DOI:
10.1039/C8SC01388H
(Edge Article)
Chem. Sci., 2018,
9, 5100-5105
Curved π-conjugated corannulene dimer diradicaloids†
Received
26th March 2018
, Accepted 12th May 2018
First published on 16th May 2018
Abstract
So far, most reported open-shell singlet diradicaloids are based on planar π-conjugated molecules. Herein, we report the bridged corannulene dimer diradicaloids, Cor-D1 and Cor-D2, both showing a three-dimensional curved π-conjugated structure. Cor-D1 has a small diradical character (y0 = 5.4%) and behaves more like a closed-shell quinoidal compound at room temperature, while Cor-D2 is a typical open-shell diradicaloid with a larger diradical character (y0 = 16.9%). Both compounds exhibited paramagnetic activity at elevated temperatures, with a singlet–triplet energy gap (ΔES–T) of −8.4 and −3.0 kcal mol−1, respectively. X-ray crystallographic analysis revealed that both molecules have a dumbbell-shaped geometry, with the two terminal corannulene bowls bent to opposite directions. The spin is largely delocalized onto the two bowls in Cor-D2 and there are multiple [CH⋯π] interactions between the neighboring bowls. Chemical oxidation/reduction to their respective dications/dianions results in global aromaticity with [4n + 2] π-electrons delocalized through the periphery of the whole framework.
Introduction
Recently there have been increasing research interests on open-shell singlet diradicaloids due to their unique magnetic activity and potential applications in organic electronics, non-linear optics and spintronics.1 So far, most reported open-shell singlet diradicaloids are based on planar π-conjugated molecules or their oligomers, and there are very rare examples based on non-planar systems.2 Our particular interest here is to exploit stable diradicaloids based on curved π-conjugated molecules, which may exhibit unique three-dimensional (3D) spin distribution nature and intra- and intermolecular magnetic exchange interactions. Bowl-shaped corannulene is a very good candidate given its curved structure. It was reported that the radical anions of corannulene and its derivatives showed effective spin delocalization throughout the π-conjugated framework but they were still highly reactive and some of them tended to form intermolecular C–C σ-bond linked dimer or polymer.3 On the other hand, stable neutral radicals-substituted corannulenes such as Cor-Ver (verzazyl), Cor-IN (iminonitroxide), Cor-PhO (phenoxyl) and Cor-tBuNO (tert-butylnitroxide) (Fig. 1a) were prepared and in all cases, the spin can be partially delocalized to the corannulene bowl.4 The phenalenyl-fused corannulene anion was synthesized and fully characterized, but the property of its neutral radical Cor-PLY (Fig. 1a) was not reported, presumably due to its poor stability.5 The corannulene-bridged phenoxyl diradical Cor-2PhO showed significant intramolecular magnetic interaction with a singlet–triplet energy gap (ΔES–T = 2JkB−1) of −810 K (−1.61 kcal mol−1).6 Calculations reveal that the unpaired electronic spin in the triplet state is delocalized onto the corannulene skeleton from the phenoxyl moieties. However, so far, corannulene-based radicals with more than one corannulene unit have never been reported. Herein, we report the first two stable π-conjugated corannulene dimer diradicaloids, Cor-D1 and Cor-D2, in which the two corannulene units are annulated with a para-quinodimethane (p-QDM) and 2,6-naphthoquinodimethane (2,6-NQDM) moiety, respectively (Fig. 1b). The central pro-aromatic quinodimethanes have irresistible wish to become diradicals by recovering aromaticty,1d and one can expect a significant contribution of the open-shell diradical form to the ground-state electronic structure. Therefore, they are ideal open-shell singlet diradicaloids with a unique 3D curved π-conjugated structure and the spin is supposed to be largely delocalized onto the two corannulene bowls (Fig. 1b).
