
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a 2D phosphorus material in a MOF-based 2D nano-reactor†
Ming
Li‡
 *a,
Chuang
Ma‡
a,
Xiong
Liu
b,
Jun
Su
c,
Xiaofeng
Cui
d and
Yunbin
He
e
*a,
Chuang
Ma‡
a,
Xiong
Liu
b,
Jun
Su
c,
Xiaofeng
Cui
d and
Yunbin
He
e
aHubei Collaborative Innovation Center for Advanced Organic Chemical Materials, Key Laboratory for the Synthesis and Application of Organic Functional Molecules, Ministry of Education, Hubei Key Laboratory of Polymer Materials, College of Chemistry and Chemical Engineering, Hubei University, Wuhan 430062, People's Republic of China. E-mail: limingljy@163.com
bCarl Zeiss Microscopy GmbH, Carl-Zeiss-Str. 22, Oberkcohen, 73447, Germany
cWuhan National Laboratory for Optoelectronics, Huazhong University of Science and Technology, Luoyu Road 1037, 430074, Wuhan, China
dSchool of Chemistry, Chemical Engineering and Life Sciences, Wuhan University of Technology, Wuhan, Hubei 430070, China
eSchool of Materials Science and Engineering, Hubei University, Wuhan 430062, China
First published on 12th June 2018
Abstract
Synthesis of two dimensional (2D) materials mainly relies on interface-templates, exfoliation of layered crystals and surfactant-assistance. Here we have realized another strategy of polymerization in a 2D nano-reactor. As an application of this strategy, a 2D nano-reactor was constructed within a new pillar–layer metal organic framework (MOF) and in this nano-reactor a moderately stable 2D phosphorus material was synthesized from white phosphorus (P4). Our research may provide a new approach for the synthesis of various 2D materials.
Introduction
2D materials have been extensively researched in recent decades since the discovery of graphene.1–6 However, strategies for the synthesis of 2D materials are quite limited, mainly relying on interface-templates,7,8 exfoliation of layered crystals9–11 and surfactant-assistance.12–14 All these methods have limitations. In order to satisfy this booming research area, the development of new synthetic strategies is quite appealing. Pioneering studies on synthesis in nano-reactors revealed that the obtained product can replicate precise morphology derived from the nano-reactor.15–17 They provided us with an inspiration to synthesize 2D materials by polymerization within 2D nano-reactors. Unfortunately, although 0D (e.g., fullerene18) and 1D (e.g., carbon tubes19–21) nano-reactors well existed, 2D nano-reactors are quite rare. Via searching for appropriate structures for application in 2D nano-reactors, we found that pillar–layer MOFs suggested by Susumu Kitagawa 10 years ago, which have 2D-channels, can be an option for the synthesis of 2D polymers.22,23 One of the advantages of using MOFs as nano-reactors is that the framework can be disassembled afterwards to release the resulting product.24,25 However, this is a long-lasting unrealized strategy, possibly due to the lack of appropriate MOFs: (1) layers of many MOFs are permeable, hindering their application in 2D nano-reactors;26 (2) except