
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering a bifunctional copper site in the cupredoxin fold by loop-directed mutagenesis†
Andrés
Espinoza-Cara
 ab,
Ulises
Zitare
c,
Damián
Alvarez-Paggi
cd,
Sebastián
Klinke
de,
Lisandro H.
Otero
de,
Daniel H.
Murgida
c and
Alejandro J.
Vila
*abe
ab,
Ulises
Zitare
c,
Damián
Alvarez-Paggi
cd,
Sebastián
Klinke
de,
Lisandro H.
Otero
de,
Daniel H.
Murgida
c and
Alejandro J.
Vila
*abe
aInstituto de Biología Molecular y Celular de Rosario (IBR, CONICET-UNR), Rosario, Argentina. E-mail: vila@ibr-conicet.gov.ar
bÁrea Biofísica, Departamento de Química Biológica, Facultad de Ciencias Bioquímicas y Farmacéuticas, Universidad Nacional de Rosario, Rosario, Argentina
cDepartamento de Química Inorgánica, Analítica y Química Física—INQUIMAE, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires—CONICET, Buenos Aires, Argentina
dFundación Instituto Leloir, IIBBA-CONICET, Buenos Aires, Argentina
ePlataforma Argentina de Biología Estructural y Metabolómica PLABEM., Buenos Aires, Argentina
First published on 28th June 2018
Abstract
Copper sites in proteins are designed to perform either electron transfer or redox catalysis. Type 1 and CuA sites are electron transfer hubs bound to a rigid protein fold that prevents binding of exogenous ligands and side reactions. Here we report the engineering of two Type 1 sites by loop-directed mutagenesis within a CuA scaffold with unique electronic structures and functional features. A copper–thioether axial bond shorter than the copper–thiolate bond is responsible for the electronic structure features, in contrast to all other natural or chimeric sites where the copper thiolate bond is short. These sites display highly unusual features, such as: (1) a high reduction potential despite a strong interaction with the axial ligand, which we attribute to changes in the hydrogen bond network and (2) the ability to bind exogenous ligands such as imidazole and azide. This strategy widens the possibility of using natural protein scaffolds with functional features not present in nature.
Introduction
Nature employs a few transition metal ions to fulfil a broad repertoire of chemically challenging biological functions.1 In particular, copper is essential for electron transfer and for catalysing redox reactions, being responsible for the chemical transformations from which living organisms draw energy, such as oxygen and nitrous oxide reduction.2–4 These functions are achieved by the unique coordination geometries and electronic structures elicited by the protein matrix and its fold around the copper site.5–7 Copper centres involved in intra- and inter-protein electron transfer include the mononuclear T1 and the binuclear CuA site, while catalytic redox centres are generally mononuclear T2 sites.3,7 The unique geometries of electron transfer sites give rise to typical spectroscopic features, readily identified by their colour. Type 1 (T1) centres can be blue (“normal T1”) or green (“perturbed T1”), and CuA sites are purple. In contrast, T2 centres show less intense colours.3Most T1 centres display a distorted tetrahedral geometry with a Cys-Met-His2 ligand set, characterized by a short, highly covalent Cu–S(Cys) bond and a uniquely long Cu–S(Met) axial bond.7,8 This ligand set is highly conserved in nature, but differences in the protein scaffold tune the relative lengths of these copper–sulphur bonds. In general, a shorter Cu–S(Met) bond results in weakening the Cu–S(Cys) bond, accompanied by a tetragonal distortion of the site, a mechanism known as the coupled distortion model.7,9 These geometries are embedded in a protein fold that imposes a long copper–thioether bond and a short copper–thiolate bond. This constraint is referred to as the rack/entatic state.9–11
T1 and CuA centres are bound to the cupredoxin fold, a β-barrel decorated with several loops connecting the different β-strands.4,12 All except one of the metal ligands are located in a loop between the last two C-terminal β-strands,13 while an additional His ligand is present in an adjacent β-strand. Loop-directed mutagenesis has been an extremely useful strategy to interrogate the effect of different structural elements on the function of these metal sites.4,13–16 The length and sequence of this loop (which changes among different proteins) have been shown to play a key role in tuning the reduction potential of these centres. In addition, the topology of the cupredoxin fold positions the copper sites close to the protein surface thus optimizing intermolecular electron transfer, at the same time preventing the binding of exogenous ligands that may elicit undesired side reactions.
Here we report the engineering15,16 of two chimeric proteins in which we have inserted the ligand loops of two T1 (blue) proteins into a cupredoxin scaffold naturally harbouring a binuclear CuA site. These chimeras do not reproduce the expected electronic structure based on the loop sequence, instead, they give rise to distorted (green) T1 sites. The CuA cupredoxin fold provides a rack that gives rise to unique copper sites, with the copper–thioether bond shorter than the copper–thiolate bond. Despite the strong interaction with the axial ligand, these centres display a high reduction potential. Both chimeric proteins exhibit low reorganization energy, thus being efficient electron transfer centres. Unexpectedly, at the same time they are able to bind imidazole as an exogenous ligand, eliciting a T2 copper site. These results show that the cupredoxin fold, despite its rigidity, can be engineered to design novel copper centres with new functionalities, expanding the chemical toolbox of natural proteins.
Results
Engineering, expression and functional characterization
Protein scaffolds can be utilized for harbouring copper sites constructed by de novo design,17–20 metal site redesign,21–24 directed evolution25,26 or combinations of these strategies. For example, CuA sites have been reconstructed into a cupredoxin scaffold of subunit II of a quinol oxidase,14 and in Type 1 proteins amicyanin27 and azurin,28 by loop-directed mutagenesis, i.e., replacing the copper-binding loop of the Type 1 centre by the corresponding loop in a cytochrome c oxidase. This strategy has led to binuclear copper centres with electronic structures similar to those of native CuA sites.4,29 One of these chimeric proteins, CuA-Az, is easy to purify, handle and crystalize and was extensively used as model of the CuA site. Nevertheless, differences between this model and CuA soluble protein truncates were observed. For example, the mutation of the axial ligand in CuA-Az exerts a smaller effect on the reduction potential than in natural truncates. This phenomenon was explained by the difference in the distance of the axial ligand between the two protein types, suggesting a different rack between T1 scaffolds and “purple” CuA scaffolds.30 In order to expand the understanding of how loops and scaffold interplay in the determination of the site structure and fine-tune reactivity we follow the reverse strategy employed for the CuA loop, i.e., the replacement of the C-terminal ligand-containing T1 loops of azurin and amicyanin into a cupredoxin scaffold harbouring a CuA site.The soluble domain of subunit II of the ba3 oxidase from Thermus thermophilus (TtCuA) was used as a stable scaffold for loop engineering30–33 since it is small and stable in solution retaining the structure observed in the whole oxidase. Through loop-directed mutagenesis the native loop present in TtCuA (CNQYCGLGHQNM) was replaced by the loops from wild-type T1 proteins amicyanin (CTPHPFM) and azurin (CTFPGHSALM). These two chimeras were expressed as stable proteins, with only one bound copper ion, confirming the success of this loop engineering strategy. The resulting proteins were named Ami-TtCuA and Az-TtCuA, respectively.
The reduction potentials (E0) of both chimeras were obtained from cyclic voltammetry (CV) experiments of protein solution samples, yielding 422 ± 5 mV and 487 ± 5 mV for Ami-TtCuA and Az-TtCuA, respectively (Fig. S1A†). Both values are much higher than the value of TtCuA (280 mV)34–37 or the related T1 proteins amicyanin (255 mV) and azurin (295 mV).
Electron transfer reorganization free energies (λ) were determined from the temperature-dependence of ET rate constants (k0ET) as obtained by protein film voltammetry. To this end, proteins were adsorbed on Au electrodes coated with self-assembled monolayers (SAMs) of 40% SH–(CH2)16–CH3/60% SH–(CH2)16–CH2OH mixtures, which are known to maximize the biocompatible adsorption of cupredoxins.38k0ET values were obtained applying Laviron's formalism to CVs recorded at variable scan rates from 50 to 400 mV s (ref. 39) (Fig. S1†). Charge transfer coefficients (α) were estimated from the FWHM of the CVs, yielding 0.51 ± 0.05 and 0.47 ± 0.05 for Ami-TtCuA and for Az-TtCuA, respectively (Fig. S1†). Under the usual approximation of negligibly small activation entropies40 (ΔG* = ΔH* = λ/4), the obtained λ values are 0.4 ± 0.1 eV and 0.3 ± 0.1 eV for Az-TtCuA and Ami-TtCuA, respectively (Fig. 1). These values are similar within experimental error to that of WT azurin (0.30 eV).41 Thus, loop engineering has a significant impact on the ET thermodynamics, while maintaining the capacity of the T1 site for performing a kinetically efficient ET reaction.
