
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiple roles of aryloxide leaving groups in enantioselective annulations employing α,β-unsaturated acyl ammonium catalysis†
Mark D.
Greenhalgh
 ,
Shen
Qu
,
Alexandra M. Z.
Slawin
and
Andrew D.
Smith
*
,
Shen
Qu
,
Alexandra M. Z.
Slawin
and
Andrew D.
Smith
*
EaStCHEM, School of Chemistry, University of St Andrews, North Haugh, St Andrews KY16 9ST, UK. E-mail: ads10@st-andrews.ac.uk
First published on 4th May 2018
Abstract
An isothiourea-catalysed Michael addition–annulation process using β-fluoroalkyl-substituted α,β-unsaturated aryl esters and a range of 2-acylbenzazoles is reported for the enantioselective synthesis of dihydropyranone and dihydropyridinone products bearing polyfluorinated stereocenters (29 examples, up to 98% yield, >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 er). The choice of aryl group of the aryl ester proved essential in determining reaction enantioselectivity and dihydropyranone:dihydropyridinone product selectivity. The aryloxide leaving group is shown to play a number of essential additional roles, operating (i) as a Brønsted base, circumventing the need for an auxiliary base; and (ii) as a Lewis base to catalyse the isomerisation of dihydropyranone products into thermodynamically-favoured dihydropyridinones. After optimisation, this isomerisation process was exploited for the selective synthesis of dihydropyridinone products using acylbenzothiazoles, and either dihydropyranone or dihydropyridinone products using acylbenzoxazoles. Finally, the phenol derivative, produced following protonation of the aryloxide, is proposed to act as a Brønsted acid, which promotes an isothiourea-catalysed kinetic resolution of benzoxazole-derived dihydropyranones.
:1 er). The choice of aryl group of the aryl ester proved essential in determining reaction enantioselectivity and dihydropyranone:dihydropyridinone product selectivity. The aryloxide leaving group is shown to play a number of essential additional roles, operating (i) as a Brønsted base, circumventing the need for an auxiliary base; and (ii) as a Lewis base to catalyse the isomerisation of dihydropyranone products into thermodynamically-favoured dihydropyridinones. After optimisation, this isomerisation process was exploited for the selective synthesis of dihydropyridinone products using acylbenzothiazoles, and either dihydropyranone or dihydropyridinone products using acylbenzoxazoles. Finally, the phenol derivative, produced following protonation of the aryloxide, is proposed to act as a Brønsted acid, which promotes an isothiourea-catalysed kinetic resolution of benzoxazole-derived dihydropyranones.
1. Introduction
Lewis base organocatalysis is now firmly established as a cornerstone of modern organic synthesis, with a variety of distinct reactivity modes allowing the construction of complex products with high levels of regio-, chemo- and stereocontrol.1 A critical feature for the widespread adoption of any synthetic procedure is the ease of access and handling of the required substrates. A current focus in the fields of tertiary amine and N-heterocyclic carbene (NHC) catalysis is the use of bench-stable ammonium/azolium enolate and α,β-unsaturated acyl ammonium/azolium precursors.2 This has resulted in the replacement of notoriously-unstable ketenes and acid chlorides in these processes with homoanhydrides, in situ formed mixed anhydrides, and aryl esters. The use of isolated, bench-stable aryl esters as starting materials is of particular interest due to the simplicity of reaction set-up and improved atom economy relative to methods using anhydrides. Chi first introduced aryl esters as azolium enolate precursors in 2012,3 demonstrating that aryl esters bearing electron withdrawing substituents were essential to achieve sufficient nucleofugality of the aryloxide leaving group (Scheme 1a). Chi subsequently applied electron-deficient aryl esters in a range of NHC-catalysed formal cycloaddition and domino cascade processes involving azolium (homo)enolate and α,β-unsaturated acyl azolium intermediates.4 In each case, the aryloxide was only considered as a simple leaving group.5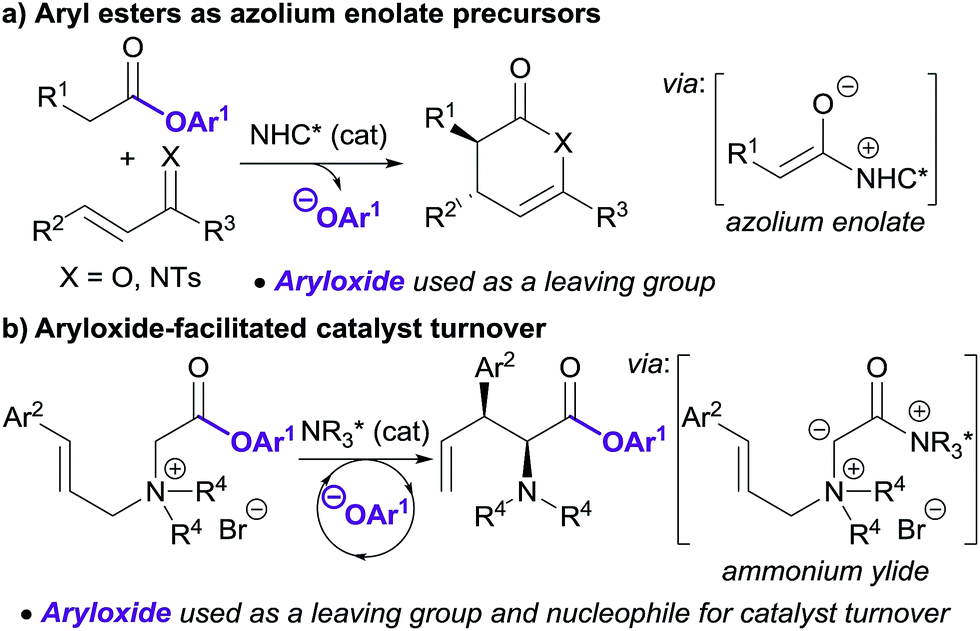 | ||
| Scheme 1 Previous uses of aryl ester substrates in NHC and tertiary amine catalysis. | ||
The use of electron-deficient aryl esters in enantioselective tertiary amine catalysis was first reported by our research group in 2014 for the 2,3-rearrangement of allylic ammonium ylides (Scheme 1b).6,7 This method represented a conceptually different approach, with the aryloxide released from the substrate also required to facilitate intermolecular catalyst turnover.8 This approach, in which the aryloxide performs a dual role, has since been successfully applied in ammonium enolate and α,β-unsaturated acyl ammonium catalysis.9
The field of enantioselective α,β-unsaturated acyl ammonium catalysis has seen a recent rise in popularity.10,11 Following seminal work in 2006 by Fu on [3 + 2] annulations using a planar-chiral DMAP catalyst,12 little attention was given to this field until publications by Lupton, Romo and ourselves using isothiourea catalysis.13 Since 2013 a range of highly enantioselective Michael addition–annulation, formal cycloaddition and complex cascade methodologies have been developed.14 For example, we reported recently an isothiourea-catalysed Michael addition–annulation process using 2-acylbenzazole pro-nucleophiles 1 and homoanhydrides 2 as α,β-unsaturated acyl ammonium precursors (Scheme 2).14e,15 In this work, the selectivity of annulation depended upon the identity of the 2-acylbenzazole substrate. 2-Acylbenzoxazole substrates (X = O) exclusively gave dihydropyranones 4, whilst 2-acylbenzothiazoles (X = S) preferentially gave the corresponding dihydropyridinone 5 (typically in ∼85:15 ratio of 5:4). Experimental and computational studies showed the selectivity of annulation to be kinetically-derived, with non-covalent C–H⋯O and S⋯O interactions present in the respective annulation transition states implicated in determining product selectivity.