 |
| | Fig. 1 (a) Structures of neutral radical-substituted corannulenes, phenalenyl-fused corannulene, and corannulene-bridged phenoxyl diradical. (b) Structures of the new p-QDM and 2,6-NQDM bridged corannulene dimer diradicaloids Cor-D1 and Cor-D2. The closed-shell quinoidal and open-shell diradical resonance forms are shown for Cor-D2. Mes: mesityl. | |
Results and discussion
Synthesis
The synthesis of π-extended corannulenes is challenging7 and our synthesis was based on regio-selective reactions starting from the 1-methoxy-corannulene 1 (ref. 8) (Scheme 1). Mono-bromination of 1 at the ortho-position by N-bromosuccinimide (NBS) in the presence of diisopropylamine gave the 1-bromo-2-methoxy-corannulene 2 in 85% yield. The bromo-group was then converted into pinacol boronate in 3 by lithiation followed by quenching with 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane. Suzuki coupling reaction between 3 and the 2,5-dibromoterephthalaldehyde (4) or 3,7-diformylnaphthalene-2,6-diyl bis(trifluoromethanesulfonate) (5)9 gave the corresponding dialdehyde 6a or 6b. Nucleophilic addition of the aldehyde groups in 6a/6b with excessive mesitylmagnesium bromide generated the diol intermediates and subsequent BF3·Et2O mediated intramolecular Friedel–Crafts alkylation afforded the dihydro-precursors 7a/7b. At this step, the existence of a methoxy group on each corannulene unit ensured a six-membered ring cyclized product only and avoided possible formation of five-membered rings.10 Oxidative dehydrogenation of 7a/7b with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in toluene at 80 °C gave the final desired products Cor-D1 and Cor-D2, respectively. The bulky mesityl groups are essential to kinetically block the reaction sites and also make the final products reasonably soluble. Both Cor-D1 and Cor-D2 are stable and can be purified by normal silica gel column chromatography.
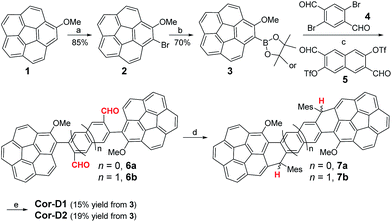 |
| | Scheme 1 Synthetic routes of Cor-D1 and Cor-D2. Reagents and conditions: (a) NBS, i-Pr2NH, DCM, rt; (b) (i) n-BuLi, −78 °C, THF; (ii) 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, −78 °C – rt; (c) Pd(PPh3)4, K2CO3, toluene/EtOH/H2O (2/1/1), 110 °C; (d) (i) mesitylmagnesium bromide, THF, rt; (ii) BF3·OEt2, DCM, rt; (e) DDQ, toluene, 80 °C. | |
Ground-state geometry
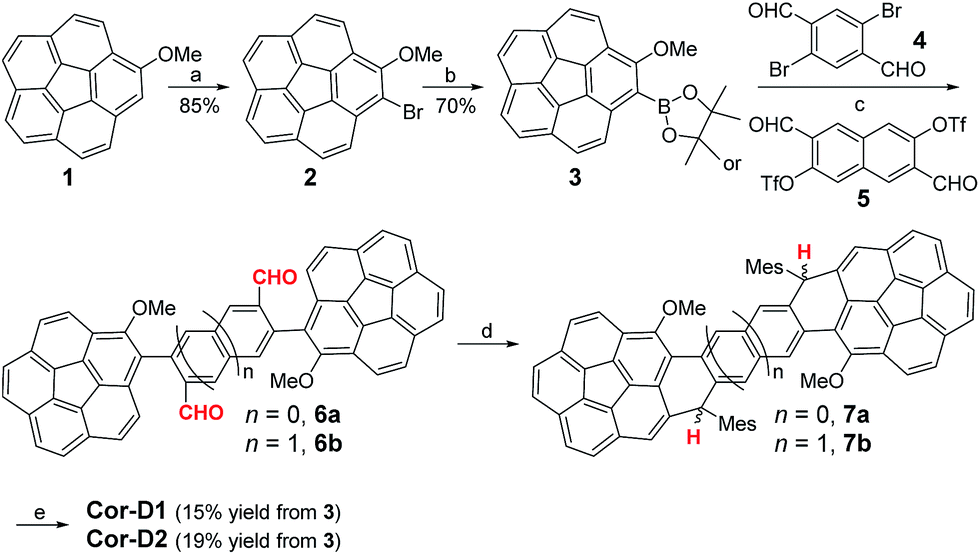
Single crystals of Cor-D1 and Cor-D2 suitable for X-ray crystallographic analysis were grown by slow solvent evaporation from chlorobenzene solution.11 Both molecules adopt a double-curved geometry, with the two corannulene bowls bent to opposite directions (Fig. 2). The bowl depth (d) of Cor-D1 is measured to be 0.89 Å, which is slightly larger than that of parent corannulene (d = 0.87 Å),12 and Cor-D2 shows even larger bowl depth of 0.94 Å. No intermolecular close contact is found between Cor-D1 molecules, and chlorobenzene molecules fill the void with [CH⋯π] interactions with backbone of Cor-D1. On the other hand, multiple [CH⋯π] interactions (distance: 2.711 Å) between neighboring