for the layer ligand, most of the MOFs need auxiliary pillar ligands (Fig. 1a top), which may result in cleavage of layer and ligand units during the insertion of molecules due to weak coordination;27 (3) general pillars such as bipyridine and triethylenediamine may be too fat to be efficiently circled by intruded monomers.28 In order to solve these problems, this study will use a slim pillar covalently bonded with the layer unit (Fig. 1a bottom) to construct a new pillar–layer MOF. We hoped that inserted monomers can circle pillars more efficiently and the MOF can be stable during polymerization.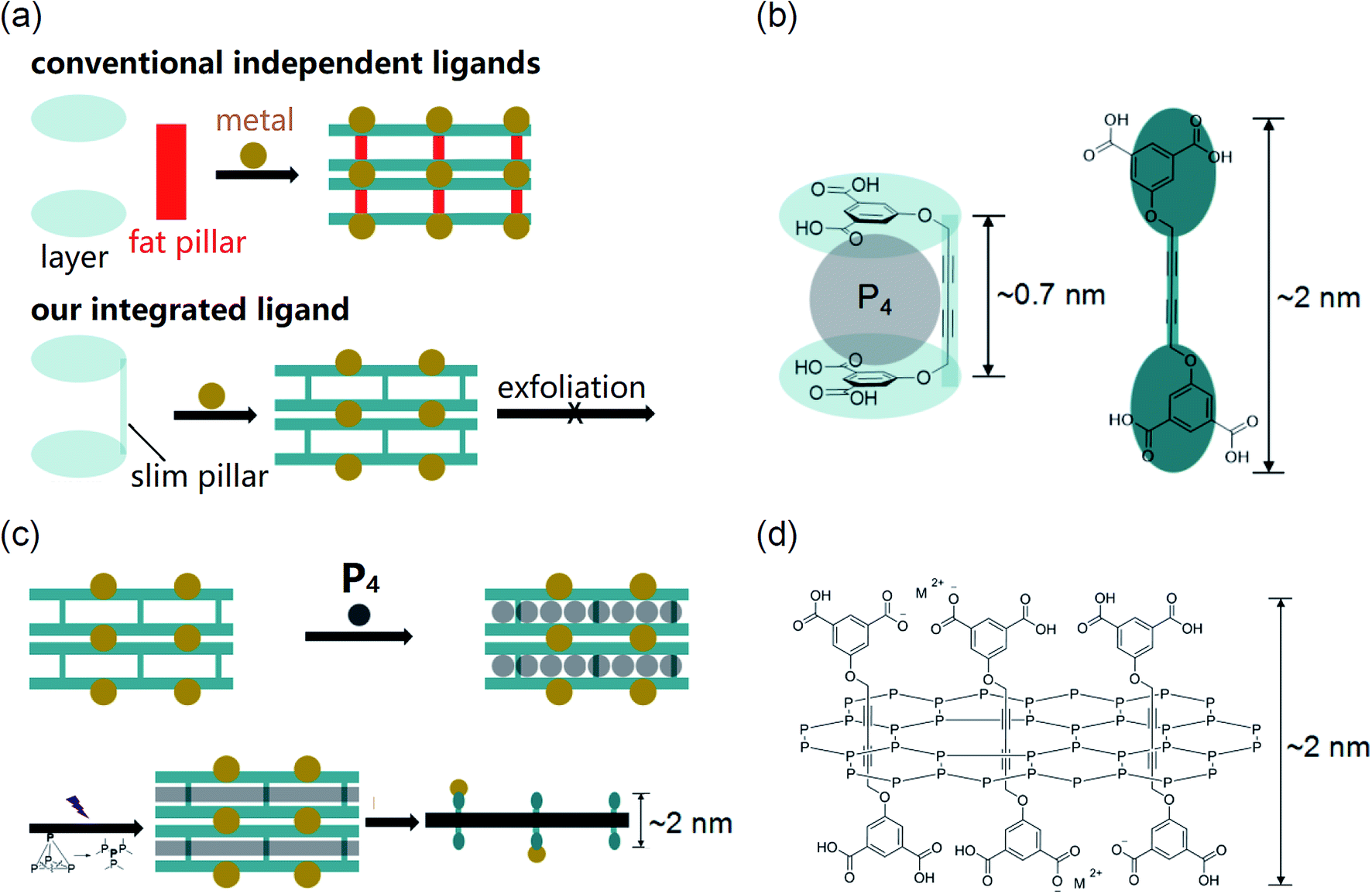 | ||
| Fig. 1 (a) Pillar–layer MOF constructed from independent ligands and our integrated ligand (because such a MOF has a 3D framework, the MOF itself has no chance to be exfoliated to 2D sheets); (b) the structure of our ligand; (c) preparation of the 2D phosphorus material within the 2D nano-reactor; (d) the proposed structure of the 2D phosphorus material (M2+: metal ions). | ||
Based on the above analysis, we excluded MOFs existing so far as we have known and developed a new MOF from an integrated ligand for the construction of a 2D nano-reactor. The slimmest diacetylene was selected as the pillar unit, which is covalently bonded with the layer unit (isophthalic acid) by an ether linkage. Due to the kink from the linkage, the ligand can bend to a suitable conformation with a void (height ∼0.7 nm, Fig. 1b left), whilst the length of the extended ligand is ∼2 nm (Fig. 1b right). For a conceptual application model of this 2D nano-reactor in the synthesis of 2D materials, we selected white phosphorus (P4) as the monomer because its void is well suitable for the uptake of a P4 molecule (Fig. 1b left). If a layer of P4 was restricted within the 2D space, poly-phosphorus networks can be produced after irradiation-induced polymerization.29 The free-standing 2D phosphorus material could be obtained through disassembling the MOF (Fig. 1c). The proposed molecular structure of this 2D material is shown in Fig. 1d. In this material, phosphorus atoms are covalently bonded, while ligands interpenetrate the 2D network. The thickness of a monolayer should be ∼2 nm if ligands are extended.
Results and discussion
Ligand 1 was simply synthesized under mild conditions. 3-Bromoprop-1-yne was substituted by dimethyl 5-hydroxyisophthalate to give known compound 3, followed by a coupling reaction to yield 2. After removal of methyl groups, ligand 1 was obtained with an overall yield of 72% (ESI Scheme S1 and Fig. S1–S5†).The complexation of ligand 1 and Co2+ in DMF/ethanol solution under heating resulted in purple-red MOF crystals (Fig. 2a). These crystals are larger than 0.1 mm and show a significant birefringence feature, indicating their nice crystallinity (ESI Fig. S6†). The single crystallography X-ray diffraction (SC-XRD) analysis was performed after storage of the sample in air (moisture ∼ 70%, room temperature) for more than a month and fortunately the quality of