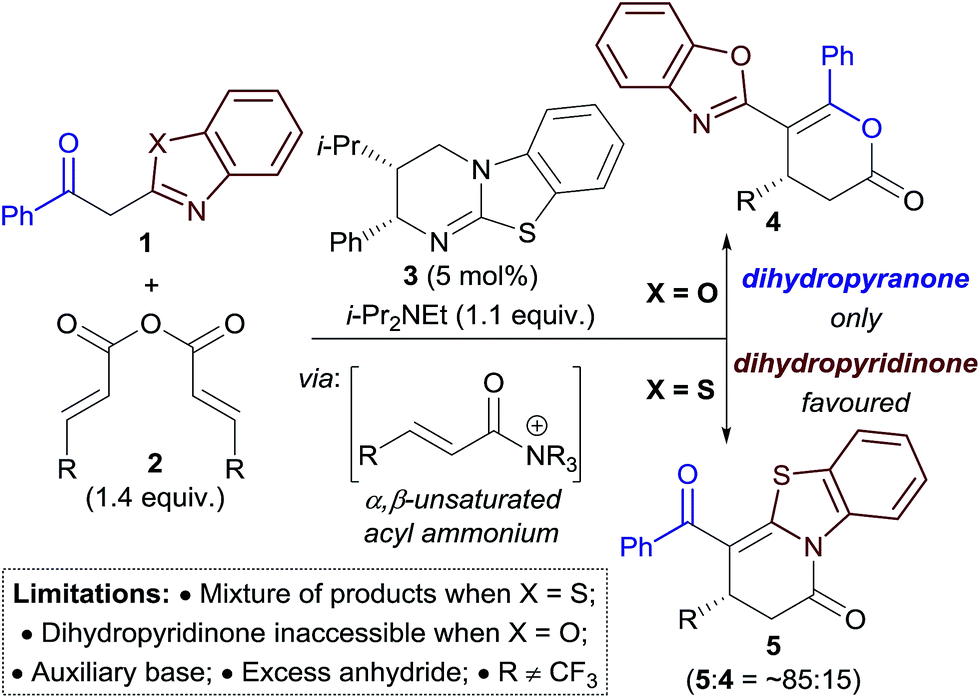 | ||
| Scheme 2 Isothiourea-catalysed Michael addition–annulation of homoanhydrides with 2-acylbenzazole derivatives. | ||
Due to general widespread interest in the formation of products containing fluorinated substituents at stereogenic centres,16 we sought to apply this method to prepare fluorinated heterocycles in enantiopure form. However, the prohibitive instability of fluorinated homoanhydrides (e.g. R = CF3) led us to investigate β-polyfluoroalkyl-substituted α,β-unsaturated aryl esters as alternative acyl ammonium precursors. Herein, we report the development of this process, during which the aryloxide leaving group (ArO−) has been identified as playing a number of additional key roles in determining catalytic efficiency and selectivity. In this manuscript we show that the in situ generated aryloxide (ArO−) acts as (i) a Brønsted base, circumventing the previous requirement for an auxiliary base; (ii) a Lewis base, which can be exploited to selectively catalyse the isomerisation of dihydropyranones into thermodynamically-favoured dihydropyridinones. Additionally, it was found that ArOH, produced upon protonation of the aryloxide, can act as a Brønsted acid that promotes an isothiourea-catalysed kinetic resolution of benzoxazole-derived dihydropyranones.
2. Results and discussion
2.1 Reaction optimisation
Initial studies focused on the Michael addition–annulation of 2-phenacylbenzothiazole 6 and β-CF3-substituted α,β-unsaturated para-nitrophenyl (PNP) ester 7 using HyperBTM 3 as catalyst. In the presence of i-Pr2NEt as auxiliary base and 5 mol% HyperBTM, dihydropyridinone 10 was obtained as the sole product in quantitative yield and 86:14 er (Table 1, entry 1). In the absence of i-Pr2NEt full conversion was still observed, however a mixture of dihydropyridinone 10 and dihydropyranone 11 was obtained as a 3:1 ratio (entry 2). This preservation of reactivity is consistent with the released aryloxide operating as the Brønsted base in this case. Significantly, the absence of i-Pr2NEt also led to vastly improved enantioselectivity (95:5 er). This difference in enantioselectivity can be attributed to a competitive base-mediated background reaction in the presence of i-Pr2NEt (entry 3). Alternative isothiourea catalysts, solvents and reaction temperatures did not improve the er,17 so attention turned to the use of different α,β-unsaturated aryl esters. 3,5-Bis(trifluoromethyl)phenyl (BCF3P) ester 8 provided a mixture of dihydropyridinone 10 and dihydropyranone 11 in a similar ratio and er to PNP ester 7 (entry 4), however 2,4,6-trichlorophenyl (TCP) ester 9 gave 10 and 11 in close to a 1:1 ratio, but with excellent enantioselectivity (97:3 er, entry 5). Lowering the catalyst loading to 1 mol% resulted in a slight drop in conversion; however both products were still obtained with excellent enantiocontrol (entry 6).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 3 (mol%) | Substrate | i-Pr2NEt (equiv.) | 10 | 11 | ||
| %a | erb | %a | erb | ||||
| a Determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as internal standard. b Determined by chiral HPLC analysis. c NA = not applicable. d ND = not determined. | |||||||
| 1 | 5 | 7 | 1.2 | 96 | 86:14 |
0 | NAc |
| 2 | 5 | 7 | 0 | 75 | 95:5 |
23 | NDd |
| 3 | 0 | 7 | 1.2 | 75 | NAc | 13 | NAc |
| 4 | 5 | 8 | 0 | 73 | 94:6 |
21 | 94:6 |
| 5 | 5 | 9 | 0 | 54 | 97:3 |
42 | 97:3 |
| 6 | 1 | 9 | 0 | 47 | 97:3 |
38 | 97:3 |
| 7 | 0 | 7, 8 or 9 | 0 | 0 | NAc | 0 | NAc |
| 8 | 0 | 7 | 0.1 | 63 | NAc | 15 | NAc |