corannulene bowls are found in the crystallographic structure of Cor-D2, and there are additional [CH⋯π] interactions between the methyl groups and the corannulene bowls (distance: 2.736 Å), both drive the molecules into a densely packed structure (Fig. 2b). The central p-QDM unit in Cor-D1 has a large bond length alternation (BLA) (Fig. 2a), indicating that the quinoidal structure is more important. However, the exo-methylene bond, C1–C2 (1.388(13) Å), is significantly longer than that of typical olefins (1.33–1.34 Å), implying certain contribution of diradical form. The central 2,6-NQDM unit in Cor-D2 also has significant BLA, with the exo-methylene C1–C2 bond being elongated (1.389(8) Å) (Fig. 2b). Natural orbital occupation number (NOON) calculations at UCAM-B3LYP/6-31G(d,p) level based on the X-ray structures suggest that Cor-D1 and Cor-D2 have a diradical character (y0) of 5.4% and 16.9%, respectively. Interestingly, a dihydro-product (Cor-D2-2H), the anti-isomer of 7b, was found during the crystal growth of Cor-D2 by slow evaporation of chlorobenzene at 60 °C. The structure was confirmed by X-ray crystallographic analysis (Fig. S60 in ESI†), and the formation can be explained by the hydrogen abstraction from solvents at the most reactive sites (C1). Variable-temperature (VT) 1H NMR spectra of Cor-D1 in CDCl3 (245–335 K) revealed splitting of the protons for both the central benzenoid ring, the OMe group, and the ortho-methyl groups in the mesityl substituents below the coalescent temperature Tc (≈280 K for the central benzene ring protons) (Fig. S1 in ESI†). Due to the bowl-shaped corannulene units, there would be two isomers of Cor-D1: one with two terminal bowls bent to the opposite directions (anti-form) and the other with two terminal bowls bent to the same directions (syn-form). Our calculations (B3LYP/6-31G(d,p)) predict that the syn-form is 0.538 kcal mol−1 higher in energy than the anti-isomer (Table S13 in ESI†), and thus the species with higher NMR integration can be correlated to the anti-isomer. The bowl inversion rate constant k at different temperatures was estimated by careful line-shape analysis13 and fitting of the data by Eyring equation (ln(k/T) = −ΔH‡/R × 1/T + ln(kB/h) + ΔS‡/R) gave thermodynamic parameters ΔH‡ = 19.8 ± 0.6 kcal mol−1 and ΔS‡ = 21.9 ± 2.3 cal (mol−1 K). Accordingly, ΔG‡ was estimated to be 13.3 ± 1.3 kcal mol−1 at 298 K (Fig. S2 and Table S1 in ESI†). This barrier is larger than the parent corannulene (∼11.5 kcal mol−1) but slightly smaller than that of the corannulene annulated with a –CH2–CE2–CH2– (E = COOCH3) ring (∼15.5 kcal mol−1).12bCor-D2 showed similar behavior with slightly lower Tc (≈270 K for central naphthalene ring protons) (Fig. S3 in ESI†) and line-shape analysis gave ΔH‡ = 19.7 ± 1.4 kcal mol−1, ΔS‡ = 22.5 ± 5.3 cal (mol−1 K−1) and ΔG‡ (298 K) = 13.0 ± 3.0 kcal mol−1 (Fig. S4 and Table S2 in ESI†). Calculations show that the anti- and syn-isomers of Cor-D2 have nearly identical energy (Table S13 in ESI†), and thus both isomers exist equally in solution.