the X-ray data was good enough for structure solving. This indicated that the MOF crystal is reasonably stable in moisture, which was also proved by PXRD measurements (ESI Fig. S7†). The X-ray structure indicates that the ligand bends with a rectangular void (Fig. 2b) containing a free lying N,N-dimethylformamide (DMF) molecule and four vertical Co2+-coordinated DMF molecules (ESI Fig. S8†). The height of the void is ∼0.72 nm, and if the van der Waals radius was considered, it is ∼0.36 nm indicating that the void accommodates well a P4 molecule (Fig. 2b). Isophthalic acid groups are coordinated with Co2+ (Fig. 2b and c) to form coordinated tight layers (Fig. 2c and d) and between layers there are 2D nano-reactors (Fig. 2e), regardless of DMF molecules. In the tight layer, neighboring ligands are not from the same 2D nano-reactor but top or lower nano-reactors (Fig. 2c). Such a manner indicates that (1) guest P4 molecules cannot permeate through tight layers but stay in an individual 2D nano-reactor and (2) stacked 2D nano-reactors filled with a poly-phosphorus 2D network can be disassembled by breaking of coordination. Distances between neighboring diacetylene pillars in a 2D nano-reactor are 0.9, 1.5 and 1.6 nm, respectively, which are suitable for P4 insertion (Fig. 2e) but are not suitable for topo-polymerization of diacetylenes.30
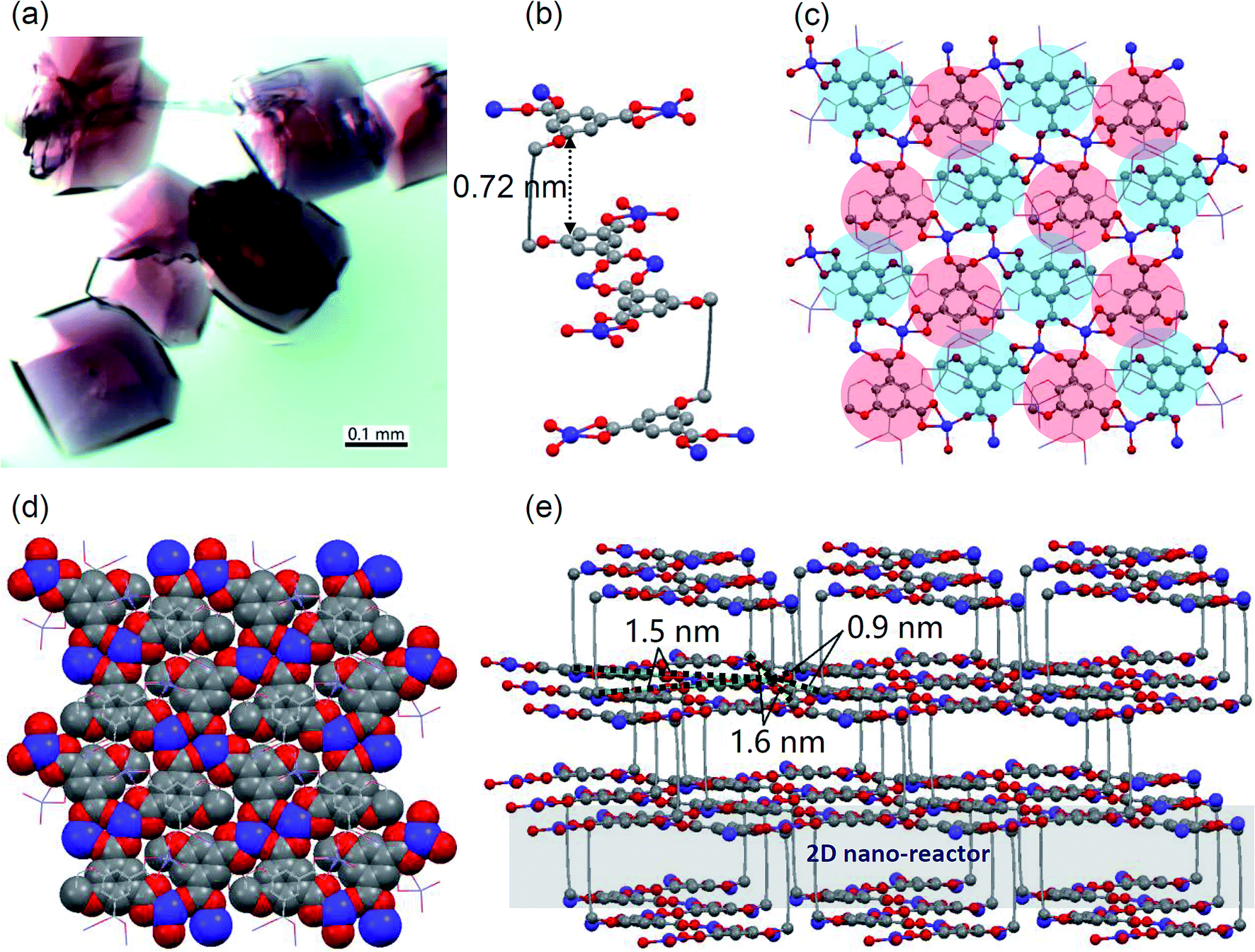 | ||
| Fig. 2 (a) The optical microscopy image of MOF crystals. SC-XRD analysis (DMF molecules and H-atoms are omitted for clarity; blue: Co, gray: C, red: O): (b) coordination between neighboring ligands; (c) ball-stick model of the coordinated tight layer (the blue disk indicates that the layer unit is from the top 2D nano-reactor, while the red disk indicates that the layer unit is from the lower 2D nano-reactor); (d) space-filling model of the coordinated tight layer; (e) 2D nano-reactors within the MOF and distances between pillars in a 2D nano-reactor. | ||
From the thermal gravimetric (TG) analysis of MOF crystals, the first slope (from 30 °C to 153 °C) is attributed to the loss of the free DMF molecule in the framework (Fig. 3a, ESI Fig. S9a†). Indeed, after the activation of MOF crystals at 140 °C for 10 h, the SC-XRD analysis demonstrated the disappearance of the free DMF molecule and maintenance of other four Co2+-coordinated DMF molecules (Fig. 3b, ESI Fig. S10†). Parameters of the crystal cell are almost the same before and after activation (ESI Fig. S9b†). Generally the cell volume should decrease after the removal of guest molecules.31 However, for our MOF crystal, its cell volume was maintained after activation (4009.4 Å3), which should be owing to our specially integrated rigid ligand. The TG analysis further reveals that coordinated DMF molecules within MOF crystals can be totally removed until ∼400 °C (Fig. 3a, ESI Fig. S9a†). As a lot of cracks appeared within crystals at such high temperature, here we used the partially activated MOF crystal which only lost the free DMF molecule as the nano-reactor.