| 9 | 0 | 8 | 0.1 | 45 | NAc | 21 | NAc |
| 10 | 0 | 9 | 0.1 | 17 | NAc | 7 | NAc |
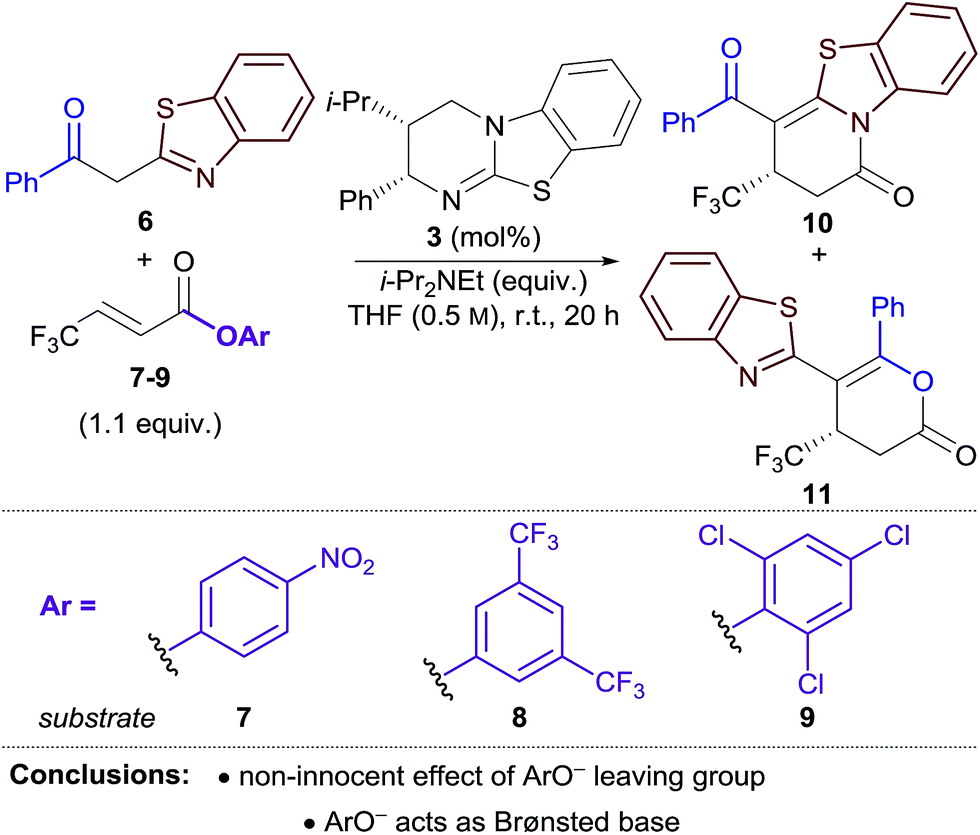
The differences in product ratio and enantioselectivity using different aryl esters provided the first indication that the aryloxide ‘leaving group’ was performing additional roles in the reaction. First, the differences in enantioselectivity were investigated. Control reactions between 2-phenacylbenzothiazole 6 and aryl esters 7–9 in the absence of HyperBTM resulted in no conversion in each case (entry 7). The addition of a substoichiometric amount of i-Pr2NEt (0.1 equiv.) successfully promoted the reaction, with high conversion obtained when using PNP and BCF3P esters 7 and 8 (66–78%) (entries 8, 9). In contrast, only modest conversion was observed when using TCP ester 9 (24%) (entry 10). These experiments indicate that the 0.1 equiv. of base served to initiate the reaction, with conversions of >10% consistent with the released aryloxide acting as a Brønsted base to propagate the reaction. The lower enantioselectivities obtained using PNP and BCF3P esters 7 and 8 in the Michael addition–annulation reaction may therefore be attributed to an enhanced base-mediated background reaction promoted by the released aryloxide.
Next, the variation in the ratio of dihydropyridinone and dihydropyranone products was probed. It was hypothesised this variation may arise from isomerisation of dihydropyranone 11 to give the thermodynamically-favoured dihydropyridinone 10 under the reaction conditions. The isomerisation of dihydropyranone 11 was therefore investigated in isolation under various conditions (Fig. 1a). In the presence of either HyperBTM 3, i-Pr2NEt or a substituted phenol derivative alone, essentially no isomerisation of dihydropyranone 11 was observed (<5% in 5 h, Fig. 1b ). However, a combination of i-Pr2NEt (2.2 equiv.) and either para-nitrophenol 12 (PNPOH, 2.2 equiv., Fig. 1b
). However, a combination of i-Pr2NEt (2.2 equiv.) and either para-nitrophenol 12 (PNPOH, 2.2 equiv., Fig. 1b ) or 3,5-bis(trifluoromethyl)phenol 13 (BCF3POH, 2.2 equiv., Fig. 1b
) or 3,5-bis(trifluoromethyl)phenol 13 (BCF3POH, 2.2 equiv., Fig. 1b ) promoted effective isomerisation (t1/2 ≈ 1 h). This is consistent with the aryloxide, formed upon deprotonation of the phenol derivative, catalysing this isomerisation. In contrast, a combination of 2,4,6-trichlorophenol 14 (TCPOH, 2.2 equiv.) and i-Pr2NEt (2.2 equiv.) (Fig. 1b
) promoted effective isomerisation (t1/2 ≈ 1 h). This is consistent with the aryloxide, formed upon deprotonation of the phenol derivative, catalysing this isomerisation. In contrast, a combination of 2,4,6-trichlorophenol 14 (TCPOH, 2.2 equiv.) and i-Pr2NEt (2.2 equiv.) (Fig. 1b ) resulted in much slower isomerisation (t1/2 ≈ 12 h). These differences in the rate of dihydropyranone isomerisation in the presence of each aryloxide are consistent with the variation in product selectivity observed during reaction optimisation (Table 1, entries 2, 4, 5). A more extensive study of aryloxide derivatives found that those bearing ortho-substituents were uniformly ineffective for the isomerisation of dihydropyranone 11.17 This trend in reactivity is synonymous with the aryloxide operating as a Lewis base in this process (Fig. 1c). Nucleophilic attack of the aryloxide on the dihydropyranone 11 would result in ring-opening to give aryl ester intermediate 15, which may undergo lactonisation to reform the dihydropyranone 11 or lactamisation to give the thermodynamically-favoured dihydropyridinone 10.