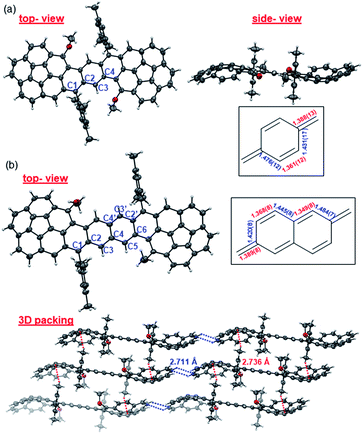 |
| | Fig. 2 X-ray crystallographic structures of (a) Cor-D1 and (b) Cor-D2 (top-view, side-view and 3D packing), together with selected bond lengths (in Å) for the central p-QDM and 2,6-NQDM units. | |
Optical, electrochemical and magnetic properties
Compound Cor-D1 is blue while Cor-D2 is green in dichloromethane (DCM). Cor-D1 shows an intense absorption band with maximum (λmax) at 612 nm (Fig. 3a), which can be correlated to HOMO → LUMO electronic transition based on time-dependent (TD) DFT calculations (see ESI†). Cor-D2 displays a similar intense absorption band with λmax at 690 nm, but with one weak shoulder at 743 nm. The appearance of such weak shoulder at long wavelength is characteristic for many open-shell singlet diradicaloids, which can be mainly correlated to the H,H → L,L double excitation.14 This is in accordance with the calculated moderate diradical character for Cor-D2. Our TD DFT calculations show that the anti- and syn-isomers show almost the same absorption spectra (ESI†) and thus one cannot simply distinguish the contribution of the individual isomers.
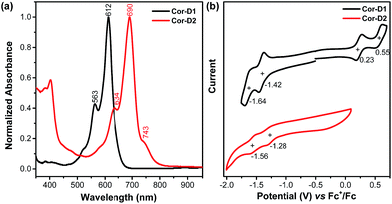 |
| | Fig. 3 (a) Normalized UV-vis absorption spectra in DCM and (b) cyclic voltammograms of Cor-D1 and Cor-D2 in 1,2-DCB at 50 °C. | |
Cor-D1 showed sharp NMR spectra in THF-d8/CS2, CDCl3 and D-toluene even at 373 K. Its solid powder displayed a weak broad electron spin resonance (ESR) signal (Fig. S5 in ESI†). Heating led to increase of the ESR intensity due to thermal population from the diamagnetic singlet state to the paramagnetic triplet state. Fitting of the IT–T data by Bleaney–Bowers equation15 gave a large singlet-triplet energy gap (ΔES–T) of −8.4 kcal mol−1 (Fig. S6 in ESI†). Such a large gap is in accordance with its very small diradical character as well as the observed sharp NMR spectra. Cor-D2 also exhibited sharp NMR spectra in THF-d8/CS2 and CDCl3 (from 245 K to 335 K). However, NMR spectral broadening in toluene-d8 was observed along with the elevated temperatures from 333 K to 373 K (Fig. S10 in ESI†). This phenomenon is consistent with its larger diradical character. The powder of Cor-D2 exhibited an intense one-line ESR signal with ge = 2.0027 at room temperature (Fig. S7 in ESI†), indicating existence of thermally populated paramagnetic species. Superconducting quantum interference device (SQUID) measurement on the powder sample of Cor-D2 revealed that the product of molar magnetic susceptibility and temperature (χMT) increased after 150 K and fitting of the data using Bleaney–Bowers equation gave a moderate ΔES–T = −3.0 kcal mol−1 (Fig. 4). The calculated spin-density distribution maps for the triplet diradical of Cor-D2 in both anti- and syn-forms show that the unpaired electronic spin is delocalized onto the two corannulene bowls, with the largest spin density distributed at the zigzag edges (inset in Fig. 4 and S15 in ESI†).