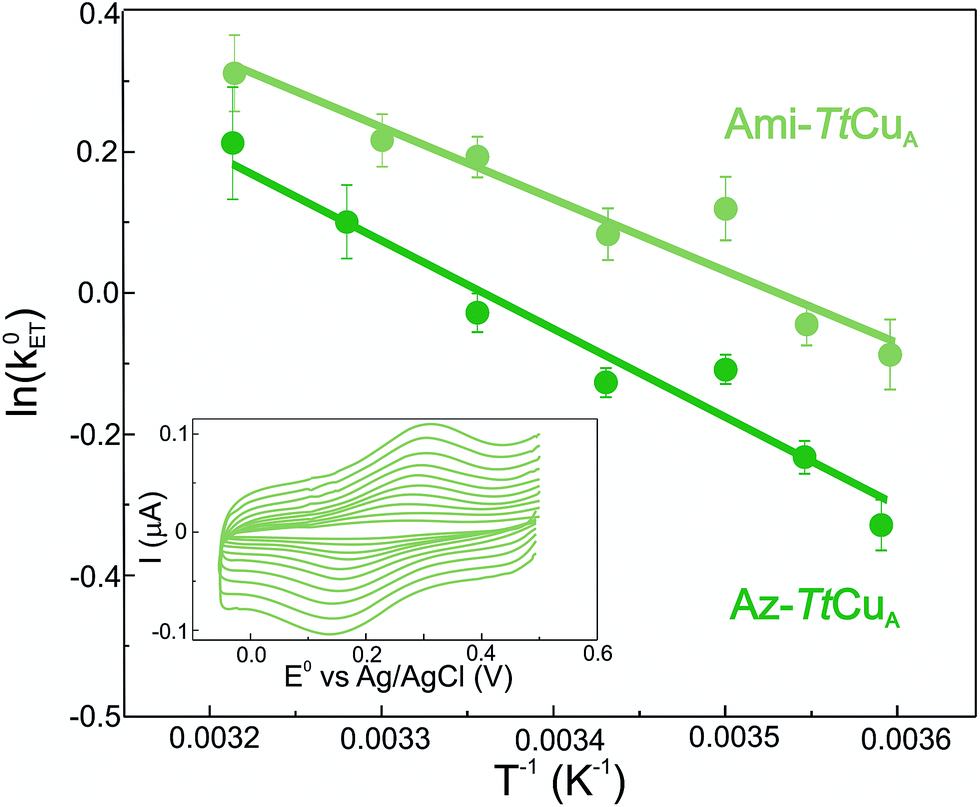 | ||
| Fig. 1 Arrhenius plots corresponding to Ami-TtCuA (light green) and Az-TtCuA (dark green) adsorbed on SAM-coated Au electrodes. Values of λ were obtained from the slopes by using ΔG* = λ/4. Inset: voltammograms of Ami-TtCuA adsorbed on SAM-coated Au electrodes, acquired at 25 °C at different scan rates from 50 to 400 mV s−1, from which kinetic parameters were extracted. | ||
Spectroscopic features of the chimeric proteins
Ami-TtCuA and Az-TtCuA are green proteins with distorted T1 sites, despite bearing ligand loops from normal T1 proteins. The absorption and CD spectra of Ami-TtCuA resemble those of Achromobacter cycloclastes nitrite reductase (AcNiR)11 while those of Az-TtCuA are similar to that of Acidithiobacillus ferrooxidans cytochrome c oxidase partner (AcoP)42 (Fig. 2). The absorption spectrum of Az-TtCuA is dominated by a broad envelope at 430 nm (23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 cm−1), while in Ami-TtCuA, two intense features at 405 (24700 cm−1) and 475 nm (21000 cm−1) are distinguished, responsible for the green colour. Both spectra show less intense absorption envelopes at 570 (17500 cm−1) and 750 nm (13300 cm−1). These spectra can be fit to seven Gaussian bands (Fig. 2a and Table S1†). The low energy region (10000–15000 cm−1) consists of three bands with lower intensity, attributable to metal-based ligand field (d → d) transitions. The positions of these bands are consistent with a tetragonal distortion with respect to the normal T1 site in plastocyanin. The high-energy, intense bands 1–4 (15000–30000 cm−1) can be assigned to ligand-to-metal charge-transfer (LMCT) transitions based on their intensity and by comparison with other T1 sites.9,11 The intensity ratio between the pseudo-σ- and π-SCys → Cu(3d(x2 − y2)) charge transfer transitions (bands 3 and 4, respectively) reflects the axial ligand field strength. Both Ami-TtCuA and Az-TtCuA display a σ/π ratio > 1, as observed for the perturbed T1 sites in NiR and cucumber basic protein (CBP), suggesting a stronger interaction with the axial ligand in both cases.
200 cm−1), while in Ami-TtCuA, two intense features at 405 (24700 cm−1) and 475 nm (21000 cm−1) are distinguished, responsible for the green colour. Both spectra show less intense absorption envelopes at 570 (17500 cm−1) and 750 nm (13300 cm−1). These spectra can be fit to seven Gaussian bands (Fig. 2a and Table S1†). The low energy region (10000–15000 cm−1) consists of three bands with lower intensity, attributable to metal-based ligand field (d → d) transitions. The positions of these bands are consistent with a tetragonal distortion with respect to the normal T1 site in plastocyanin. The high-energy, intense bands 1–4 (15000–30000 cm−1) can be assigned to ligand-to-metal charge-transfer (LMCT) transitions based on their intensity and by comparison with other T1 sites.9,11 The intensity ratio between the pseudo-σ- and π-SCys → Cu(3d(x2 − y2)) charge transfer transitions (bands 3 and 4, respectively) reflects the axial ligand field strength. Both Ami-TtCuA and Az-TtCuA display a σ/π ratio > 1, as observed for the perturbed T1 sites in NiR and cucumber basic protein (CBP), suggesting a stronger interaction with the axial ligand in both cases.
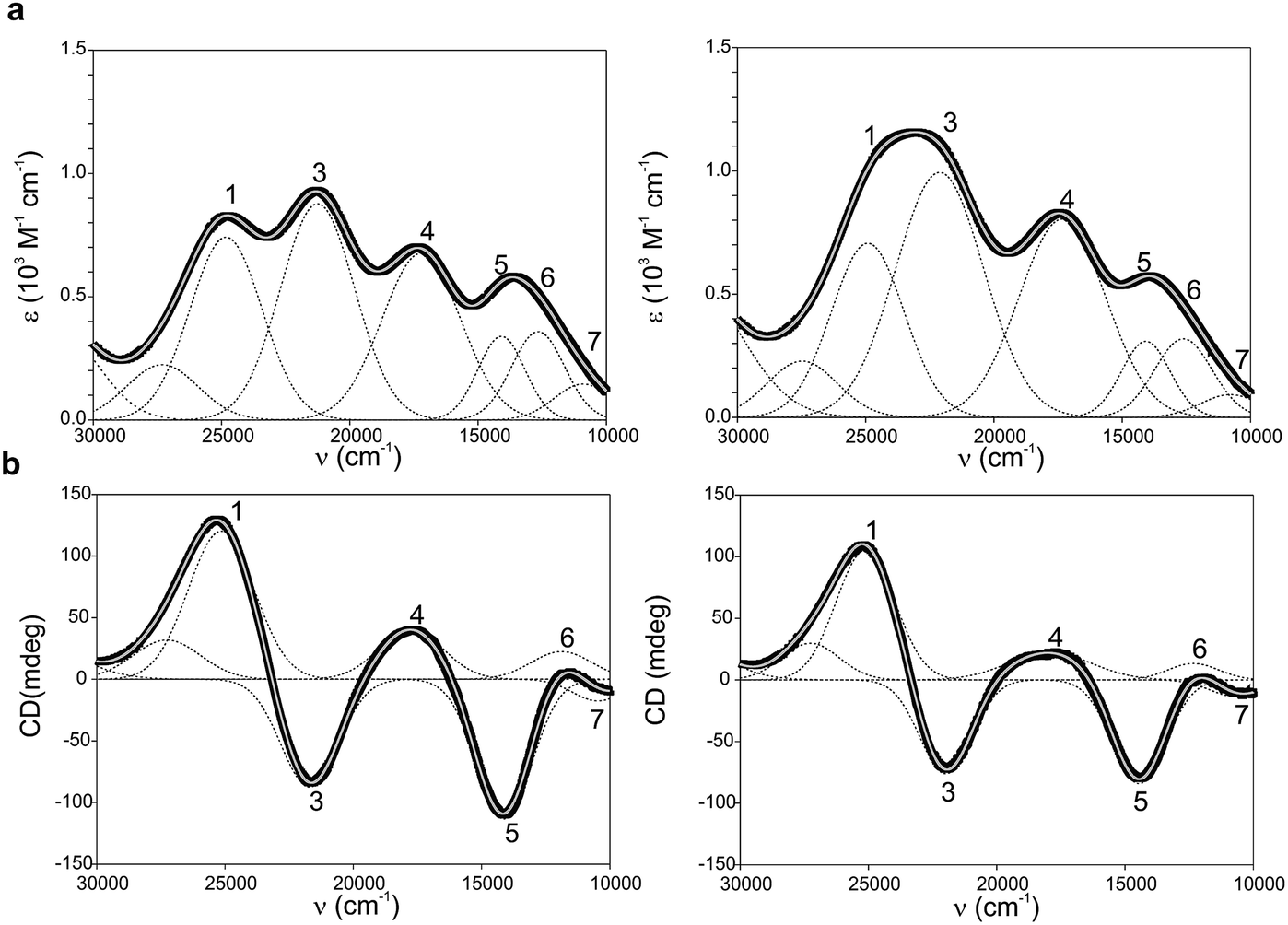 | ||
| Fig. 2 (a) Electronic absorption spectra of Ami-TtCuA (left) and Az-TtCuA (right). (b) Circular dichroism spectra of Ami-TtCuA and Az-TtCuA. The spectra were obtained in 100 mM Tris–HCl and 100 mM NaCl, pH 7.0 at 298 K. Gaussian resolution of bands (grey) in the absorption spectra is based on a simultaneous linear least-squares fit of the absorption and CD data for each protein, and the simulated spectra are shown in grey. The bands have been labelled 1–7 for both proteins; the numbering scheme is chosen to be consistent with the band assignment in plastocyanin (Pc), Cucumber Basic Protein (CBP), pseudoazurin (PAz) and Achromobacter cycloclastes nitrite reductase (AcNiR) (see Table S1† for assignments). Bands at higher energies than band 1 are not labelled as they have no numbered counterpart in the band assignment of the other perturbed T1 proteins. | ||
The X-band EPR spectra of Ami-TtCuA and Az-TtCuA are slightly rhombic, with smaller gz and larger Az values than T1 blue centres (Fig. 2b and Table S2†). The decrease in gz is indicative of an increased ligand field in the chimeric proteins compared to normal blue centres, close to values reported for green nitrite reductase.11 The rhombicity in Ami-TtCuA (Δg = 0.047) and Az-TtCuA (0.028) is in the range of those reported for the perturbed T1 sites in NiR (0.04) and CBP (0.041), consistent with the large absorbance of the pseudo-σ SCys → Cu(3d(x2 − y2)) charge transfer transition of these mutants and with a stronger axial ligand field.9,11
Resonance Raman (RR) spectra of T1 centers recorded under LMCT SCys–Cu excitation are dominated by fundamental vibrational bands in the 300–450 cm−1 spectral region, whose intensities reflect the contribution of Cu–SCys stretching to the normal mode composition (Fig. S2† and Table S3†). The average position of these bands weighted by their relative intensities is an empirical estimate of the Cu–SCys local oscillator stretching frequency 〈νCu–SCys〉.43 For canonical blue T1 sites this value is typically around 400 cm−1 (Table S4†).44 The RR spectrum of Ami-TtCuA exhibits a general downshift of all the fundamental bands in the Cu–SCys stretching region with respect to prototypical blue copper centers, with 〈νCu–SCys〉 = 378 cm−1. Such a downshift is indicative of a distorted T1 site with a weakened Cu–SCys bond.45,46 A similar downshift is observed for Az-TtCuA (〈νCu–SCys〉 = 379 cm−1), thus suggesting that both chimeras present distorted metal centres within the range reported for T1 sites.