) resulted in much slower isomerisation (t1/2 ≈ 12 h). These differences in the rate of dihydropyranone isomerisation in the presence of each aryloxide are consistent with the variation in product selectivity observed during reaction optimisation (Table 1, entries 2, 4, 5). A more extensive study of aryloxide derivatives found that those bearing ortho-substituents were uniformly ineffective for the isomerisation of dihydropyranone 11.17 This trend in reactivity is synonymous with the aryloxide operating as a Lewis base in this process (Fig. 1c). Nucleophilic attack of the aryloxide on the dihydropyranone 11 would result in ring-opening to give aryl ester intermediate 15, which may undergo lactonisation to reform the dihydropyranone 11 or lactamisation to give the thermodynamically-favoured dihydropyridinone 10.
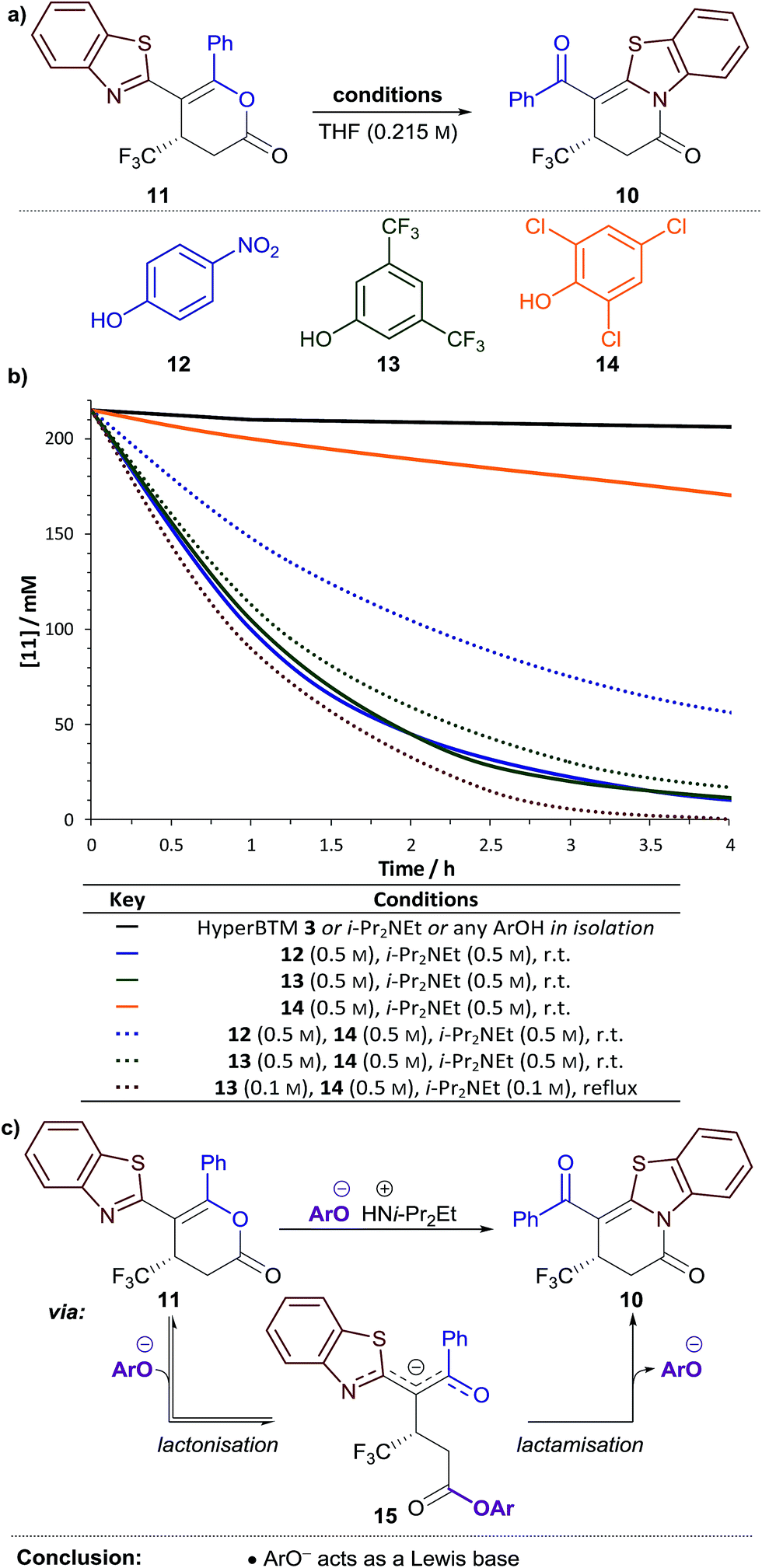 | ||
| Fig. 1 Isomerisation of dihydropyranone 11 to dihydropyridinone: (a) general reaction scheme; (b) temporal change in the concentration of 11 under different reaction conditions; (c) proposed mechanism for isomerisation. | ||
We envisioned that this isomerisation process could be applied following the Michael addition–annulation reaction to provide a single dihydropyridinone product. The highest enantioselectivity was obtained using TCP ester 9, however isomerisation was most efficient using aryloxides derived from PNPOH 12 or BCF3POH 13. Therefore the addition of either PNPO− or BCF3PO− at the end of the Michael addition–annulation process would be required. As a stoichiometric amount of TCPOH 14 would also be present at this stage in the process, the efficiency of dihydropyranone isomerisation using either PNPO− or BCF3PO− in the presence of an equivalent of TCPOH 14 was tested. The rate of dihydropyranone isomerisation using a combination of BCF3POH 13 and i-Pr2NEt (2.2 equiv. of each) (Fig. 1b ) was essentially unaffected by the additional TCPOH 14 (2.2 equiv.); however a significant retardation in the rate of isomerisation was observed when using PNPOH 12 and i-Pr2NEt (2.2 equiv. of each) (Fig. 1b
) was essentially unaffected by the additional TCPOH 14 (2.2 equiv.); however a significant retardation in the rate of isomerisation was observed when using PNPOH 12 and i-Pr2NEt (2.2 equiv. of each) (Fig. 1b ). This confirmed the combination of BCF3POH 13 and i-Pr2NEt to be optimal for use in a telescoped Michael addition–annulation–isomerisation sequence. Further studies found that substoichiometric BCF3POH 13 and i-Pr2NEt could be used to affect efficient isomerisation by heating the reaction at reflux (Fig. 1b
). This confirmed the combination of BCF3POH 13 and i-Pr2NEt to be optimal for use in a telescoped Michael addition–annulation–isomerisation sequence. Further studies found that substoichiometric BCF3POH 13 and i-Pr2NEt could be used to affect efficient isomerisation by heating the reaction at reflux (Fig. 1b ).
).