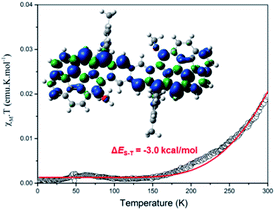 |
| | Fig. 4
χ
M
T–T curve in the SQUID measurement for the solid of Cor-D2 and the red line is the fitted curve by Bleaney–Bows equation. Inset is the calculated spin density distribution map of its triplet biradical. | |
Cyclic voltammetry measurements of Cor-D1 and Cor-D2 were carried out in dry 1,2-dichlorobenzene (1,2-DCB) under heating (50 °C) which can suppress molecular aggregation and give better resolved redox waves (Fig. 3b). Cor-D1 exhibited two oxidation waves with half-wave potential Eox1/2 = 0.23 and 0.55 V, and two reduction waves with half-wave potential Ered1/2 = −1.42 and −1.64 V (vs. Fc+/Fc). The HOMO and LUMO energy levels were calculated to be −5.03 and −3.38 eV from the onset of the first oxidation/reduction wave, respectively, and an electrochemical energy gap (EECg) of 1.65 eV was estimated. Cor-D2 showed two reduction waves at Ered1/2 = −1.28 and −1.56 V, with a LUMO energy level of −3.52 eV. Due to strong aggregation, clear oxidation wave could not be observed. Both compounds can be chemically oxidized into radical cation and dication by NO·SbF6 in dry DCM, and reduced into radical anion and dianion by sodium anthracenide (NaAn) in anhydrous THF (Fig. 5 and S11–S12 in ESI†). In both cases, the radical anion and radical cation show similar band structure, indicating a similar electronic structure, but the corresponding bands of radical anion are red-shifted compared to the radical cation (λmax = 1490 nm vs. 1417 nm in Cor-D1, and λmax = 1808 nm vs. 1700 nm in Cor-D2). The dianion and dication exhibit similar band structure and trend (λmax = 892 nm vs. 844 nm in Cor-D1, and λmax = 1030 nm vs. 960 nm in Cor-D2). Clear 1H NMR spectra of the dianions of Cor-D1 and Cor-D2 can be recorded by in situ reduction with NaAn in anhydrous THF-d8 and most of the aromatic protons appear above 7 ppm despite their electron-rich character (Fig. S13–S14 and S59 in ESI†), indicating that they are strongly aromatic compounds.