The 1H NMR spectra of Az-TtCuA and Ami-TtCuA revealed hyperfine-shifted resonances (Fig. 3, Table S5†), with chemical shifts typical of T1 copper proteins. The signal line widths and their Curie-type temperature behaviour are indicative of an electron relaxation time of 10−10 s (typical of T1 sites and 10-fold longer than CuA centres).47,48 Assignments for Ami-TtCuA were achieved by recording spectra in D2O and by saturation transfer experiments (Fig. S3† and Table S5†), and resonances of Az-TtCuA were assigned by spectral comparison. Most chemical shifts resemble those previously reported for other T1 sites, except for the ε-CH3 of Met155, located at 37.7 ppm in Ami-TtCuA. Met ε-CH3 resonances have been identified only in distorted T1 sites, at 12.1 (PAz) and 8.1 ppm (Rc)49 (Table S6†). The current value can only be accounted for by a strong Fermi contact contribution to the observed shift, i.e., disclosing a net electron spin density on the axial Met ligand. Thus, the increased axial ligand field could be induced by a strong bonding interaction with this residue.
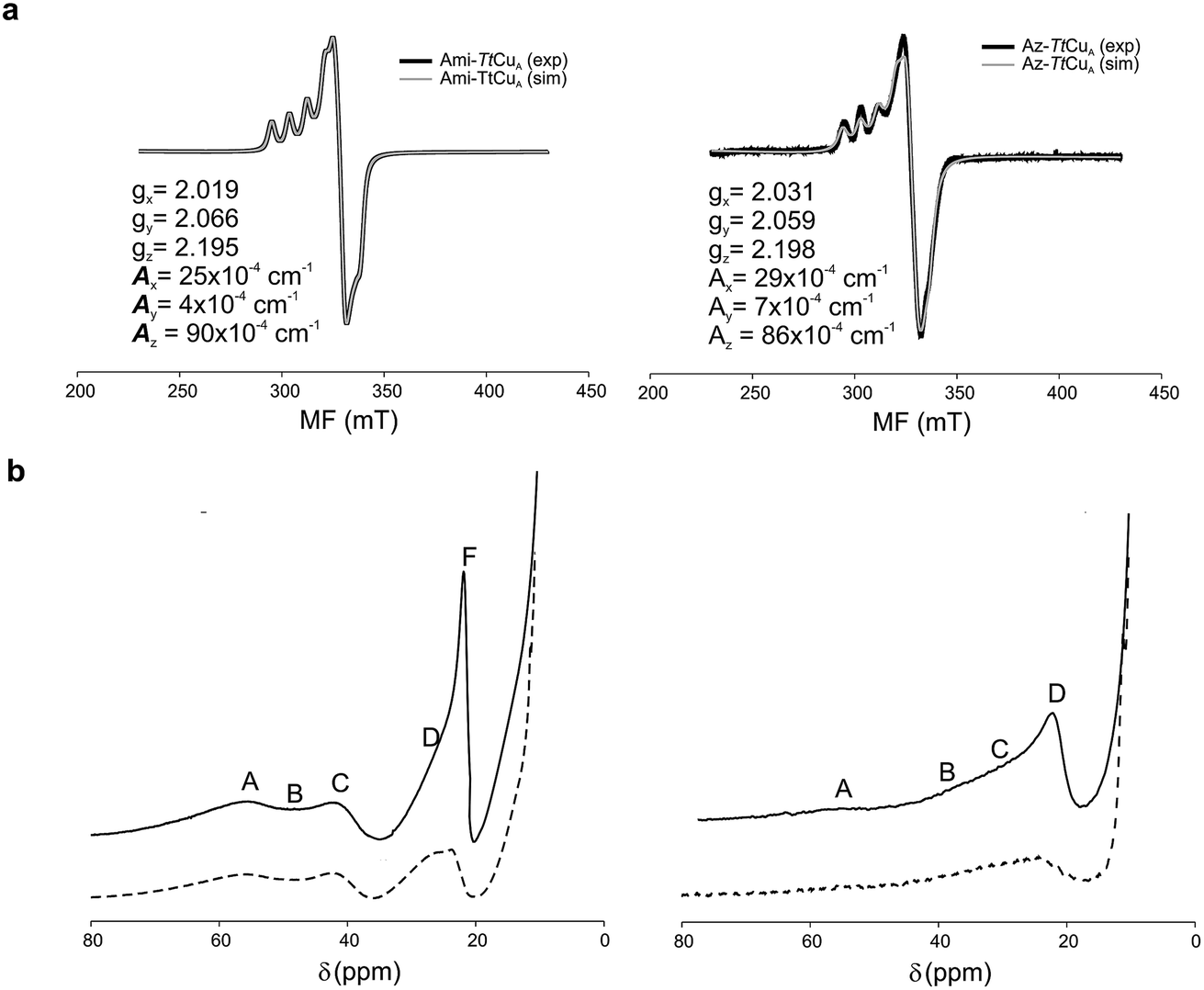 | ||
| Fig. 3 (a) X-band EPR data and simulations of Ami-TtCuA (left) and Az-TtCuA (right). The spectra were obtained in 100 mM potassium phosphate and 100 mM KCl at pH 7.0. The parameters used for the simulations are given in the bottom spectra, and the simulated spectra are shown in grey. (b) 1H NMR (600 MHz) spectra of Ami-TtCuA (left) and Az-TtCuA (right) were obtained in 100 mM potassium phosphate and 100 mM KCl at pH 7.0 at 298 K (full line) and in the same buffer in D2O at pD* 7.0 at 298 K (dotted line). The signals are labelled with capital letters, and their positions and tentative assignments are shown in Tables S5 and S6† respectively. | ||
The structure of Ami-TtCuA reveals an unprecedented copper centre
The crystal structure of Ami-TtCuA was solved in its oxidized form (Table S7†). It reveals that the β-barrel structure is preserved upon loop replacement (Fig. 4). Differences are confined to the replaced loop, which displays a backbone conformation identical to that observed in amicyanin (Fig. 4c).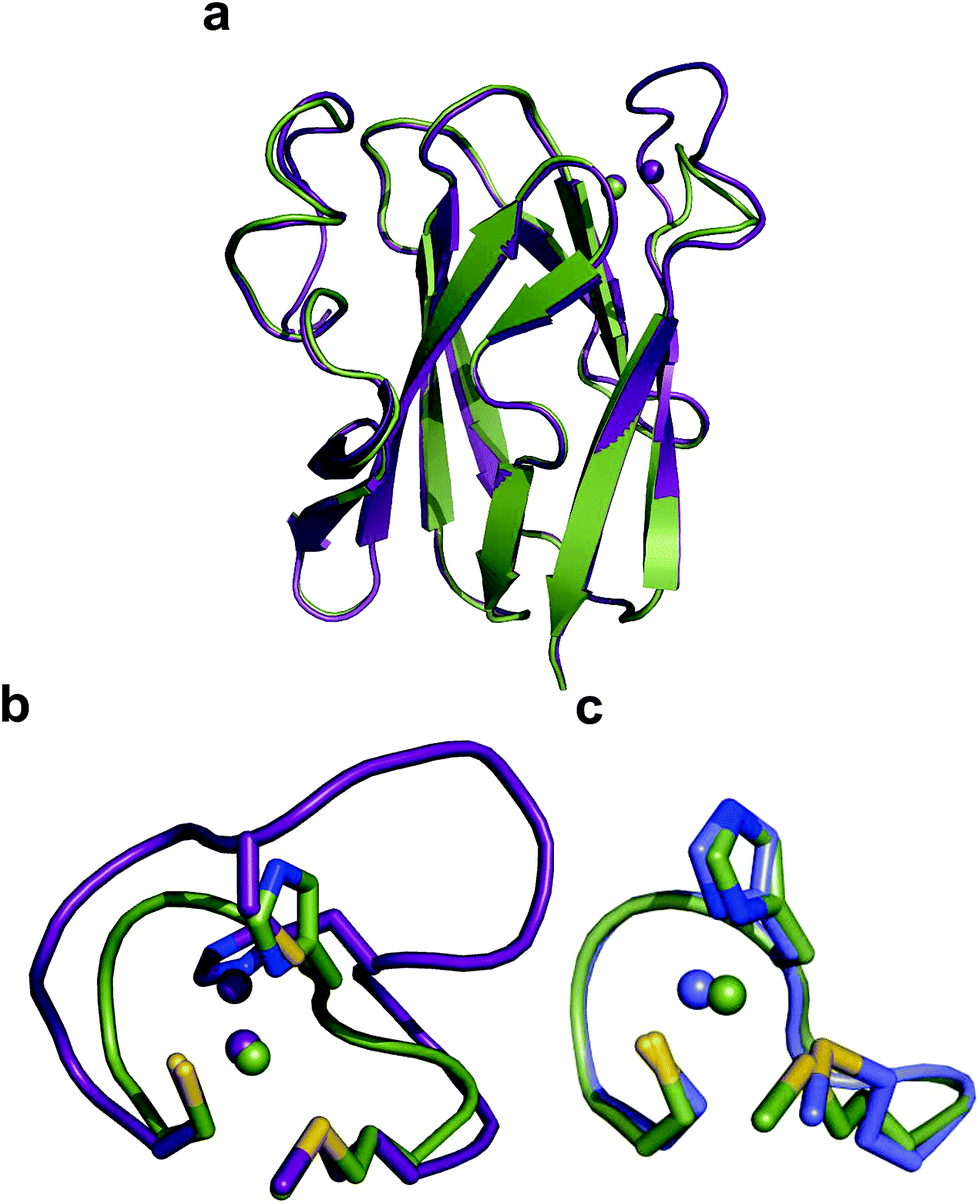 | ||
| Fig. 4 Structure of Ami-TtCuA. (a) Superposition of the structures of TtCuA (PDB code 2CUA, purple) and Ami-TtCuA (5U7N, green). The Cα RMSD between the two structures is 0.295 Å for 108 atoms. (b) Details of the superposition of the copper binding loops. (c) Overlay of the active sites of amicyanin (1AAC, blue) and Ami-TtCuA (5U7N, green). The side chains of the coordinating residues are shown as stick models, copper atoms as spheres, and the backbone of the C-terminal ligand-containing loops as secondary structure traces. | ||
The copper ion is bound to His114, Cys149, His152 and Met155, the canonical ligand set of a T1 centre. However, there are significant differences in the copper site geometry. First, the Cu–S bond with the axial Met has shortened to 2.35 Å, while the Cu–S bond with the Cys ligand is 2.41 Å. This situation contrasts the situation met in most T1 sites, in which the Cu–S Met bond is longer than that the Cu–S Cys bond (Fig. 5, S4 and Tables S8–S10†). Second, the position of the Cu(II) ion is significantly displaced from the plane defined by the equatorial His-Cys-His ligands with respect to other T1 copper proteins (Fig. 5b and c). These features give rise to an unprecedented copper site geometry that accounts for the unusual spectroscopic features.