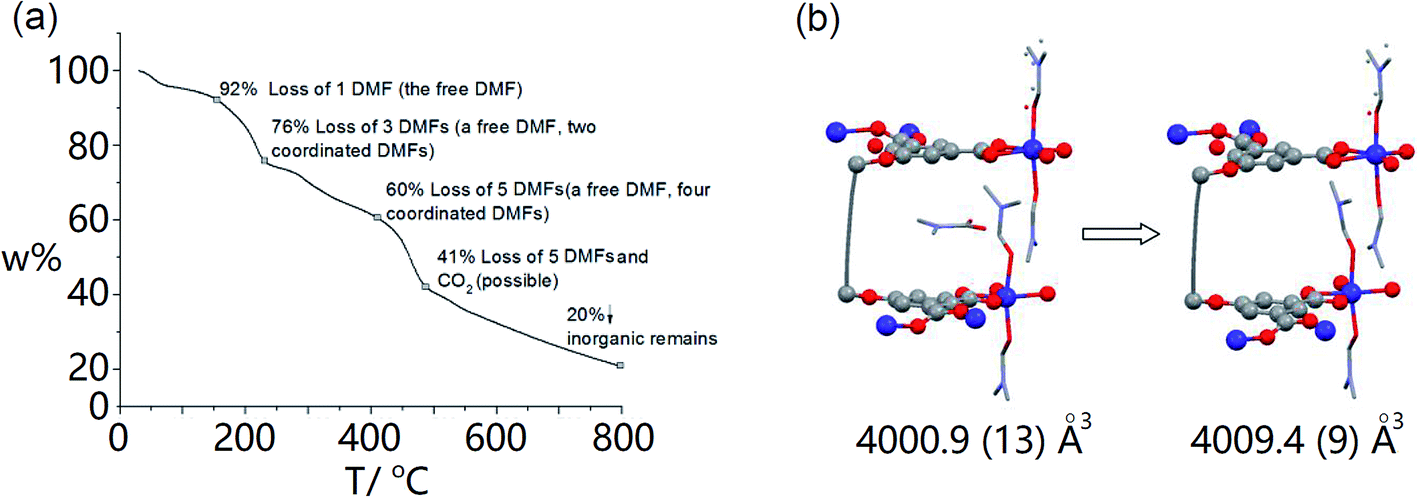 | ||
| Fig. 3 (a) TG analysis of MOF crystals; (b) SC-XRD analysis (crystal structure and cell parameters) of the MOF crystal before and after activation (the free DMF molecule in the void has conformational disorder due to the rotation of the C–N bond.). | ||
The uptake of P4 was conducted in a glass tube equipped with an argon and vacuum system (ESI Fig. S11†). Firstly, activated crystals were immersed in the melting P4 in the dark under intermittent vacuum. P4 molecules not only diffused into empty voids but also exchanged with coordinated DMF molecules during the removal of DMF molecules by the vacuum.32 After uptake of P4, MOF crystals were washed with chloroform and ethanol to remove remaining P4 on the surfaces (×3 times). Washed MOF crystals were then irradiated with UV-light (365 nm, 36 W) in air at room temperature for three days. The whole process was monitored by powder X-ray diffraction (PXRD), SEM, infrared (IR), Raman and X-ray photoelectron spectroscopies (XPS). Except for some neglectable shifts, peaks of the PXRD patterns of MOF crystals, MOF crystals filled with P4 (MOF@P4) and irradiated MOF@P4 are almost in the same region, which indicates that the main structure of the MOF is stable during P4 intrusion and polymerization (ESI Fig. S12†). Both SEM images of MOF@P4 and irradiated MOF@P4 exhibit smooth/flat crystal surfaces and homogenous distribution of P, C, Co and O elements (ESI Fig. S13 and 14†). As phosphorus on the surface has been washed away, such distribution reveals that phosphorus was encapsulated in the MOF. For IR spectra, peaks referring to coordinated DMF (3070 and 1424 cm−1) decreased after uptake of P4, indicating the removal of DMF (remaining tiny peaks referring to traces of DMF restricted in crystals did not decrease after irradiation, indicating DMF didn't react during irradiation). Other peaks are almost remained the same, demonstrating that the MOF itself didn't react during P4 insertion and irradiation (ESI Fig. S15†). Non-polar components such as phosphorus and diacetylene were characterized by Raman spectroscopy (Fig. 4a). After filling of P4, typical peaks of P4 at 364, 466 and 600 cm−1 appeared.21 Because P4 is highly self-combustible in air, the appearance of P4 peaks reveals that P4 must be encapsulated in MOF crystals.33 After irradiation, peaks of P4 disappeared but a broad region (355–500 cm−1) referring to poly-phosphorus appeared,34 while there was no visible peak referring to the P–C bond,35 indicating that all P4 has converted to poly-phosphorus. The typical peak of a pillar unit (diacetylene) at 2253 cm−1 remained in all spectra36 of the MOF, MOF@P4 and irradiated MOF@P4. The C 1s envelope in XPS measurements reveals that all peaks referring to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C, C
C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–C, C–O and COO remained the same with no new peaks referring to the P–C bond (ESI Fig. S16†).35 These spectra all exclude significant chemical bonding between poly-phosphorus and MOF groups. XPS spectra of the P 2p envelope of MOFs@P4 and irradiated MOFs@P4 all show an elemental phosphorus P (0) peak (Fig. 4b and c), which also indicates that P4 has been encapsulated in MOF crystals. A P (oxid.) peak besides the P (0) peak can also be observed in XPS measurements, which can be oxidized phosphorus, while IR spectra show no visible P (oxid.) signal (ESI Fig. S15†).37 This difference indicates that partial oxidation only occurred at the surface (e.g., P4 molecules present at the open edges of MOFs21), because IR spectra show data from the whole material (including deep in the material) but XPS spectra only show surface-data. Moreover, irradiation of the sample in air didn't decrease the proportion of elemental phosphorus in MOFs (Fig. 3d), which is further evidence that oxidation only occurred at the surface upon the first exposure of the sample to air.
