu, M. Garcia, V. Bondada, A. Joshi and J. W. Geddes, J. Biol. Chem., 2008, 283, 3409–3417 CrossRef PubMed.
- L. F. Liu, Z. H. Qian, Q. Qin, M. Shi, H. Zhang, X. M. Tao and W. P. Zhu, Genet. Mol. Res., 2015, 14, 7481–7489 CrossRef PubMed.
- T. R. O'Donovan, G. C. O'Sullivan and S. L. McKenna, Autophagy, 2011, 7, 509–524 CrossRef.
- F. R. Khuri, J. Nemunaitis, I. Ganly, J. Arseneau, I. F. Tannock, L. Romel, M. Gore, J. Ironside, R. H. MacDougall, C. Heise, B. Randlev, A. M. Gillenwater, P. Bruso, S. B. Kaye, W. K. Hong and D. H. Kirn, Nat. Med., 2000, 6, 879–885 CrossRef PubMed.
- L. Ravagnan, S. Gurbuxani, S. A. Susin, C. Maisse, E. Daugas, N. Zamzami, T. Mak, M. Jaattela, J. M. Penninger, C. Garrido and G. Kroemer, Nat. Cell Biol., 2001, 3, 839–843 CrossRef PubMed.
- L. Y. Li, X. Luo and X. Wang, Nature, 2001, 412, 95–99 CrossRef PubMed.
- G. Kroemer, L. Galluzzi, P. Vandenabeele, J. Abrams, E. S. Alnemri, E. H. Baehrecke, M. V. Blagosklonny, W. S. El-Deiry, P. Golstein, D. R. Green, M. Hengartner, R. A. Knight, S. Kumar, S. A. Lipton, W. Malorni, G. Nunez, M. E. Peter, J. Tschopp, J. Yuan, M. Piacentini, B. Zhivotovsky and G. Melino, Cell Death Differ., 2009, 16, 3–11 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, NMR, ES-MS, photophysical properties, crystal structure and data, confocal images, cell viability and IC50 values. CCDC 1578910. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01142g |
| This journal is © The Royal Society of Chemistry 2018 |