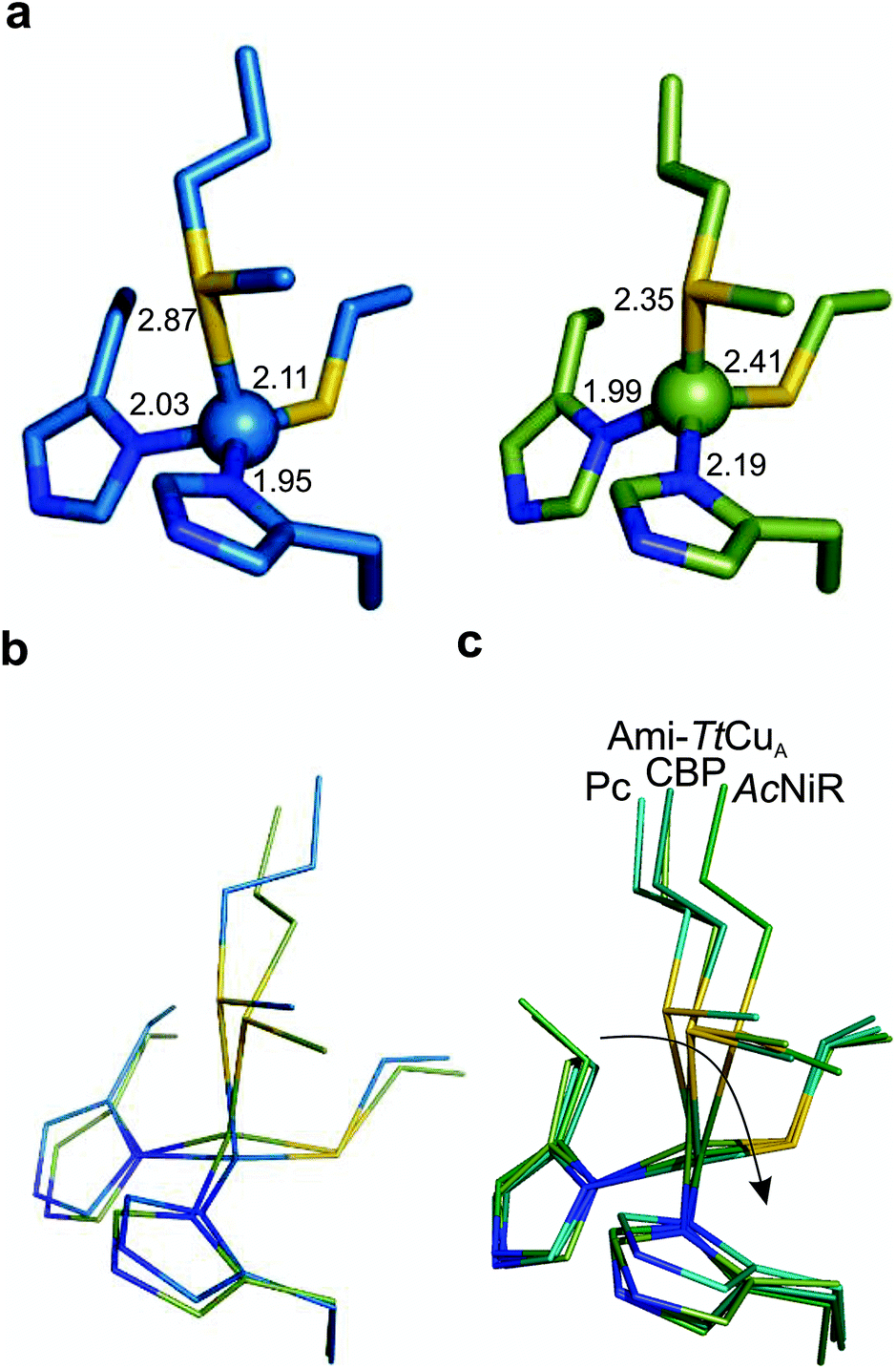 | ||
| Fig. 5 Structure of the Ami-TtCuA copper site. (a) Copper sites of amicyanin (1AAC, left) and Ami-TtCuA (5U7N, right). (b) Overlay of the Cu sites of amicyanin (1AAC, blue) and Ami-TtCuA (5U7N, green). Note that the Ami-TtCuA site adopts a slightly tetragonally distorted conformation with the copper atom displaced from the N–S–N plane. (c) Coupled distortion model exemplified by an overlay of the active sites of plastocyanin (1PLC), cucumber basic protein (2CBP), Ami-TtCuA (5U7N) and nitrite reductase (1NIF). The arrow indicates the direction of the distortion. | ||
The relative orientation of the sulphur-containing ligands Cys149 and Met155 (located at the base of the loop) is almost identical to that in TtCuA (Fig. 4b), while differing compared to amicyanin (Fig. 4c). The trans conformation of Met155 in Ami-TtCuA resembles that of Met160 in TtCuA rather than the gauche conformation of the axial Met ligand in amicyanin.50 As seen for other loop contraction mutants the β-barrel governs the orientation of these two ligands, instead of the loop sequence or length.51
The interaction of metal ligands with second sphere residues is also expected to be dictated by the β-barrel. Indeed, the sulphur atom of Cys149 retains a strong hydrogen bonding interaction with the backbone NH of Gly115, instead of the conserved S(Cys)–NH interaction with an Asn residue conserved in all T1 sites except in rusticyanin (Table S11†). This hydrogen bond is shortened from 3.50 to 3.25 Å in the chimeric protein. A second hydrogen bonding interaction with the backbone NH of His152 is created which is absent in TtCuA but mimics a similar interaction present in amicyanin. As a result, the chimeric protein presents a hydrogen bonding pattern which differs from those found in canonical T1 copper sites.
Active site accessibility
Addition of imidazole to both chimeric proteins gave rise to an orange colour, which was reversible upon dialysis against imidazole-free buffer (Fig. 6). A stepwise titration of imidazole to the two chimeric proteins allowed us to estimate binding constants of 2.91 ± 0.05 and 1.28 ± 0.05 mM for Ami-TtCuA and Az-TtCuA, respectively (Fig. S5†). The absorption and CD spectra of the imidazole adducts resemble those of the T2 copper site of nitrosocyanin. A Gaussian deconvolution of these spectra allows band assignment by comparison with nitrosocyanin52 (Fig. S6 and Table S12†). These proteins exhibit bands at 24500, 19500 and 14500 cm−1. The bands at 24500 and 19500 cm−1 can be assigned to Sthiolate(σ) → Cu(3d(x2 − y2)) and Sthiolate(π)→Cu(3d(x2 − y2)) ligand-to-metal charge transfer transitions, respectively. The σ/π ratio is larger than 1, as observed in T2 centres with thiolate ligands. The EPR spectra of these adducts display parameters similar to those of nitrosocyanin (Fig. 6b and Table S2†). The addition of azide elicited spectral changes similar to those found upon imidazole binding (Fig. S7†), while cyanide and thiocyanide led to the removal of the metal ion.