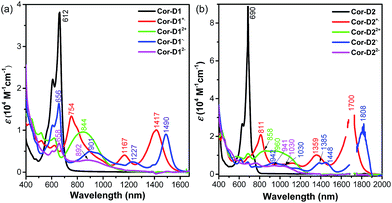 |
| | Fig. 5 UV-vis-NIR absorption spectra of the neutral, radical cation, dication, radical anion and dianion of Cor-D1 (a) and Cor-D2 (b), ε is the molar extinction coefficient. | |
To better understand the electronic structure, anisotropy of the induced current-density (ACID) plot16 and nucleus independent chemical shift (NICS)17 calculations were conducted at (U)B3LYP/6-31G(d,p) level (see Fig. 6 and more images in Fig. S16–S25 in ESI†). The neutral anti-Cor-D1 shows clear diamagnetic/paramagnetic ring current flow along the outer/inner ring of each corannulene bowl, like the parent corannulene (Fig. S26 in ESI†), while there is no ring current circuit at the central p-QDM unit, indicating its non-aromatic character. In accordance with this, the average NICS(1)zz value18 of the central benzene ring is near zero (−0.34 ppm). However, its dianion shows diamagnetic ring current along the periphery of the whole π-conjugated framework. At the same time, the average NICS(1)zz value of the central benzene ring becomes largely negative (−30.66 ppm), and even the inner cyclopenta-ring in the each corannulene unit shows significantly negative NICS(1)zz value (−14.75 ppm), indicating a strong global aromatic system. Apparently, 38 π electrons can be counted along the periphery, which satisfies Hückel's [4n + 2] aromaticity rule. Its dication exhibits similar global aromaticity with 34 π electrons delocalized along the periphery. The NICS(1)zz value of the central benzene ring (−12.45 ppm) is less negative than that in dianion, and that of the cyclopenta-ring becomes quite positive (+34.13 ppm), indicating relatively weak global aromaticity. In the anti-Cor-D2, the two corannulene bowls remain their individual aromatic character similar to that in Cor-D1, but diamagnetic ring current was found in the central naphthalene unit (NICS(1)zz: −10.18 ppm), indicating significant contribution of the open-shell diradical form. Its dianion also show global aromaticity with 42 π-electrons delocalized along the periphery and the NICS(1)zz value in the central naphthalene ring is shifted to −26.98 ppm. Its dication does not display global aromaticity as Cor-D12+, and the localized aromaticity at the central naphthalene unit dominates (NICS(1)zz: −12.74 ppm), making the two corannulene units somehow segregated. The radical anions and radical cations of both compounds with unpaired electron show partial global aromatic character (Fig. S22–S25 in ESI†).
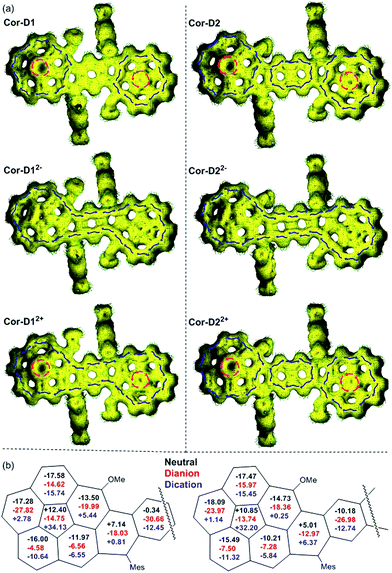 |
| | Fig. 6 Calculated ACID plots (a) and NICS values (b) of the neutral, dication and dianion of anti-Cor-D1 and Cor-D2. For ACID calculations, the magnetic field is perpendicular to the molecular plane and points out through the paper. Isovalue is 0.02, and the blue and red arrows indicate the diamagnetic and paramagnetic ring current flow, respectively. The NICS values are the average of NICS(1)zz and NICS(−1)zz at each individual ring. | |
Conclusions
In summary, rigid, p-QDM and 2,6-NQDM bridged corannulene dimers Cor-D1 and Cor-D2 were synthesized in stable form, which represent a new type of curved π-conjugated open-shell singlet diradicaloids. Cor-D1 has a small diradical character and behaves more like a closed-shell hydrocarbon at room temperature, while Cor-D2 possesses a moderate diradical character. Both compounds display magnetic activity at elevated temperatures. Electronic spins are distributed onto both corannulene bowls in Cor-D2, and stronger intermolecular interactions and more ordered packing are observed in its single crystals. Notably, their dianions show prominent global aromaticity with [4n + 2] π electrons delocalized along the periphery. Our studies reveal the unique electronic structure of such kind of bowl-shaped π-conjugated polycyclic hydrocarbons, which implies their potential applications for organic electronic and spintronic devices in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge financial support from the MOE Tier 3 programme (MOE2014-T3-1-004), MOE Tier 1 grant (R-143-000-623-112) and China “973” program (2015CB856500). We thank Dr Tan Geok Kheng for her help on X-ray crystallographic analysis.