Combining the Michael addition–annulation and isomerisation processes, 2-phenylbenzothiazole, β-trifluoromethyl-substituted α,β-unsaturated TCP ester 9 and HyperBTM 3 (5 mol%) were reacted in THF at room temperature for 20 h, followed by the addition of BCF3POH 13 (20 mol%) and i-Pr2NEt (20 mol%) and heating at reflux for a further 4 h. This sequence provided dihydropyridinone 10 as the sole reaction product in 95% yield and 96:4 er (Scheme 3).
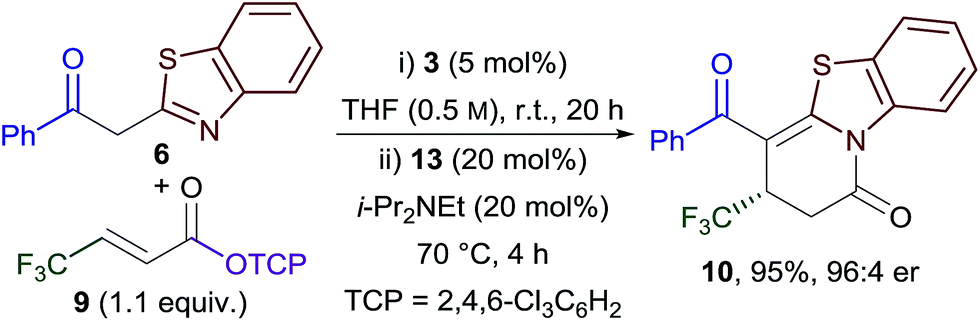 | ||
| Scheme 3 Optimised Michael addition–annulation–isomerisation reaction. | ||
2.2 Reaction scope: benzothiazoles
The generality of this method was investigated for a range of 2-acylbenzothiazole derivatives and β-fluoroalkyl-substituted α,β-unsaturated TCP esters (Table 2). Substitution of the phenacyl group with both electron-donating and moderately electron-withdrawing groups provided dihydropyridinones 16–18 in excellent yield and with high enantioselectivity (Table 2a).18 Incorporation of a strong electron-withdrawing group (NO2) provided 19 in excellent yield, but with diminished enantioselectivity (87:13 er). ortho-Substitution of the aryl group was also tolerated, with 2-iodophenyl- and 1-naphthyl-functionalised products 20 and 21 obtained in excellent yield and enantiocontrol. The scope was extended to include heteroaromatic and alkyl-substituted ketones, with 22–24 all obtained in excellent yield and enantiocontrol. Next, variation of the benzothiazole unit was investigated (Table 2b). Substitution with fluoro, bromo, and methoxy groups was tolerated to give 25–27 in equally high yield and enantiocontrol. In addition, the use of 2-phenacylthiazole proved effective in giving dihydropyridinone 28 in high yield and enantiocontrol. The scope of the process was extended to different β-fluoroalkyl-substituted α,β-unsaturated TCP esters (Table 2c). Difluoromethyl substituents, which have experienced significant recent interest in drug design,16b,c,19 were successfully incorporated. A small scope including different (hetero)aryl- and alkyl-substituted ketones was demonstrated giving dihydropyridinones 29–32 in excellent yield and with good to high enantioselectivity. The incorporation of a pentafluoroethyl group at the stereogenic centre was also successful, with 33 obtained in excellent yield and enantiocontrol. The series of dihydropyridinones 10, 29 and 33, bearing different polyfluoroalkyl groups at the stereogenic centre, reveals a trend of improved enantioselectivity with increasing fluorine substitution.
| a 10 mol% 3 used. |
|---|
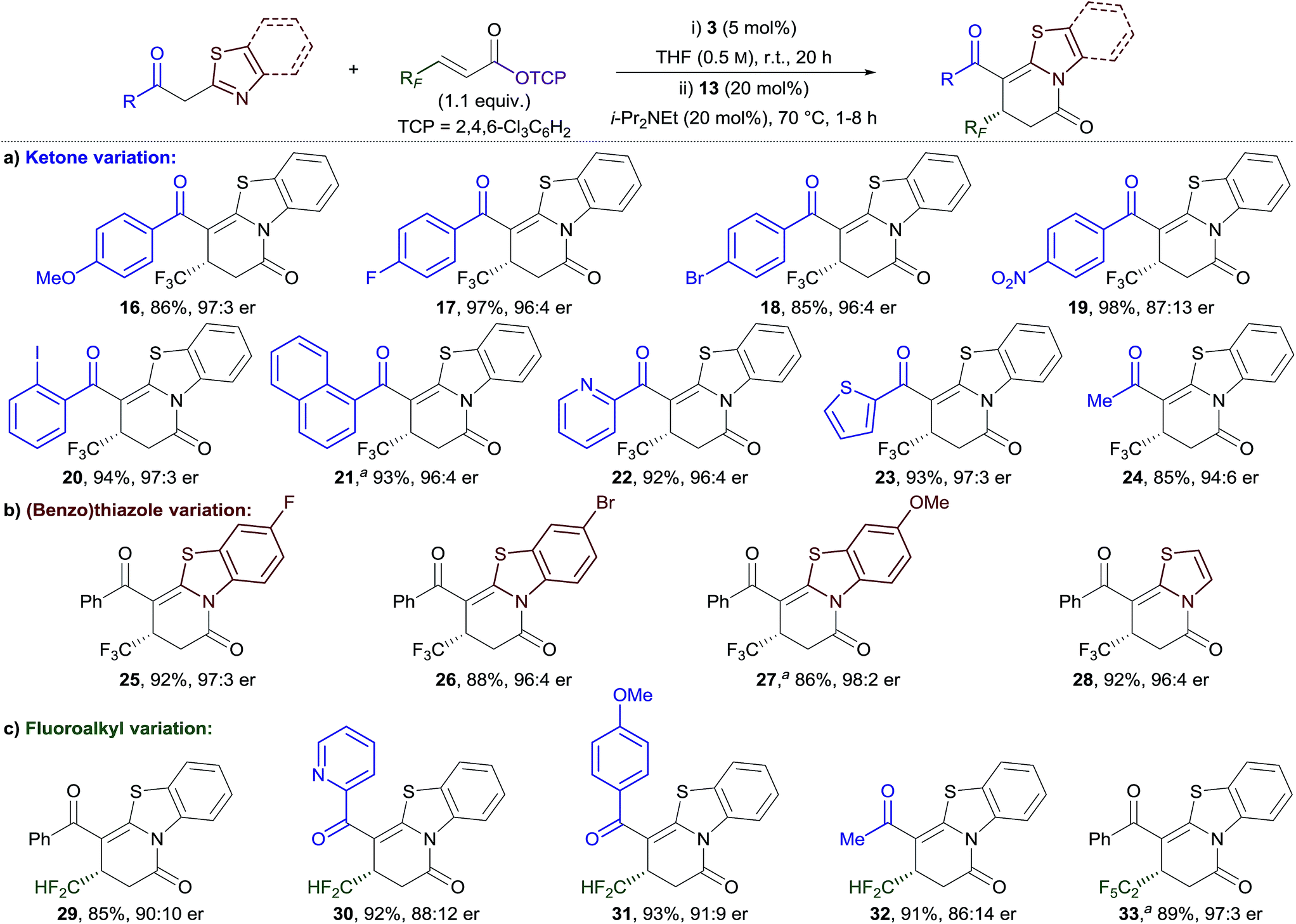
|
2.3 Reaction scope: benzoxazoles
In our previously-reported Michael addition–annulation methodology,14e the reaction of 2-phenacylbenzoxazole with homoanhydrides provided dihydropyranones as the sole reaction products. We postulated that the newly developed isomerisation process could allow selective access to either dihydropyranone or dihydropyridinone products, broadening the scope of this process. In the absence of an isomerisation step, the Michael addition–annulation between 2-phenacylbenzoxazole and β-trifluoromethyl-substituted α,β-unsaturated TCP ester 9 using HyperBTM 3 (10 mol%) at room temperature provided dihydropyranone 34 as the sole reaction product with exceptional enantioselectivity (>99:1 er) (Table 3, left). The scope of the acyl group was further investigated, with 3-pyridyl and 3-thienyl substituents providing dihydropyranones 35 and 36 in good yield and excellent enantiocontrol. Substitution of the acyl group with electron-withdrawing groups resulted in the formation of dihydropyranones 37 and 38 in improved yield, and with excellent enantioselectivity, albeit with reduced selectivity for the dihydropyranone product (∼90:10 dihydropyranone:dihydropyridinone). Interestingly, in these examples, the minor dihydropyridinone products 42 and 43 were obtained with lower enantioselectivity (∼92:8 er) in comparison to the major dihydropyranone products (>99:1 er). This effect is discussed in more detail in Section 2.5.