C, C–C, C–O and COO remained the same with no new peaks referring to the P–C bond (ESI Fig. S16†).35 These spectra all exclude significant chemical bonding between poly-phosphorus and MOF groups. XPS spectra of the P 2p envelope of MOFs@P4 and irradiated MOFs@P4 all show an elemental phosphorus P (0) peak (Fig. 4b and c), which also indicates that P4 has been encapsulated in MOF crystals. A P (oxid.) peak besides the P (0) peak can also be observed in XPS measurements, which can be oxidized phosphorus, while IR spectra show no visible P (oxid.) signal (ESI Fig. S15†).37 This difference indicates that partial oxidation only occurred at the surface (e.g., P4 molecules present at the open edges of MOFs21), because IR spectra show data from the whole material (including deep in the material) but XPS spectra only show surface-data. Moreover, irradiation of the sample in air didn't decrease the proportion of elemental phosphorus in MOFs (Fig. 3d), which is further evidence that oxidation only occurred at the surface upon the first exposure of the sample to air.
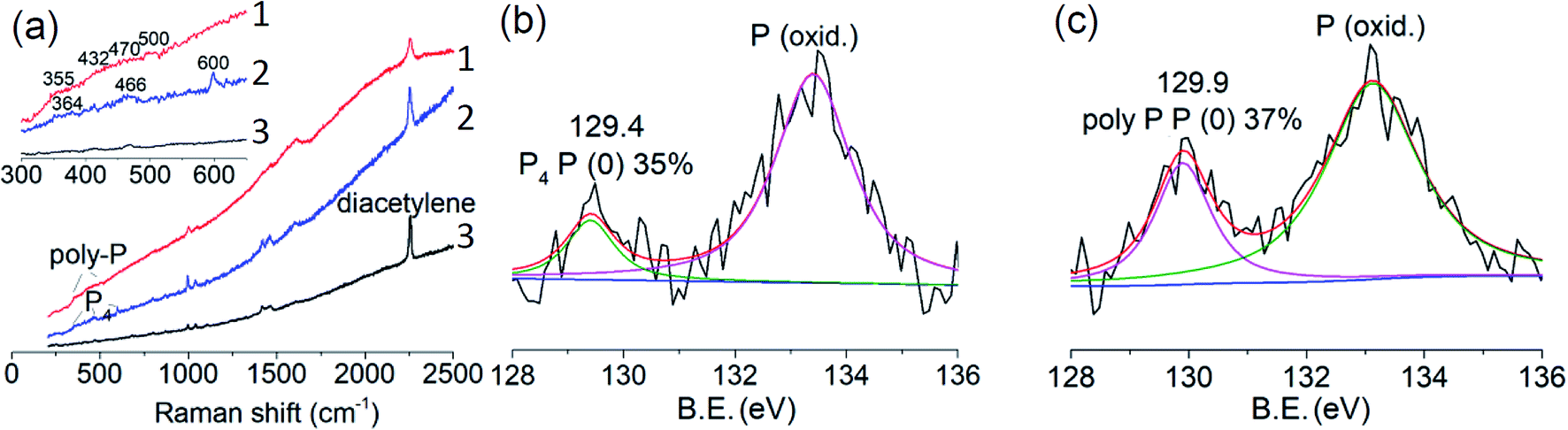 | ||
| Fig. 4 (a) Raman spectra of the MOF (curve 1), MOFs@P4 (curve 2) and irradiated MOFs@P4 (curve 3), respectively; (b) XPS spectrum of the P 2p envelope of MOFs@P4; (c) XPS spectrum of the P 2p envelope of irradiated MOFs@P4. | ||
After irradiation, coordination in crystals was disassembled by water under ultra-sonication for a few seconds at room temperature. The dissembling process could be visualized by SEM imaging. As shown in Fig. 5a, stacked thinner sheets with nm-thickness were delaminated from irradiated crystals, similar to opening a book. This exfoliation property unambiguously proves that the 2D polymeric material formed in MOF crystals; otherwise, there is no chance for such exfoliation.38 Since polymerization of the ligand (diacetylene unit) itself is impossible (as shown in Fig. 2e), then there must be polymerization of P4. The EDX spectrum further demonstrated that elements in this material are in complete accord with the expectation, including P, Co, C and O (inset in Fig. 5a; Cu is from the TEM grid). For ultra-sonication for a longer time (15 min), a thin sheet can be extracted from the supernatant, as shown in the SEM image in Fig. 5b. The EDX mappings also reveal expected elements P, C, O and Co distributed within this sheet (insets in Fig. 5b, and ESI Fig. S17†). Compared to partially exfoliated crystals in Fig. 5a, Co remaining on this sheet is very limited, which may be owing to the fact that many Co ions have been extracted into water during exfoliation. TEM imaging showing stacked layers with some natural rolling-up at the margin also proves that thin sheets are derived from exfoliation (Fig. 5c), whose chemical compositions include expected phosphorus and also very limited Co as well (ESI Fig. S18†). The XPS measurements for the bulk sample of these films majorly show elements of P, C and O besides traces of Co (ESI Fig. S19†). The selected area electron diffraction (SAED) spectrum reveals the reasonable internal order within the thin sheet, which represents d-spacings of 2.1 Å, 1.9 Å, 1.8 Å, 1.3 Å and 1.2 Å (the inset in Fig. 5c). HRTEM imaging also reveals ordered domains with d-spacings of 2.5 Å, 2.1 Å and 1.8 Å (Fig. 5d). These d-spacing values generally appear in diffraction spectra39 and the HRTEM image40 of black phosphorus, indicating that small domains of this 2D material may have a black phosphorus phase, in accordance with three characteristic regions in the Raman spectrum at 355 cm−1 (Ag−1), 432 cm−1 (B2g) and 470 cm−1 (Ag2) (Fig. 4a). Until now, except for a few examples, many 2D covalent materials have shown no internal order or their ordered domain is small. The partial internal order of our product reveals that preparation in a 2D-nanoreactor has the advantage of endowing internal order to the resulting 2D covalent material. Anyway, further research is required on how to obtain products with long-range internal order, e.g., tuning the MOF to perfectly fit the structure of the designed 2D material and controlling reaction conditions.