Notes and references
-
(a) M. Abe, Chem. Rev., 2013, 113, 7011 CrossRef PubMed;
(b) Z. Sun, Z. Zeng and J. Wu, Acc. Chem. Res., 2014, 47, 2582 CrossRef PubMed;
(c)
M. Nakano, Excitation Energies and Properties of Open-Shell Singlet Molecules, Springer, New York, 2014 Search PubMed;
(d) Z. Zeng, X. Shi, C. Chi, J. T. López Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578 RSC;
(e) T. Kubo, Chem. Lett., 2015, 44, 111 CrossRef;
(f) P. Hu and J. Wu, Can. J. Chem., 2017, 95, 223 CrossRef;
(g) T. Y. Gopalakrishna, W. Zeng, X. F. Lu and J. Wu, Chem. Commun., 2018, 54, 2389 RSC.
-
(a) J. Ma, J. Liu, M. Baumgarten, Y. Fu, Y.-Z. Tan, K. S. Schellhammer, F. Ortmann, G. Cuniberti, H. Komber, R. Berger, K. Müllen and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 3280 CrossRef PubMed;
(b) H. Zhang, H. Phan, T. S. Herng, T. Y. Gopalakrishna, W. Zeng, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 13484 CrossRef PubMed.
-
(a) J. Janata, J. Gendell, C.-Y. Ling, W. Barth, L. Backs, H. B. Mark Jr and R. G. Lawton, J. Am. Chem. Soc., 1967, 89, 3506 CrossRef;
(b) A. Weitz, E. Shabtai, M. Rabinovitz, M. S. Bratcher, C. C. McComas, M. D. Best and L. T. Scott, Chem.–Eur. J., 1998, 4, 234 CrossRef;
(c) I. Aprahamian, R. E. Hoffman, T. Sheradsky, D. V. Preda, M. Bancu, L. T. Scott and M. Rabinovitz, Angew. Chem., Int. Ed., 2002, 41, 3702 CrossRef;
(d) I. Aprahamian, D. V. Preda, M. Bancu, A. P. Belanger, T. Sheradsky, L. T. Scott and M. Rabinovitz, J. Org. Chem., 2006, 71, 290 CrossRef PubMed.
-
(a) Y. Morita, S. Nishida, T. Kobayashi, K. Fukui, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, Org. Lett., 2004, 6, 1397 CrossRef PubMed;
(b) S. Nishida, Y. Morita, T. Kobayashi, K. Fukui, A. Ueda, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, Polyhedron, 2005, 24, 2200 CrossRef;
(c) Y. Morita, A. Ueda, S. Nishida, K. Fukui, T. Ise, D. Shiomi, K. Sato, T. Takui and K. Nakasuji, Angew. Chem., Int. Ed., 2008, 47, 2035 CrossRef PubMed;
(d) A. Ueda, K. Ogasawara, S. Nishida, K. Fukui, K. Sato, T. Takui, K. Nakasuji and Y. Morita, Aust. J. Chem., 2010, 63, 1627 CrossRef.
- S. Nishida, Y. Morita, A. Ueda, T. Kobayashi, K. Fukui, K. Ogasawara, K. Sato, T. Takui and K. Nakasuji, J. Am. Chem. Soc., 2008, 130, 14954 CrossRef PubMed.
- A. Ueda, S. Nishida, K. Fukui, T. Ise, D. Shiomi, K. Sato, T. Takui, K. Nakasuji and Y. Morita, Angew. Chem., Int. Ed., 2010, 49, 1678 CrossRef PubMed.