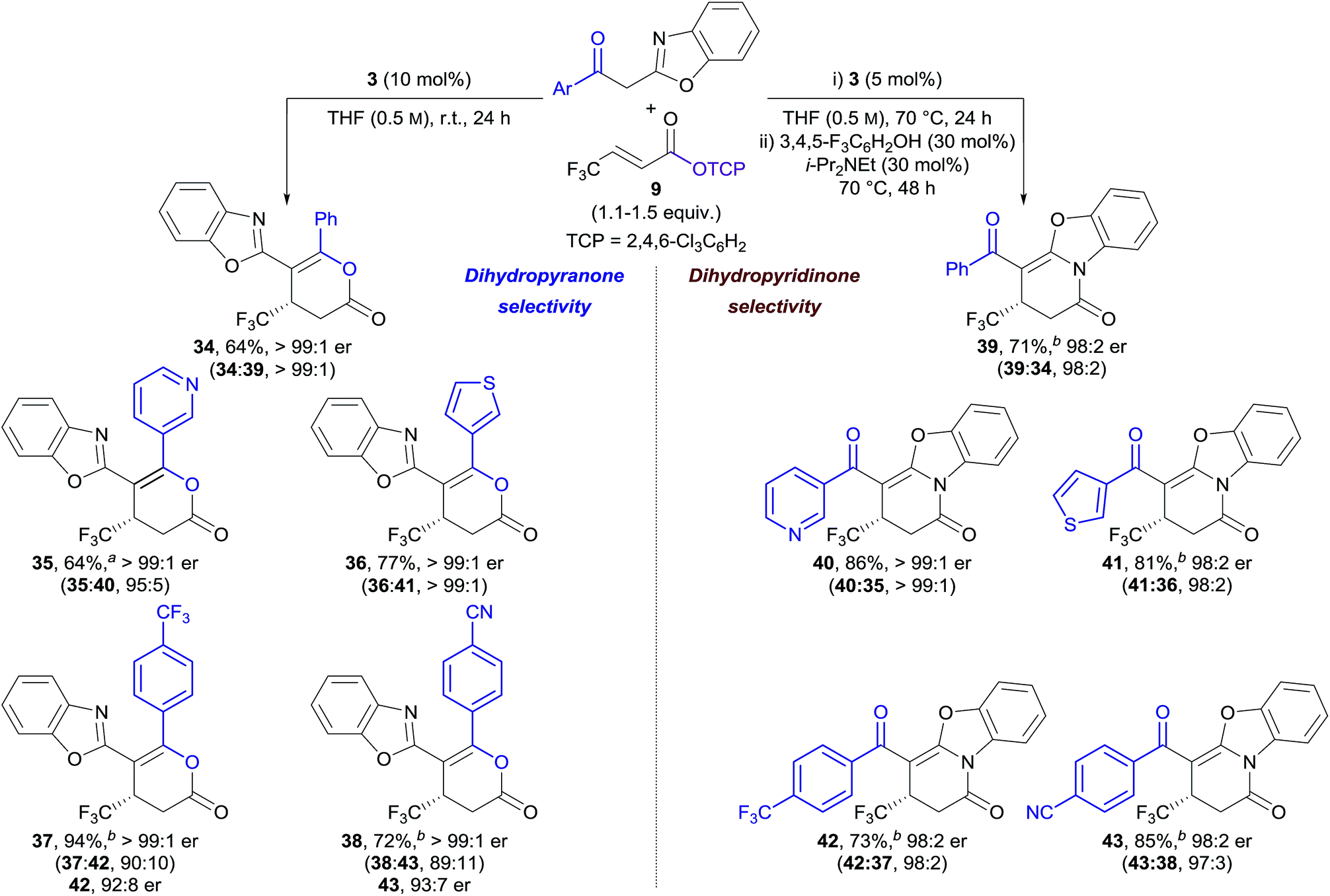
The applicability of the telescoped Michael addition–annulation–isomerisation sequence was next investigated for the synthesis of dihydropyridinones. In this case, a combination of 3,4,5-trifluorophenol (30 mol%) and i-Pr2NEt (30 mol%), in addition to longer reaction times, proved optimal for complete conversion to the corresponding dihydropyridinones (Table 3, right). The isothiourea catalyst loading could be reduced to 5 mol% by heating the Michael addition–annulation step at reflux. Under these conditions the Michael addition–annulation proceeded in higher yield over shorter reaction times with excellent enantioselectivities (∼98:2 er). While reduced dihydropyranone:dihydropyridinone ratios were obtained, this product ratio was considered inconsequential due to the subsequent isomerisation step. The same five 2-acylbenzoxazole derivatives were applied in the Michael addition–annulation–isomerisation sequence giving dihydropyridinones 39–43 in good yield, excellent dihydropyridinone selectivity, and with high enantioselectivity (≥98:2 er) in each case.20
2.4 Extension of the isomerisation protocol
Finally, we were interested to see if the isomerisation protocol could be applied to our previously-reported Michael addition–annulation reaction using 2-acylbenzothiazoles and homoanhydrides.14e Although good to excellent yields and enantioselectivities had been reported, product isolation was complicated by the concurrent formation of dihydropyridinone and dihydropyranone products as a kinetically-determined and sometimes inseparable mixture (typically ∼85:15 ratio). As the original Michael addition–annulation process using homoanhydrides required a small excess of i-Pr2NEt (1.3 equiv.), isomerisation was attempted by the addition of 3,5-bis(trifluoromethyl)phenol 13 (40 mol%) after 6 h, followed by heating the reaction at reflux. This method proved successful, with the generality of the process demonstrated for aryl, alkyl, heteroaryl and alkenyl-substituted derivatives (Table 4). Dihydropyridinones 44–47 were obtained as the exclusive reaction products in excellent yield and with comparable enantioselectivity to the previously-reported method. This simple protocol improves the synthetic utility of the original method, and highlights the potential for the more widespread application of aryloxides as Lewis base catalysts.21