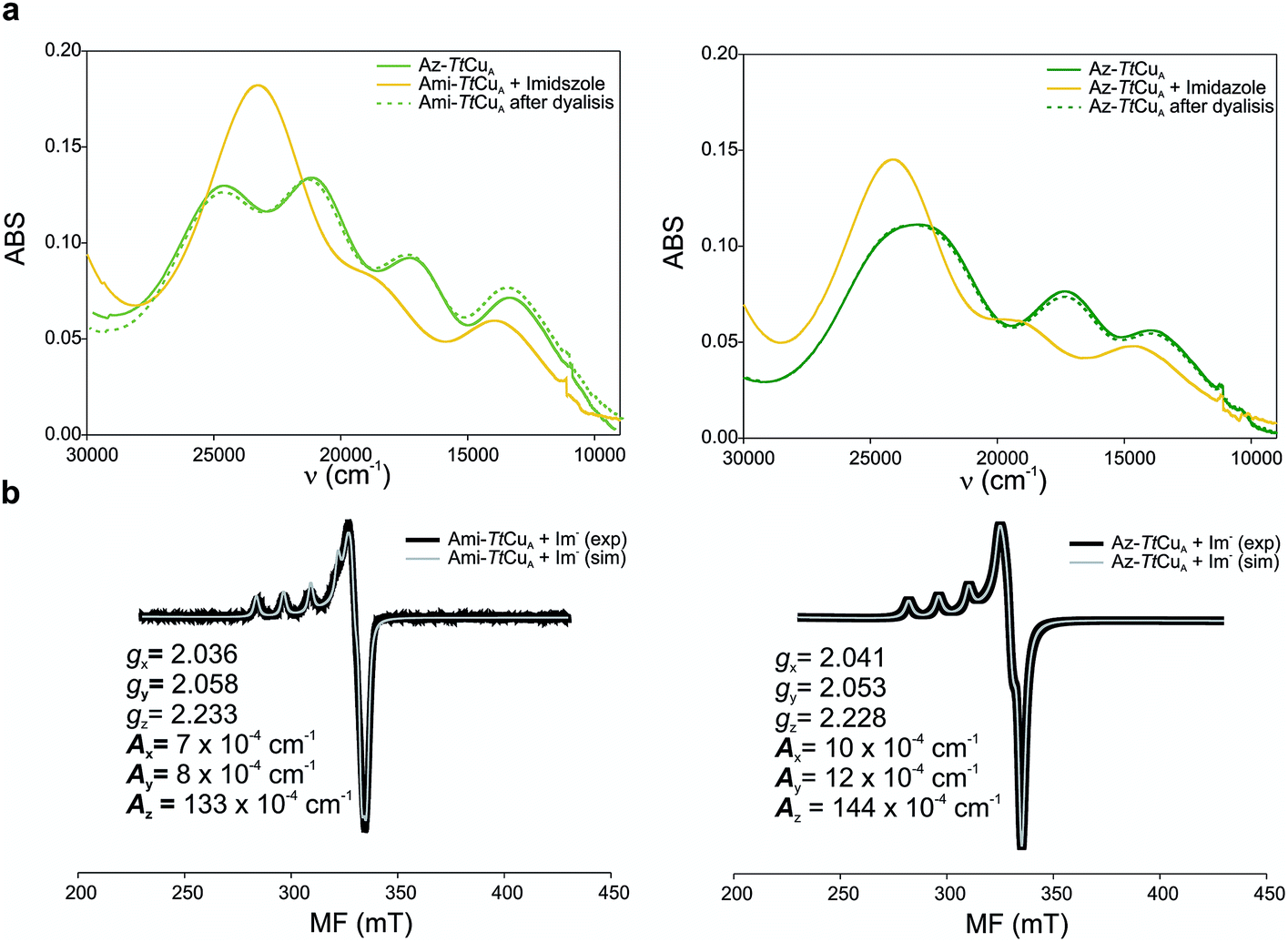 | ||
| Fig. 6 (a) Reversible imidazole binding to Ami-TtCuA. UV-Vis spectra of Ami-TtCuA (left) and Az-TtCuA (right) as expressed (green full line), with excess of imidazole (yellow full line) and after overnight dialysis (dotted green line). The spectra were obtained in Tris (100 mM) and NaCl (100 mM) and excess of imidazole (50 mM) pH 7.0 at 298 K. (b) X-band EPR spectra of imidazole adducts of Ami-TtCuA (right) and Az-TtCuA (left). The parameters used for the simulations of the EPR spectra are given in the bottom spectra. | ||
Exogenous ligand binding is unusual in T1 sites, since the protein architecture occludes the metal sites within the protein matrix.7 Attempts to crystallize the imidazole adducts or soaking experiments were unsuccessful. We also performed 1H NMR experiments of the imidazole adduct, but we could not locate any hyperfine-shifted signals, in agreement with the relaxation properties corresponding to a T2 site (that broaden the signals beyond the detection limit). The inspection of the crystal structure of Ami-TtCuA reveals two cavities next to the active site elicited upon loop replacement that can account for imidazole binding (Fig. 7). These cavities arise from the absence of several residues and their corresponding hydrogen bonding interactions that occlude the copper site in amicyanin, which cannot be reproduced within the Ami-TtCuA scaffold.
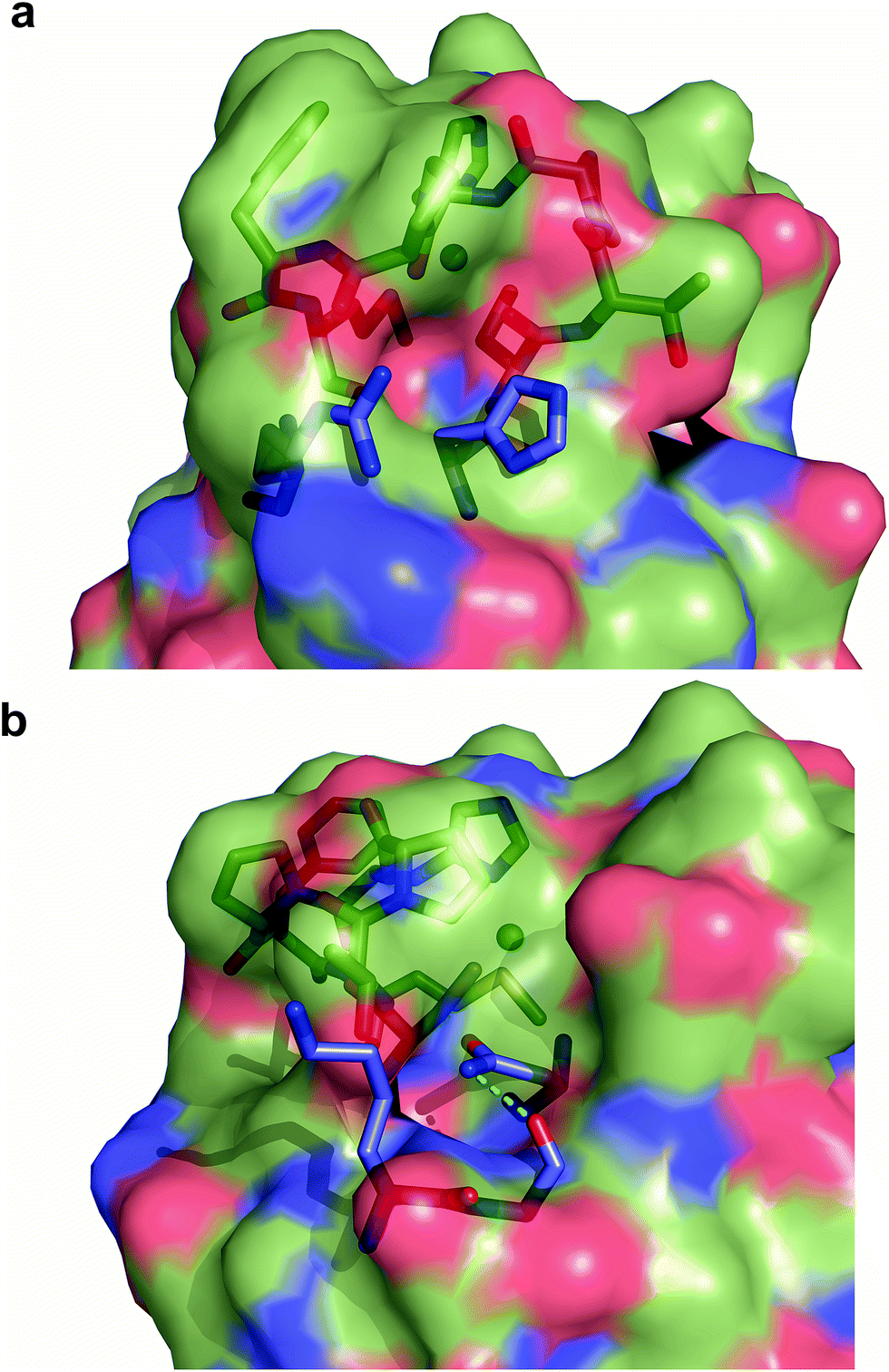 | ||
| Fig. 7 Cavities within Ami-TtCuA. Surface representation of Ami-TtCuA overlaid with a stick representation of amicyanin showing the residues that are missing in Ami-TtCuA, which provide hindrance to the active site in amicyanin. (a) Arg91 and His91 are absent in Ami-TtCuA and create a cavity. (b) Lys68, Gly69 and Asn54 are absent in Ami-TtCuA and create another cavity. | ||
Discussion
Naturally evolved scaffolds can be used to design and engineer various new site structures and functions. The cupredoxin fold is a useful model to exploit in protein redesign, particularly by loop directed mutagenesis since most copper ligands are present in a single loop. Most of the experiments have been performed on T1-containing scaffolds,14,53–58 encompassing loop replacement, contraction and lengthening experiments. The amicyanin loop represents the shortest natural loop in T1 sites. Indeed, an engineered loop being one residue shorter than the one from amicyanin has been shown to be the minimal loop length able to bind a T1 site.55 Loop-directed mutagenesis models of the CuA copper site show different properties than natural CuA truncates,30,59 suggesting that the cupredoxin scaffolds of the CuA site have differences in the outer sphere residues and the protein rack defined by the β-barrel. In this work we aimed to evaluate the influence of the scaffold upon the site structure and reactivity. With this aim in mind, we proceeded to transplant the native loops from azurin and amicyanin into a “purple” scaffold, TtCuA, obtaining two functional chimeric proteins.Introducing the ligand loops of amicyanin and azurin into TtCuA produced stable chimeric cupredoxins. On one hand, these chimeras allow us to explore the effect of shortening the CuA loop involving the removal of one Cys ligand. On the other hand, it allows the analysis of the impact of introducing different loop lengths into the CuA scaffold. The spectroscopic features of the Ami-TtCuA and Az-TtCuA chimeras are indicative of highly perturbed T1 centres. The electronic spectra of both mutants have σ/π band ratios and gz values similar to other T1 distorted copper centres. In addition, these sites present: (a) a strong LMCT Met–Cu band, (b) Az values in the EPR spectra higher than those known for other T1 proteins, suggesting a reduced spin localization onto the Cys ligand, (c) an unprecedented electron spin density in the Met ligand in Ami-TtCuA, and (d) a smaller Cu–SCys stretching frequency than normal T1 sites. Overall, these features reveal an unusually strong axial Met–Cu interaction in both chimeras, which is confirmed by the crystal structure of Ami-TtCuA. The electronic structure of Ami-TtCuA reveals a distortion level slightly larger than that of green nitrite reductase, while the spectral features of Az-TtCuA resemble those of the distorted T1 site of AcoP from Acidithiobacillus ferrooxidans.42 Despite the lack of a crystal structure for Az-TtCuA and for AcoP, the different spectroscopies allow us to compare both chimeras. Ami-TtCuA is characterized by a more intense Met–Cu(II) LMCT band and a larger rhombicity factor than Az-TtCuA, suggesting that the latter chimera is characterized by a weaker Cu–S(Met) bonding, consistent with a smaller distortion.