 | ||
| Fig. 5 (a) SEM image and the EDX spectrum (inset) of an irradiated MOFs@P4 with partial exfoliation; (b) SEM image of a thin sheet and the EDX mapping of the P element (inset); (c) TEM image and SAED spectrum (inset) of thin sheets; (d) HRTEM image of the thin sheet. | ||
The monolayer character of thin sheets was visualized by AFM imaging (Fig. 6a), corresponding to the predicted monolayer thickness of ∼2 nm (Fig. 1b and d). Larger area surveys reveal that the thickness of these irregular shaped sheets is quite homogenous at ∼2 nm with lateral sizes of ∼1.5 μm (ESI Fig. S20†). A large area (>0.2 mm) XPS mapping further reveals that the existence of phosphorus species (red spots) with homogenous lateral sizes is similar to the AFM imaging (Fig. 6b). The intensity ratio between the phosphorus atom and the penetrated ligand is about 6 (ESI Fig. S21†), in accordance with the structure shown in Fig. 1d revealing that statistically there are at least 6 phosphorus atoms surrounding a ligand. The high resolution XPS shows that the P 2p envelope is made up of P (0) and P (oxid.) species (inset in Fig. 6b). The oxidized phosphorus species should have resulted from oxidation in air. Deeper oxidation can be proved by the absolutely oxidized phosphorus species after storing the sample in air for 15 d (ESI Fig. S22†). This deeper oxidation of the exfoliated sample is different from the crystal of MOF@P4 (Fig. 3b and c), because for the exfoliated sample, all phosphorous was exposed to air while for the crystal of MOF@P4, most of the phosphorus was stored in the framework. Actually, even red phosphorus can be oxidized from the surface41 and C–O species can be observed in the XPS spectra of other 2D materials such as graphene42 and graphdiyne.43 As all our exfoliation experiments were conducted in air, the existence of P–P can still give a conclusion that this 2D phosphorus material has some higher stability compared to white phosphorus and black phosphorene, which is totally unstable in air. This higher stability may be due to the protection effect from penetrated ligands.
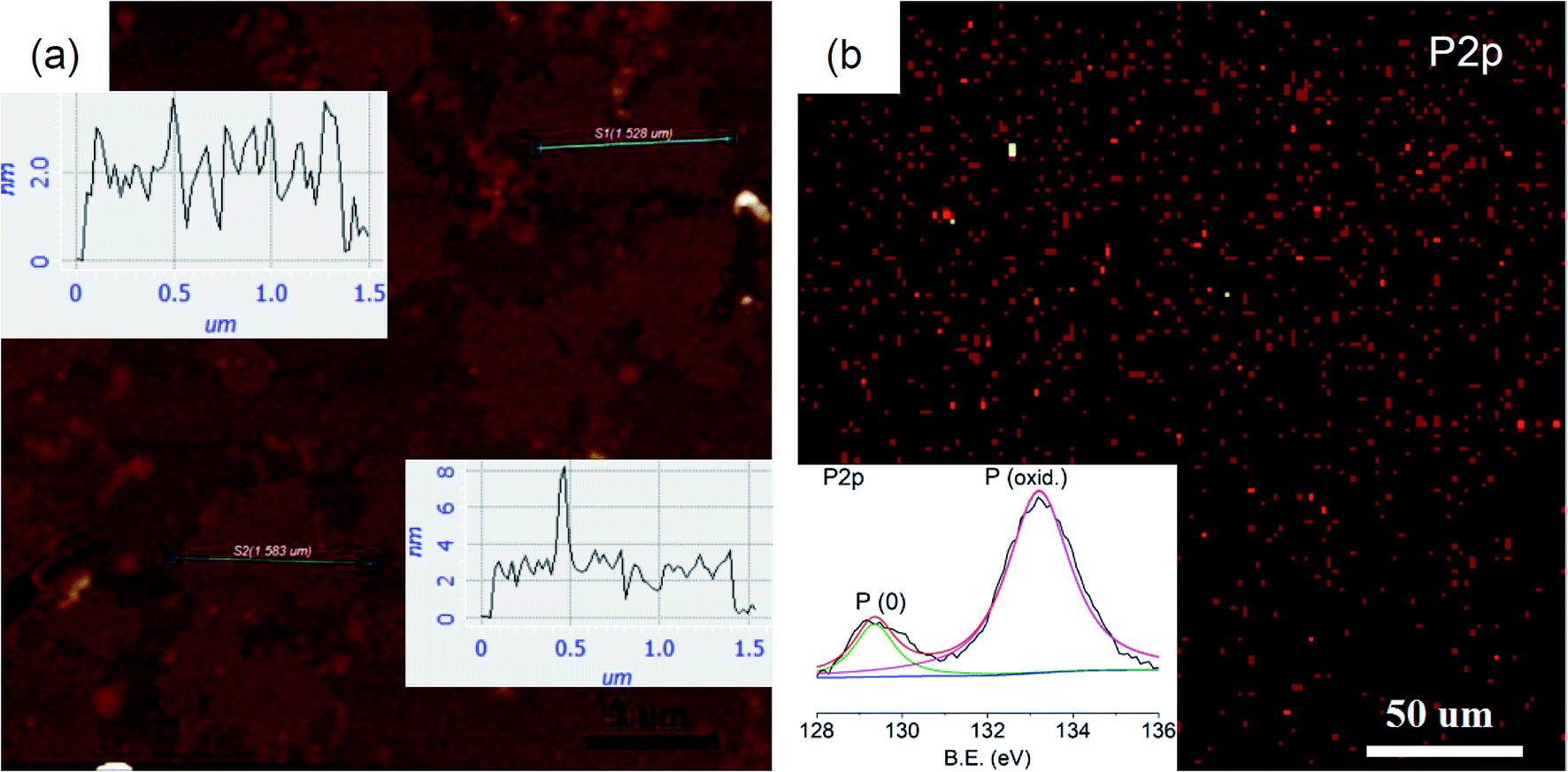 | ||
| Fig. 6 (a) AFM image and height profiles (insets) of thin sheets; (b) XPS mapping and spectrum (inset) of P 2p of thin sheets. | ||
Conclusions