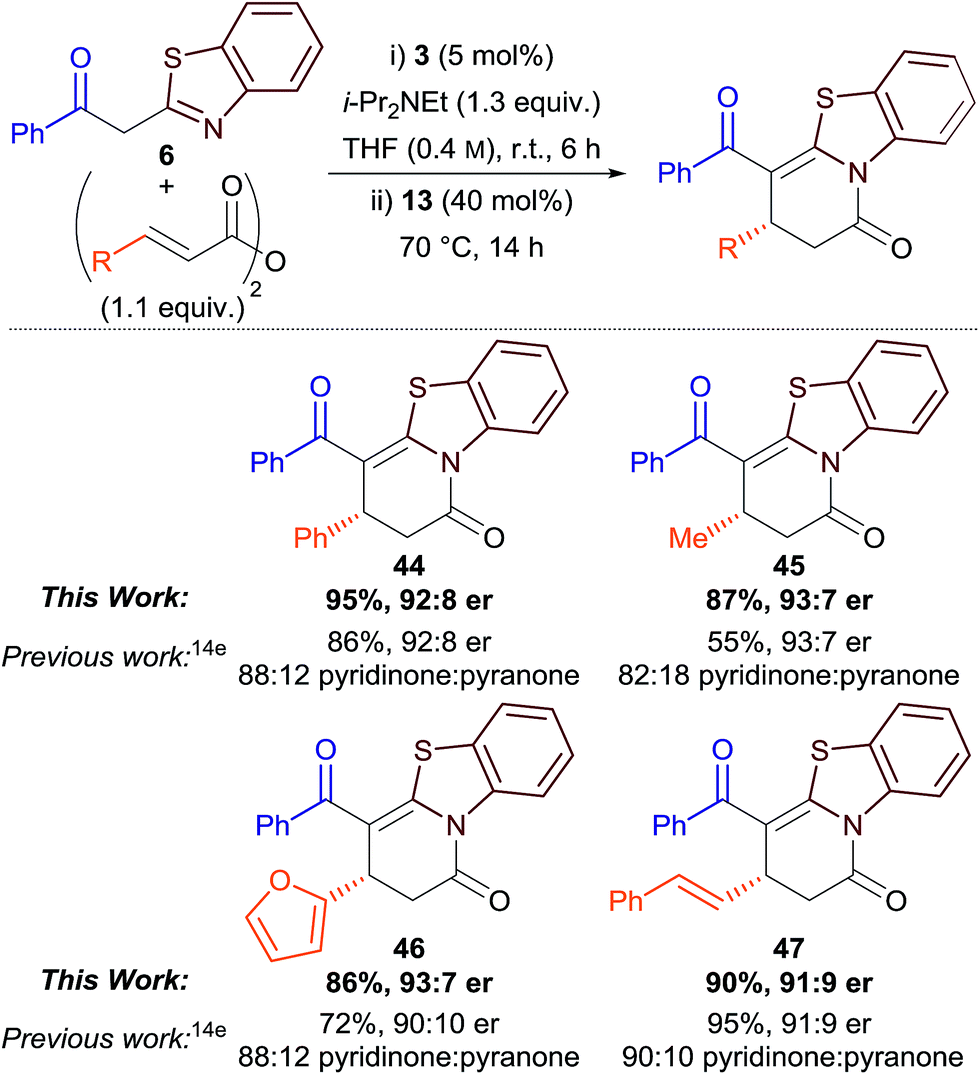
|
2.5 Kinetic resolution
Whilst exploring the selective synthesis of benzoxazole-derived dihydropyranones outlined in Section 2.3 (Table 3, left column), dihydropyranones 37 and 38 were obtained in essentially enantiopure form (>99:1 er), whilst the minor dihydropyridinone products, 42 and 43, were obtained with significantly lower enantioenrichment (∼92:8 er). These differences in product er prompted further investigation. Reaction of racemic dihydropyranone (±)-37 with HyperBTM 3 (10 mol%) gave a mixture of enantioenriched (R)-dihydropyridinone 42 and (S)-dihydropyranone 37 (Table 5, entry 1). This demonstrates that HyperBTM 3 is capable of affecting the isomerisation of benzoxazole-derived dihydropyranone 37. This is in contrast to the isomerisation studies using benzothiazole-derived dihydropyranone 11, in which HyperBTM was inactive (see Fig. 1b ). The observed formation of enantioenriched (R)-dihydropyridinone 42 and (S)-dihydropyranone 37 in this process suggests it can be simplistically described as a kinetic resolution.22 While the selectivity factor metric, s, is commonly used to report the efficiency of kinetic resolutions; in this case s was found to be dependent on reaction conversion, and was therefore not considered a valid descriptor.17 The enantioselectivity of this process, however, can be used to rationalise the differences in enantioenrichment observed between the dihydropyranone and dihydropyridinone products formed in the Michael addition–annulation process (Table 3). Significantly, (2S,3R)-HyperBTM 3 produces dihydropyranone (S)-37 as the major product in the Michael addition–annulation reaction, but is more efficient at catalysing the isomerisation of (R)-37 to give dihydropyridinone 42. This larger rate constant for the isomerisation of (R)-37 leads to further enrichment of the dihydropyranone product in (S)-37, whilst consequentially producing dihydropyridinone 42 with a lower level of enantioenrichment.