The overall structure of Ami-TtCuA is remarkably similar to that of TtCuA. Loop shortening changes the active-site environment dramatically, going from a two-copper to a one-copper site, including changes in the hydrogen bond pattern. The loop structure in Ami-TtCuA is similar to that of amicyanin (Fig. 4c), suggesting that the purple cupredoxin scaffold does not influence the loop conformation. A similar situation was met when the amicyanin loop was engineered into the azurin scaffold.55 Nevertheless, the active-site geometries in Ami-TtCuA are different from that of amicyanin. The structure of Ami-TtCuA reveals the shortest axial copper–thioether bond reported to date in a T1 copper centre. In agreement with the coupled distorted model, this shortening is accompanied by an elongation of the Cu–SCys thiolate bond that results in a tetragonal distortion observed in the X-ray structure and reflected in the spectroscopic features. This arrangement can be accounted for by the relative orientation of the Cys and Met ligands, which adopt positions virtually the same as those in TtCuA (Fig. 4b), indicating that the scaffold determines the active site structure. This results in a shorter axial Cu–SMet bond of 2.35 Å in TtCuA compared to that found amicyanin (2.90 Å) and all other cupredoxins. The importance of the cupredoxin scaffold in defining the orientation of the Cys and Met ligands located at the start and end respectively of the engineered loop has been already documented in elegant loop mutant studies of azurin,55,58 plastocyanin, pseudoazurin51 and nitrite reductase.57 These studies describe the role of T1-containing scaffolds upon the metal site. Here we show that the CuA-containing scaffold plays a similar role in defining the position of the Cys and Met ligands, even when engineering a shortened loop that lacks the extra Cys required for the CuA site.
Both chimeras display reorganization energies comparable to those of natural T1 sites,41 suggesting that these centres are able to act efficiently performing electron transfer. However, in contrast to native T1 sites, which are not usually accessible to exogenous ligands, both chimeric proteins are able to bind imidazole in the mM range, eliciting T2 copper centres whose spectroscopic features closely resemble those of nitrosocyanin. The lack of an intense LMCT Met–Cu(II) band, as observed for the unbound proteins, suggests that the Met ligand may have been displaced by the binding of the exogenous ligands to the metal ion. Previous reports of binding of exogenous ligands to mutant T1 centres correspond to mutations of copper ligands,60–66 to T1 centres lacking an axial ligand or to loop-engineered variants.58 The variants described herein are able to bind either imidazole or azide, with higher binding affinities than those reported for azurin loop mutants.63,66 These results suggest that the bifunctional nature of T1 sites can be further expanded.
The copper sites of both mutants show reduction potentials (422 and 487 mV), much higher than those of amicyanin and azurin (255 and 295 mV, respectively). The coordinating Cys in Ami-TtCuA forms a hydrogen bond with the backbone NH of Gly115, which is stronger than the hydrogen bond with an Asn residue present in most T1 sites. This strong hydrogen bond is expected to stabilize the Cu(I) form, accounting for the higher reduction potential in this chimera. This observation is supported by a series of mutagenesis studies in T1 sites altering this hydrogen bond network.67–71 Since this hydrogen bond is conserved in CuA scaffolds,72 the high reduction potential of Az-TtCuA, may also be attributed to this feature. The lack of a crystal structure for the latter protein does not allow us to assess in detail the difference between the reduction potential of both chimeras. However, previous studies allow us to provide some possible explanations: (i) a stronger interaction with the axial ligands in T1 sites is known to decrease the reduction potential by destabilizing the Cu(II) state,73 so the stronger S(Met)–Cu interaction suggested by the different spectroscopies in Ami-TtCuA could account for the lower reduction potential in this variant, (ii) a Phe residue present in the azurin loop is replaced by a Pro in the amicyanin loop, and the Phe114Pro replacement in azurin has been shown to increase the reduction potential,22,67 and (iii) possible changes in the nearby dipoles elicited by changes in the hydrogen bond network.56 Overall, we conclude that the high redox potentials are mostly due to second sphere contributions.
Conclusions
We have designed two chimeric copper proteins containing unique T1 sites. First, they present a shorter Cu–S(Met) bond than the Cu–S(Cys) bond, in contrast to the observation in normal or distorted T1 sites. Surprisingly, these features are compatible with an efficient electron transfer site, based on its reduction potential and reorganization energy. Second, they are able to bind exogenous ligands, opening the possibilities of performing catalysis. Third, this β-barrel scaffold can host purple, green or red copper sites, highlighting the versatility of this fold. The latter modulation has also been obtained by using non-natural amino acids in azurin,74 while our approach relies on using natural amino acids and loop engineering. Finally, we conclude that the protein rack provided by the β-barrel to bind a purple CuA site is different than the one required for T1 centres. This approach to obtain novel copper sites can be combined with the established knowledge in rational metalloprotein engineering to design copper centres endowed with unique functionalities. In particular, the high reduction potentials, combined with the ability of binding exogenous ligands within a stable protein scaffold, open new avenues to design water-soluble catalysts able to perform challenging tasks such as water splitting and NO sensing.75Methods
Site-directed mutagenesis and plasmid construction
The two protein chimeras were constructed by loop-directed mutagenesis. The plasmid pET9a-TtCuA (KanR)31 containing the wild type TtCuA gene was used as the template to add mutations. This was performed by the amplification of the entire plasmid using two mutagenic primers and selecting by DpnI digestion. Primers used were 5′-GAGTACCGCATCATCTGCACCCCGCACCCGTTTATGTTCGGCACGATCGTG and 5′-CACGATCGTGCCGAACATAAACGGGTGCGGGGTGCAGATGATGCGGTACTC for the amicyanin mutant; and 5′-GAGTACCGCATCATCTGCACCTTTCCGGGCCACAGCGCGCTGATGTTCGGCACGATCGTG and 5′-CACGATCGTGCCGAACATCAGCGCGCTGTGGCCCGGAAAGGTGCAGATGATGCGGTACTC for the azurin mutant. PCR reactions were performed on a Perkin Elmer Gene Amp PCR System 2400 equipment, with 100 μL reaction mixtures containing 0.3 mM of each dNTP, 0.2 nmol μL−1 of each primer, 1 mM MgSO4, 400 ng of the plasmid used as the template, 10 μL of the 10× buffer provided with the polymerase and 2 units of Pfx Platinum DNA polymerase (Invitrogen). After an initial step of 30 s at 95 °C, 18 cycles of the following steps were performed: 30 s at 95°, 1 min at 55 °C, and 5 min at 68 °C followed by a final extension step of 5 min at 68 °C. The template plasmid was digested by the addition of 1 U of DpnI and incubation for 1 h at 37 °C and the reaction mixture was then transformed on E. coli JM109 cells. The presence of the desired mutations on the DNA plasmid preparation from recovered transformants was corroborated by sequencing (University of Maine Sequencing Facility).The fragment from the coding region of Ami-TtCuA and Az-TtCuA genes was cut with BamHI and NdeI and ligated back into the pET28a plasmid (KanR) yielding the pET28a-Ami-TtCuA and pET28a-Ami-TtCuA plasmids that were used for protein expression yielding the proteins with a His6 tag.
Protein expression, purification and characterization
Ten mL of culture medium (LB and 50 mg mL−1 kanamycin) was inoculated from a freshly streaked plate of BL21(DE3) cells containing the pET28a-Ami-TtCuA or pET28a-Ami-TtCuA plasmids. After overnight incubation, this culture was used to inoculate a 1 L flask of culture medium. After approximately 3–4 h of incubation at 37 °C with shaking, the OD600 of the cell culture reached 0.6–0.8. At this point, the cell culture was induced by the addition of isopropyl-β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.4 mM and incubated for 4 h at 37 °C. Cells were harvested by pelleting via centrifugation at 5000g for 15 min. Typically, 1 L of culture produced 8–10 g of cells.Pelleted cells were resuspended in lysis buffer, lysed using a cell disruptor. The resulting cell debris was pelleted by centrifugation. The supernatant that carries both protein chimeras was purified from the lysate by chromatography on a nickel-NTA affinity column. The His6 tag was cleaved by 3 hr incubation with thrombin protease (Sigma) at room temperature, and a second metal affinity chromatography step was used to remove the cleaved His6 tag. The protein was then passed through a molecular exclusion column to eliminate the undesired protease and undesired protein contaminants. Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) (8–12% gradient stained with Coomassie brilliant blue R-250) showed a single band. Following purification the protein was dialyzed into a 100 mM Tris buffer, at pH 7.0 and then 3 mM tris(2-carboxyethyl)phosphine (TCEP) was added to prevent any disulphide cross-links or cysteine oxidation. The protein yield was ∼100 mg per liter of culture medium. Before use, apo-Ami-TtCuA and apo-Az-TtCuA reduced protein samples were then dialyzed into the desired buffer overnight to remove the TCEP. The concentration of both apo proteins was routinely determined using the molar absorption coefficient of 5500 M−1 cm−1 at 280 nm.