In summary, we have realized a new strategy to synthesize 2D materials: polymerization in 2D nano-reactors. As a conceptual application of this strategy, a 2D nano-reactor was firstly constructed within an air- and moisture-stable pillar–layer MOF and then a 2D phosphorus material was directly synthesized from P4 by using this 2D nano-reactor. The MOF was synthesized from a new integrated ligand with a slimmest pillar unit (diacetylene). Further applications of this 2D-nano-reactor to synthesize more 2D materials such as 2D-metals (indium) are under investigation. Our research may provide a new approach for the synthesis of a variety of other 2D materials and a facile method to safely-store and transport highly self-combustible white phosphorus.33,44Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support to this work by the NSFC (21504023) and the Science Foundation of Hubei (2015CFB272) is gratefully acknowledged. Authors thank Xing Lu for his kind help on XRD measurement, Lei Zhang and Keyu Zheng for sharing a lab. M. Li is the project leader, conceived completely the project, did part of the experiments and wrote the paper. C. Ma mainly did experiments. X. Liu, J. Su and X. Cui provided SEM, TEM and NMR matters, respectively. Y. He shared a lab.Notes and references
- M. Acerce, E. K. Akdoğan and M. Chhowalla, Nature, 2017, 549, 370–373 CrossRef PubMed.
- F. Shahzad, M. Alhabeb, C. B. Hatter, B. Anasori, S. Man Hong, C. M. Koo and Y. Gogotsi, Science, 2016, 353, 1137–1140 CrossRef PubMed.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef PubMed.
- S. Balendhran, S. Walia, H. Nili, S. Sriram and M. Bhaskaran, Small, 2015, 11, 640–652 CrossRef PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef PubMed.
- Y. Li, H. Liu, H. Wang, J. Qiu and X. Zhang, Chem. Sci., 2018, 9, 4132–4141 RSC.
- H. Sahabudeen, H. Qi, B. A. Glatz, D. Tranca, R. Dong, Y. Hou, T. Zhang, C. Kuttner, T. Lehnert, G. Seifert, U. Kaiser, A. Fery, Z. Zheng and X. Feng, Nat. Commun., 2016, 7, 13461 CrossRef PubMed.
- J. Liu, W. Zan, K. Li, Y. Yang, F. Bu, W. Bao and Y. Xu, J. Am. Chem. Soc., 2017, 139, 11666–11669 CrossRef PubMed.
- W. Liu, X. Luo, Y. Bao, Y. P. Liu, G.-H. Ning, I. Abdelwahab, L. Li, C. T. Nai, Z. G. Hu, D. Zhao, B. Liu, S. Y. Quek and K. P. Loh, Nat. Chem., 2017, 9, 563–570 CrossRef PubMed.
- Z. Wang, A. Błaszczyk, O. Fuhr, S. Heissler, C. Wöll and M. Mayor, Nat. Commun., 2017, 8, 14442 CrossRef PubMed.
- P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774–778 CrossRef PubMed.
- L. Cao, Z. Lin, F. Peng, W. Wang, R. Huang, C. Wang, J. Yan, J. Liang, Z. Zhang, T. Zhang, L. Long, J. Sun and W. Lin, Angew. Chem., Int. Ed., 2016, 55, 4962–4966 CrossRef PubMed.