-
(a) Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843 CrossRef PubMed;
(b) A. Sygula and P. W. Rabideau, J. Am. Chem. Soc., 1999, 121, 7800 CrossRef;
(c) E. A. Jackson, B. D. Steinberg, M. Bancu, A. Wakamiya and L. T. Scott, J. Am. Chem. Soc., 2007, 129, 484 CrossRef PubMed;
(d) A. Sygula, F. R. Fronczek, R. Sygula, P. W. Rabideau and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 3842 CrossRef PubMed;
(e) B. D. Steinberg, E. A. Jackson, A. S. Filatov, A. Wakamiya, M. A. Petrukhina and L. T. Scott, J. Am. Chem. Soc., 2009, 131, 10537 CrossRef PubMed;
(f) T.-C. Wu, H.-J. Hsin, M.-Y. Kuo, C.-H. Li and Y.-T. Wu, J. Am. Chem. Soc., 2011, 133, 16319 CrossRef PubMed;
(g) A. Butterfield, B. Gilomen and J. S. Siegel, Org. Process Res. Dev., 2012, 16, 664 CrossRef;
(h) L. T. Scott, E. A. Jackson, Q. Zhang, B. D. Steinberg, M. Bancu and B. Li, J. Am. Chem. Soc., 2012, 134, 107 CrossRef PubMed;
(i) K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739 CrossRef PubMed;
(j) V. Rajeshkumar and M. C. Stuparu, Chem. Commun., 2016, 52, 9957 RSC.
-
(a) A. Sygula, A. Sygula and L. Kobryn, Org. Lett., 2008, 10, 3927 CrossRef PubMed;
(b) G. H. Grube, E. L. Elliott, R. J. Steffens, C. S. Jones, K. K. Baldridge and J. S. Siegel, Org. Lett., 2003, 5, 713 CrossRef PubMed.
- P. Hu, S. Lee, K. H. Park, S. Das, T. S. Herng, T. P. Gonçalves, K.-W. Huang, J. Ding, D. Kim and J. Wu, J. Org. Chem., 2016, 81, 2911 CrossRef PubMed.
- K. Ota, T. Tanaka and A. Osuka, Org. Lett., 2014, 16, 2974 CrossRef PubMed.
- CCDC no. for Cor-D1, Cor-D2 and Cor-D2-2H are 1570628, 1819179, 1819178, respectively.†.
-
(a) P. U. Biedermann, S. Pogodin and I. Agranat, J. Org. Chem., 1999, 64, 3655 CrossRef PubMed;
(b) T. J. Seiders, K. K. Baldridge, G. H. Grube and J. S. Siegel, J. Am. Chem. Soc., 2001, 123, 517 CrossRef PubMed.
-
(a) H. Shanan-Atidi and K. H. Bar-Eli, J. Phys. Chem., 1970, 74, 961 CrossRef;
(b) F. P. Gasparro and N. H. Kolodny, J. Chem. Educ., 1977, 54, 258 CrossRef;
(c) B. Kersting, J. R. Telford, M. Meyer and K. N. Raymond, J. Am. Chem. Soc., 1996, 118, 5712 CrossRef.
- S. D. Motta, F. Negri, D. Fazzi, C. Castiglioni and E. V. Canesi, J. Phys. Chem. Lett., 2010, 1, 3334 CrossRef.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451 CrossRef.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758 CrossRef PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef PubMed.
- We use the average of NICS(1)zz and NICS(-1)zz to evaluate the (anti)aromaticity character of each individual rings. A list of NICS(1)zz and NICS(-1)zz values are shown in Fig. S21–22 in ESI.†
(a) A. Reisi-Vanani and A. A. Rezaei, J. Mol. Graphics Modell., 2015, 61, 85 CrossRef PubMed;
(b) A. C. Forse, J. M. Griffin, V. Presser, Y. Gogotsi and C. P. Grey, J. Phys. Chem. C, 2014, 118, 7508 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization data of all new compounds; details for all physical characterization and theoretical calculations; and additional spectroscopic data. CCDC 1570628, 1819178 and 1819179. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01388h |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Tullimilli Y.
Gopalakrishna
a,
Tun Seng
Herng
c,
Hoa
Phan
a,
Wangdong
Zeng
a,
Jun
Ding
b,
Tullimilli Y.
Gopalakrishna
a,
Tun Seng
Herng
c,
Hoa
Phan
a,
Wangdong
Zeng
a,
Jun
Ding