Protein metallation was carried out incubating reduced and dialyzed proteins with up to a three-fold excess of Cu(II) using an aqueous Cu(SO4) solution. Upon reconstitution with Cu(II), both chimeric proteins exhibited a green colour and remained stable indefinitely at 4 °C. Cu(II) excess was removed via dialysis or washing using Centricon-10 units (Millipore).
UV-Vis, Circular Dichroism and EPR spectroscopic measurements
Electronic spectra were recorded at room temperature using a Jasco V-680 spectrophotometer. Circular Dichroism spectroscopy was used to test the global folding state of proteins. CD spectra were obtained from 300 nm to 1100 nm on a Jasco J-810 spectropolarimeter with quartz cuvettes of 1 cm and an enzyme concentration of 1 mM at a constant temperature of 25 °C.Simultaneous Gaussian fitting of the low-temperature absorption, MCD, and CD spectra was performed using the Peak-Fit program (Systat Software Inc.).
EPR measurements were performed at X-band with a Bruker EMX Plus spectrometer equipped with a universal high-sensitivity cavity (HSW10819 model) at a constant temperature of 163 K. Spectra were acquired under non-saturating conditions. EPR spectra were simulated with the EasySpin toolbox for MATLAB.76
NMR spectroscopy
Samples (∼3–5 mM) for NMR experiments were concentrated using Centricon-10 (Millipore) units. A small amount of reduced protein (∼25%) was enough to perform the exchange NMR experiments. D2O solutions were prepared by exchanging the solvent in Centricon-10 units. NMR spectra were recorded on a Bruker Avance II spectrometer operating at a proton frequency of 600.13 MHz (1H frequency). Saturation-transfer experiments in solutions containing Cu(II)- and Cu(I)-Ami-TtCuA were performed using an on–off scheme where on values varied from 80 to 25 ppm.Resonance Raman spectroscopy
RR spectra were acquired in backscattering geometry by using a Jobin Yvon XY 800 Raman microscope equipped with a CCD detector. Elastic scattering was rejected with Notch filters. A 633 nm solid state laser (5 mW) was focused into 2 μL of frozen protein solution (77 K) contained in a Linkam THMS 300 thermostat. Spectra were acquired with 0.4 cm−1 increment per data point.Electrochemistry
The reduction potential (Eo) values were determined by cyclic voltammetry (CV) of protein samples in solution, as the midpoint of the peak-to-peak separation: Eo = (Eap + Ecp)/2, where Eap and Ecp are the anodic and cathodic peak potentials, respectively. The electron transfer reorganization free energies (λ) were obtained from CV experiments performed on protein samples adsorbed on gold electrodes coated with biocompatible films. The films employed consisted of a mixed self-assembled monolayer (SAM) of 60% 1-mercapto-16-hexadecanol and 40% 1-hexadecanethiol composition, to maximize the amount of adsorbed protein. The thickness of the SAM guarantees a low electronic coupling between the redox site and the electrode surface, yielding a tunnelling-controlled electron transfer reaction. The rate constants of heterogeneous electron transfer at zero over-potential (k0ET) were determined using Laviron's working curve,39 which evaluates the peak separation as a function of the scan rate in CVs, for separations of the anodic and cathodic peaks of up to 200 mV. The working curve is valid for 0.3 < α < 0.7, which is suitable for the reported CVs considering the symmetric peaks of the voltammograms. k0ET values were determined as a function of temperature between 5 and 39 °C, which allows us to obtain λ directly from Arrhenius plots, since under these experimental conditions ΔG* = λ/4 is valid.Crystallography
Ami-TtCuA was crystallized by mixing a total of 1 μL of the protein stock (34 mg mL−1 in 10 mM Tris, 25 mM NaCl, pH 7.5) with an equal amount of crystallization solution consisting of 60% (v/v) 2-methyl-2,4-pentanediol and equilibrated at 22 °C against 500 μL of the latter solution by sitting drop vapor diffusion configuration. Sharp edge bars of approximate size 0.15 × 0.15 × 0.40 mm appeared within three weeks. Crystals were flash cooled in liquid nitrogen using Hampton Research loops (Aliso Viejo, USA).X-ray data collection, structure resolution and refinement
Native X-ray diffraction data were collected at 100 K on a Bruker D8 QUEST microfocus diffractometer equipped with a PHOTON 100 CMOS detector. The collected frames were converted into a readable format with SFRMTOOLS,77 and then processed to 2.30 Å resolution in the P212121 space group with XDS78 and AIMLESS,79 separating 5% of the reflections for cross-validation. The Ami-TtCuA structure was solved by molecular replacement with Phaser80 using the coordinates of the CuA domain of the Thermus thermophilus ba3-type cytochrome c oxidase as the search model (PDB code 2CUA81). A total of eight copies were successfully found in the asymmetric unit with proper crystal packing. Several cycles of manual model building and restrained refinement applying NCS were then performed with Coot82 and Buster,83 respectively, which allowed for the complete trace of the chimeric loop in all chains. The final model was validated with MolProbity84 and then deposited in the Protein Data Bank under the code 5U7N. Detailed information on the crystallographic data collection and refinement statistics is shown in Table S7.†Conflicts of interest
There are not conflicts of interest.Acknowledgements
During this work A. E. C. and U. Z. were doctoral fellows and D. A. P. was a postdoctoral fellow at the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET). A. J. V., L. H. O., S. K. and D. H. M. are staff members of CONICET. This research was financially supported by the Agencia Nacional De Promoción Científica y Tecnológica (ANPCyT).References
- Encyclopedia of inorganic and bioinorganic chemistry, ed. R. A. Scott, John Wiley & Sons, Ltd, Chichester, UK, 2011 Search PubMed.
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef PubMed.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659–3853 CrossRef PubMed.
- J. Liu, S. Chakraborty, P. Hosseinzadeh, Y. Yu, S. Tian, I. Petrik, A. Bhagi and Y. Lu, Chem. Rev., 2014, 114, 4366–4469 CrossRef PubMed.
- E. I. Solomon, Inorg. Chem., 2006, 45, 8012–8025 CrossRef PubMed.
- E. I. Solomon, M. J. Baldwin and M. D. Lowery, Chem. Rev., 1992, 92, 521–542 CrossRef.
- E. I. Solomon, R. K. Szilagyi, S. DeBeer George and L. Basumallick, Chem. Rev., 2004, 104, 419–458 CrossRef PubMed.
- C. Dennison, Coord. Chem. Rev., 2005, 249, 3025–3054 CrossRef.
- L. B. LaCroix, D. W. Randall, A. M. Nersissian, C. W. G. Hoitink, G. W. Canters, J. S. Valentine and E. I. Solomon, J. Am. Chem. Soc., 1998, 120, 9621–9631 CrossRef.
- P. Comba, Coord. Chem. Rev., 2000, 200–202, 217–245 CrossRef.
- L. B. LaCroix, S. E. Shadle, Y. Wang, B. A. Averill, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1996, 118, 7755–7768 CrossRef.
- Y. Lu, S. M. Berry and T. D. Pfister, Chem. Rev., 2001, 101, 3047–3080 CrossRef PubMed.
- C. Dennison, Nat. Prod. Rep., 2008, 25, 15–24 RSC.
- J. van der Oost, P. Lappalainen, A. Musacchio, A. Warne, L. Lemieux, J. Rumbley, R. B. Gennis, R. Aasa, T. Pascher, B. G. Malmström and M. Saraste, EMBO J., 1992, 11, 3209–3217 Search PubMed.
- I. D. Petrik, J. Liu and Y. Lu, Curr. Opin. Chem. Biol., 2014, 19, 67–75 CrossRef PubMed.
- Y. Lu, Angew. Chem., Int. Ed. Engl., 2006, 45, 5588–5601 CrossRef PubMed.
- M. L. Zastrow and V. L. Pecoraro, Coord. Chem. Rev., 2013, 257, 2565–2588 CrossRef PubMed.
- F. Yu, V. M. Cangelosi, M. L. Zastrow, M. Tegoni, J. S. Plegaria, A. G. Tebo, C. S. Mocny, L. Ruckthong, H. Qayyum and V. L. Pecoraro, Chem. Rev., 2014, 114, 3495–3578 CrossRef PubMed.
- C. S. Mocny and V. L. Pecoraro, Acc. Chem. Res., 2015, 48, 2388–2396 CrossRef PubMed.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Chem. Rev., 2017, 118, 142–231 CrossRef PubMed.
- Y. Lu, N. Yeung, N. Sieracki and N. M. Marshall, Nature, 2009, 460, 855–862 CrossRef PubMed.
- N. M. Marshall, D. K. Garner, T. D. Wilson, Y.-G. Gao, H. Robinson, M. J. Nilges and Y. Lu, Nature, 2009, 462, 113–116 CrossRef PubMed.
- N. Yeung, Y.-W. Lin, Y.-G. Gao, X. Zhao, B. S. Russell, L. Lei, K. D. Miner, H. Robinson and Y. Lu, Nature, 2009, 462, 1079–1082 CrossRef PubMed.
- P. Hosseinzadeh, S. Tian, N. M. Marshall, J. Hemp, T. Mullen, M. J. Nilges, Y.-G. Gao, H. Robinson, D. A. Stahl, R. B. Gennis and Y. Lu, J. Am. Chem. Soc., 2016, 138, 6324–6327 CrossRef PubMed.
- I. S. MacPherson, F. I. Rosell, M. Scofield, A. G. Mauk and M. E. P. Murphy, Protein Eng., Des. Sel., 2010, 23, 137–145 CrossRef PubMed.
- D. M. Mate and M. Alcalde, Biotechnol. Adv., 2015, 33, 25–40 CrossRef PubMed.