- Q. Lu, M. Zhao, J. Chen, B. Chen, C. Tan, X. Zhang, Y. Huang, J. Yang, F. Cao, Y. Yu, J. Ping, Z. Zhang, X.-J. Wu and H. Zhang, Small, 2016, 12, 4669–4674 CrossRef PubMed.
- D. Xu, X. Liu, H. Lv, Y. Liu, S. Zhao, M. Han, J. Bao, J. He and B. Liu, Chem. Sci., 2018, 9, 4451–4455 RSC.
- K. Miura, M. Kawase, R. Ashida, I. Gerlach and T. Yamamoto, Chem. Eng. Sci., 2007, 62, 5655–5660 CrossRef.
- D. M. Vriezema, M. Comellas Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1490 CrossRef PubMed.
- J. Lee, S. M. Kim and I. S. Lee, Nano Today, 2014, 9, 631–667 CrossRef.
- W. Cai, L. Bao, S. Zhao, Y. Xie, T. Akasaka and X. Lu, J. Am. Chem. Soc., 2015, 137, 10292–10296 CrossRef PubMed.
- A. N. Khlobystov, ACS Nano, 2011, 5, 9306–9312 CrossRef PubMed.
- J. Zhang, D. Zhao, D. Xiao, C. Ma, H. Du, X. Li, L. Zhang, J. Huang, H. Huang, C.-L. Jia, D. Tománek and C. Niu, Angew. Chem., 2017, 129, 1876–1880 CrossRef.
- M. Hart, E. R. White, J. Chen, C. M. McGilvery, C. J. Pickard, A. Michaelides, A. Sella, M. S. P. Shaffer and C. G. Salzmann, Angew. Chem., 2017, 129, 8256–8260 CrossRef.
- N. Yanai, T. Uemura, M. Ohba, Y. Kadowaki, M. Maesato, M. Takenaka, S. Nishitsuji, H. Hasegawa and S. Kitagawa, Angew. Chem., Int. Ed., 2008, 47, 9883–9886 CrossRef PubMed.
- T. Uemura, N. Yanai and S. Kitagawa, Chem. Soc. Rev., 2009, 38, 1228–1236 RSC.
- T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC.
- N. Ding, H. Li, X. Feng, Q. Wang, S. Wang, L. Ma, J. Zhou and B. Wang, J. Am. Chem. Soc., 2016, 138, 10100–10103 CrossRef PubMed.
- A. Dutta, A. G. Wong-Foy and A. J. Matzger, Chem. Sci., 2014, 5, 3729–3734 RSC.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef PubMed.
- H.-N. Wang, X. Meng, C. Qin, X.-L. Wang, G.-S. Yang and Z.-M. Su, Dalton Trans., 2012, 41, 1047–1053 RSC.
- M. Donath, F. Hennersdorf and J. J. Weigand, Chem. Soc. Rev., 2016, 45, 1145–1172 RSC.
- M. Li, M. Song, G. Wu, Z. Tang, Y. Sun, Y. He, J. Li, L. Li, H. Gu, X. Liu, C. Ma, Z. Peng, Z. Ai and D. J. Lewis, Small, 2017, 13, 1604077 CrossRef PubMed.
- C. J. Kepert and M. J. Rosseinsky, Chem. Commun., 1999, 375–376 RSC.
- H. J. Park and M. P. Suh, Chem.–Eur. J., 2008, 14, 8812–8821 CrossRef PubMed.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef PubMed.
- Y. Lu, P. Zhou, K. Lei, Q. Zhao, Z. Tao and J. Chen, Adv. Energy Mater., 2017, 7, 1601973 CrossRef.
- J. Sun, G. Zheng, H.-W. Lee, N. Liu, H. Wang, H. Yao, W. Yang and Y. Cui, Nano Lett., 2014, 14, 4573–4580 CrossRef PubMed.
- Z. Tang, M. Li, M. Song, L. Jiang, J. Li, Y. He and L. Zhou, Org. Electron., 2017, 49, 174–178 CrossRef.
- P. A. Connor and A. J. McQuillan, Langmuir, 1999, 15, 2916–2921 CrossRef.
- L. Niu, J. N. Coleman, H. Zhang, H. Shin, M. Chhowalla and Z. Zheng, Small, 2016, 12, 272–293 CrossRef PubMed.
- J. R. Brent, N. Savjani, E. A. Lewis, S. J. Haigh, D. J. Lewis and P. O'Brien, Chem. Commun., 2014, 50, 13338–13341 RSC.
- G. Lee, J. Y. Lee, G. H. Lee and J. Kim, J. Mater. Chem. C, 2016, 4, 6234–6239 RSC.
- J. Brunner, M. Thüler, S. Veprek and R. Wild, J. Phys. Chem. Solids, 1979, 40, 967–971 CrossRef.
- H. Au, N. Rubio and M. S. P. Shaffer, Chem. Sci., 2018, 9, 209–217 RSC.
- S.-S. Wang, H.-B. Liu, X.-N. Kan, L. Wang, Y.-H. Chen, B. Su, Y.-L. Li and L. Jiang, Small, 2017, 13, 1602265 CrossRef PubMed.
- D. Yang, J. Zhao, L. Yu, X. Lin, W. Zhang, H. Ma, A. Gogoll, Z. Zhang, Y. Wang, X.-J. Yang and B. Wu, J. Am. Chem. Soc., 2017, 139, 5946–5951 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1820169 and 1819935. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01439f |
| ‡ These authors are co-first authors. |
| This journal is © The Royal Society of Chemistry 2018 |