- C. Dennison, E. Vijgenboom, S. de Vries, J. van der Oost and G. W. Canters, FEBS Lett., 1995, 365, 92–94 CrossRef PubMed.
- M. Hay, J. H. Richards and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 461–464 CrossRef.
- P. Hosseinzadeh and Y. Lu, Biochim. Biophys. Acta, 2016, 1857, 557–581 CrossRef PubMed.
- G. N. Ledesma, D. H. Murgida, H. K. Ly, H. Wackerbarth, J. Ulstrup, A. J. Costa-Filho and A. J. Vila, J. Am. Chem. Soc., 2007, 129, 11884–11885 CrossRef PubMed.
- L. A. Abriata, L. Banci, I. Bertini, S. Ciofi-Baffoni, P. Gkazonis, G. A. Spyroulias, A. J. Vila and S. Wang, Nat. Chem. Biol., 2008, 4, 599–601 CrossRef PubMed.
- L. A. Abriata, G. N. Ledesma, R. Pierattelli and A. J. Vila, J. Am. Chem. Soc., 2009, 131, 1939–1946 CrossRef PubMed.
- M.-E. Zaballa, L. A. Abriata, A. Donaire and A. J. Vila, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9254–9259 CrossRef PubMed.
- K. A. Gray, D. B. Knaff, M. Husain and V. L. Davidson, FEBS Lett., 1986, 207, 239–242 CrossRef PubMed.
- G. Battistuzzi, M. Borsari, J. A. Cowan, C. Eicken, L. Loschi and M. Sola, Biochemistry, 1999, 38, 5553–5562 CrossRef PubMed.
- G. Battistuzzi, M. Bellei, M. Borsari, G. W. Canters, E. de Waal, L. J. C. Jeuken, A. Ranieri and M. Sola, Biochemistry, 2003, 42, 9214–9220 CrossRef PubMed.
- G. Battistuzzi, M. Borsari, L. Loschi and M. Sola, J. Biol. Inorg Chem., 1997, 2, 350–359 CrossRef.
- K. Fujita, N. Nakamura, H. Ohno, B. S. Leigh, K. Niki, H. B. Gray and J. H. Richards, J. Am. Chem. Soc., 2004, 126, 13954–13961 CrossRef PubMed.
- E. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1979, 101, 19–28 CrossRef.
- D. H. Murgida and P. Hildebrandt, J. Phys. Chem. B, 2002, 106, 12814–12819 CrossRef.
- D. E. Khoshtariya, T. D. Dolidze, M. Shushanyan, K. L. Davis, D. H. Waldeck and R. van Eldik, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2757–2762 CrossRef PubMed.
- M. Roger, F. Biaso, C. J. Castelle, M. Bauzan, F. Chaspoul, E. Lojou, G. Sciara, S. Caffarri, M.-T. Giudici-Orticoni and M. Ilbert, PLoS One, 2014, 9, e98941 CrossRef PubMed.
- D. F. Blair, G. W. Campbell, W. K. Cho, A. M. English, H. A. Fry, V. Lum, K. A. Norton, J. R. Schoonover and S. I. Chan, J. Am. Chem. Soc., 1985, 107, 5755–5766 CrossRef.
- C. R. Andrew, H. Yeom, J. S. Valentine, B. G. Karlsson, G. van Pouderoyen, G. W. Canters, T. M. Loehr, J. Sanders-Loehr and N. Bonander, J. Am. Chem. Soc., 1994, 116, 11489–11498 CrossRef.
- C. R. Andrew and J. Sanders-Loehr, Acc. Chem. Res., 1996, 29, 365–372 CrossRef.
- J. Han, T. M. Loehr, Y. Lu, J. S. Valentine, B. A. Averill and J. Sanders-Loehr, J. Am. Chem. Soc., 1993, 115, 4256–4263 CrossRef.
- A. P. Kalverda, J. Salgado, C. Dennison and G. W. Canters, Biochemistry, 1996, 35, 3085–3092 CrossRef PubMed.
- S. J. Kroes, J. Salgado, G. Parigi, C. Luchinat and G. W. Canters, J. Biol. Inorg Chem., 1996, 1, 551–559 CrossRef.
- A. Donaire, B. Jiménez, C. O. Fernández, R. Pierattelli, T. Niizeki, J.-M. Moratal, J. F. Hall, T. Kohzuma, S. S. Hasnain and A. J. Vila, J. Am. Chem. Soc., 2002, 124, 13698–13708 CrossRef PubMed.
- L. M. Cunane, Z. W. Chen, R. C. Durley and F. S. Mathews, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1996, 52, 676–686 CrossRef PubMed.
- M. Velarde, R. Huber, S. Yanagisawa, C. Dennison and A. Messerschmidt, Biochemistry, 2007, 46, 9981–9991 CrossRef PubMed.
- L. Basumallick, R. Sarangi, S. DeBeer George, B. Elmore, A. B. Hooper, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2005, 127, 3531–3544 CrossRef PubMed.
- S. Yanagisawa and C. Dennison, J. Am. Chem. Soc., 2003, 125, 4974–4975 CrossRef PubMed.
- S. Yanagisawa and C. Dennison, J. Am. Chem. Soc., 2004, 126, 15711–15719 CrossRef PubMed.
- C. Li, S. Yanagisawa, B. M. Martins, A. Messerschmidt, M. J. Banfield and C. Dennison, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7258–7263 CrossRef PubMed.
- C. Li, M. J. Banfield and C. Dennison, J. Am. Chem. Soc., 2007, 129, 709–718 CrossRef PubMed.
- K. Sato, S. J. Firbank, C. Li, M. J. Banfield and C. Dennison, Chemistry, 2008, 14, 5820–5828 CrossRef PubMed.
- K. Sato, C. Li, I. Salard, A. J. Thompson, M. J. Banfield and C. Dennison, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5616–5621 CrossRef PubMed.
- H. J. Hwang, S. M. Berry, M. J. Nilges and Y. Lu, J. Am. Chem. Soc., 2005, 127, 7274–7275 CrossRef PubMed.
- T. Den Blaauwen, M. Van de Kamp and G. W. Canters, J. Am. Chem. Soc., 1991, 113, 5050–5052 CrossRef.
- T. den Blaauwen, C. W. Hoitink, G. W. Canters, J. Han, T. M. Loehr and J. Sanders-Loehr, Biochemistry, 1993, 32, 12455–12464 CrossRef PubMed.
- T. Den Blaauwen and G. W. Canters, J. Am. Chem. Soc., 1993, 115, 1121–1129 CrossRef.
- M. Vidakovic and J. P. Germanas, Angew. Chem., Int. Ed. Engl., 1995, 34, 1622–1624 CrossRef.
- G. van Pouderoyen, C. R. Andrew, T. M. Loehr, J. Sanders-Loehr, S. Mazumdar, H. A. Hill and G. W. Canters, Biochemistry, 1996, 35, 1397–1407 CrossRef PubMed.
- A. C. F. Gorren, T. den Blaauwen, G. W. Canters, D. J. Hopper and J. A. Duine, FEBS Lett., 1996, 381, 140–142 CrossRef PubMed.
- N. Bonander, B. G. Karlsson and T. Vänngård, Biochemistry, 1996, 35, 2429–2436 CrossRef PubMed.
- S. Yanagisawa, M. J. Banfield and C. Dennison, Biochemistry, 2006, 45, 8812–8822 CrossRef PubMed.
- M. van Gastel, Y. Nagano, R. Zondervan, G. W. Canters, L. J. C. Jeuken, G. C. M. Warmerdam, E. C. de Waal and E. J. J. Groenen, J. Phys. Chem. B, 2002, 106, 4018–4021 CrossRef.
- S. Dong, J. A. Ybe, M. H. Hecht and T. G. Spiro, Biochemistry, 1999, 38, 3379–3385 CrossRef PubMed.
- K. M. Lancaster, S. DeBeer George, K. Yokoyama, J. H. Richards and H. B. Gray, Nat. Chem., 2009, 1, 711–715 CrossRef PubMed.
- K. M. Lancaster, M.-E. Zaballa, S. Sproules, M. Sundararajan, S. DeBeer, J. H. Richards, A. J. Vila, F. Neese and H. B. Gray, J. Am. Chem. Soc., 2012, 134, 8241–8253 CrossRef PubMed.
- L. A. Abriata and A. J. Vila, J. Inorg. Biochem., 2014, 132, 18–20 CrossRef PubMed.
- J. A. Guckert, M. D. Lowery and E. I. Solomon, J. Am. Chem. Soc., 1995, 117, 2817–2844 CrossRef.
- K. M. Clark, Y. Yu, N. M. Marshall, N. A. Sieracki, M. J. Nilges, N. J. Blackburn, W. A. van der Donk and Y. Lu, J. Am. Chem. Soc., 2010, 132, 10093–10101 CrossRef PubMed.
- S. Tian, J. Liu, R. E. Cowley, P. Hosseinzadeh, N. M. Marshall, Y. Yu, H. Robinson, M. J. Nilges, N. J. Blackburn, E. I. Solomon and Y. Lu, Nat. Chem., 2016, 8, 670–677 CrossRef PubMed.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef PubMed.
- T. Gruene, SFRMTOOLS, University of Goettingen, Germany, 2014 Search PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef PubMed.
- P. R. Evans and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1204–1214 CrossRef PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef PubMed.
- P. A. Williams, N. J. Blackburn, D. Sanders, H. Bellamy, E. A. Stura, J. A. Fee and D. E. McRee, Nat. Struct. Biol., 1999, 6, 509–516 CrossRef PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef PubMed.
- G. Bricogne, E. Blanc, M. Brandl, C. Flensburg, P. Keller, W. Paciorek, P. Roversi, A. Sharff, O. S. Smart, C. Vonrhein, and T. O. Womack, BUSTER, Global Phasing Ltd., Cambridge, United Kingdom, 2014 Search PubMed.
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01444b |
| This journal is © The Royal Society of Chemistry 2018 |