). The observed formation of enantioenriched (R)-dihydropyridinone 42 and (S)-dihydropyranone 37 in this process suggests it can be simplistically described as a kinetic resolution.22 While the selectivity factor metric, s, is commonly used to report the efficiency of kinetic resolutions; in this case s was found to be dependent on reaction conversion, and was therefore not considered a valid descriptor.17 The enantioselectivity of this process, however, can be used to rationalise the differences in enantioenrichment observed between the dihydropyranone and dihydropyridinone products formed in the Michael addition–annulation process (Table 3). Significantly, (2S,3R)-HyperBTM 3 produces dihydropyranone (S)-37 as the major product in the Michael addition–annulation reaction, but is more efficient at catalysing the isomerisation of (R)-37 to give dihydropyridinone 42. This larger rate constant for the isomerisation of (R)-37 leads to further enrichment of the dihydropyranone product in (S)-37, whilst consequentially producing dihydropyridinone 42 with a lower level of enantioenrichment.
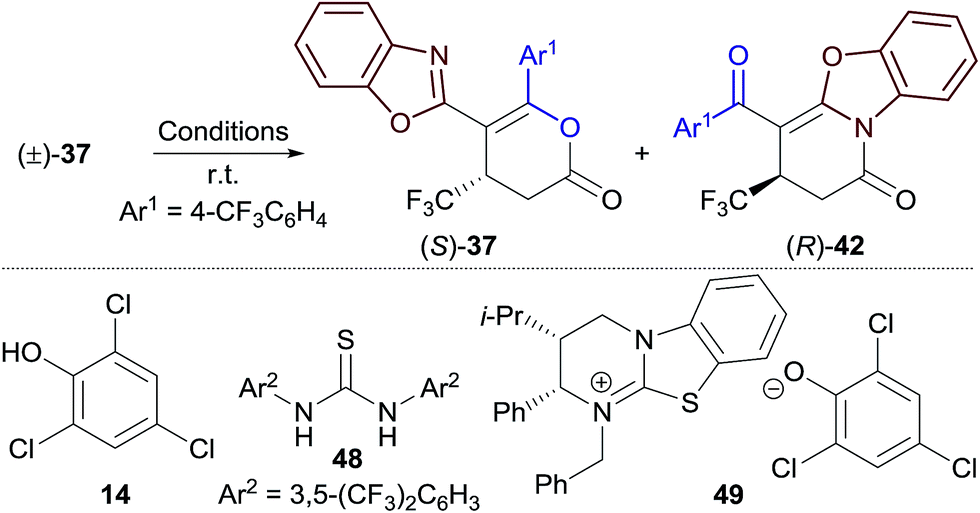
To better simulate this kinetic resolution under the reaction conditions of the Michael addition–annulation process, the isomerisation of (±)-37 was next investigated using a combination of HyperBTM 3 (10 mol%) and trichlorophenol 14 (1 equiv.) (entry 2). Improved enantioenrichment of both 37 (92:8 er) and 42 (85:15 er) was observed at a similar reaction conversion, indicating that the phenol additive has a beneficial effect on the kinetic resolution process. It was hypothesised that trichlorophenol could be either: (i) operating as a Brønsted acid/hydrogen bond donor to activate the dihydropyranone to nucleophilic attack by HyperBTM 3; or (ii) deprotonated by HyperBTM 3 to produce an isothiouronium aryloxide ion pair, where the aryloxide acts as a nucleophile and enantioselectivity is induced by the chiral counterion HyperBTM-H+. To test the first hypothesis alternative non-nucleophilic Brønsted acids/hydrogen bond donors were applied. A combination of HyperBTM 3 and either benzoic acid or Schreiner's thiourea 4823 resulted in similar or improved enantioselectivity relative to the use of trichlorophenol 14 (entries 3, 4). The beneficial effect of using benzoic acid in the planar-chiral DMAP catalysed dynamic kinetic resolution of azlactones has been previously noted, however the origin of this effect was not discussed.24 The second scenario was simulated using N-benzylisothiouronium trichlorophenoxide 49, which catalysed the isomerisation, but gave both dihydropyridinone 42 and dihydropyranone 37 as racemates (entry 5).
These experiments are consistent with the phenol additive providing Brønsted acid activation of the dihydropyranone, and HyperBTM acting as a chiral nucleophile (Scheme 4). Nucleophilic attack of HyperBTM 3 onto racemic dihydropyranone 50 would produce two diastereomeric zwitterionic acyl isothiouronium intermediates 51, which may undergo lactonisation to reform dihydropyranone 50, or undergo lactamisation to give dihydropyridinone product 52. The nucleophilic addition of HyperBTM to dihydropyranone 50 is expected to be reversible as zwitterionic acyl isothiouronium intermediate 51 is a proposed intermediate in the Michael addition–annulation process, in which dihydropyranone 50 is originally generated (see Scheme 5). The enantioselectivity observed within this process may therefore originate either from the preferential nucleophilic addition of HyperBTM 3 to (R)-50, and/or through the differential rates of lactamisation from each diastereomeric zwitterionic acyl isothiouronium intermediate 51. We cannot currently differentiate these possibilities.25
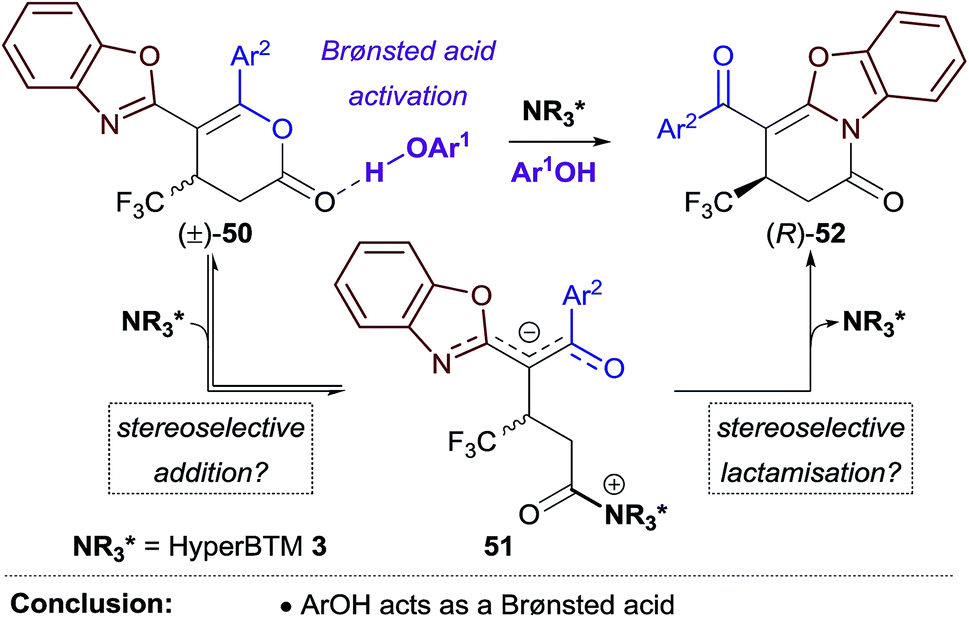 | ||
| Scheme 4 Proposed mechanism for the kinetic resolution of (±)-50. | ||
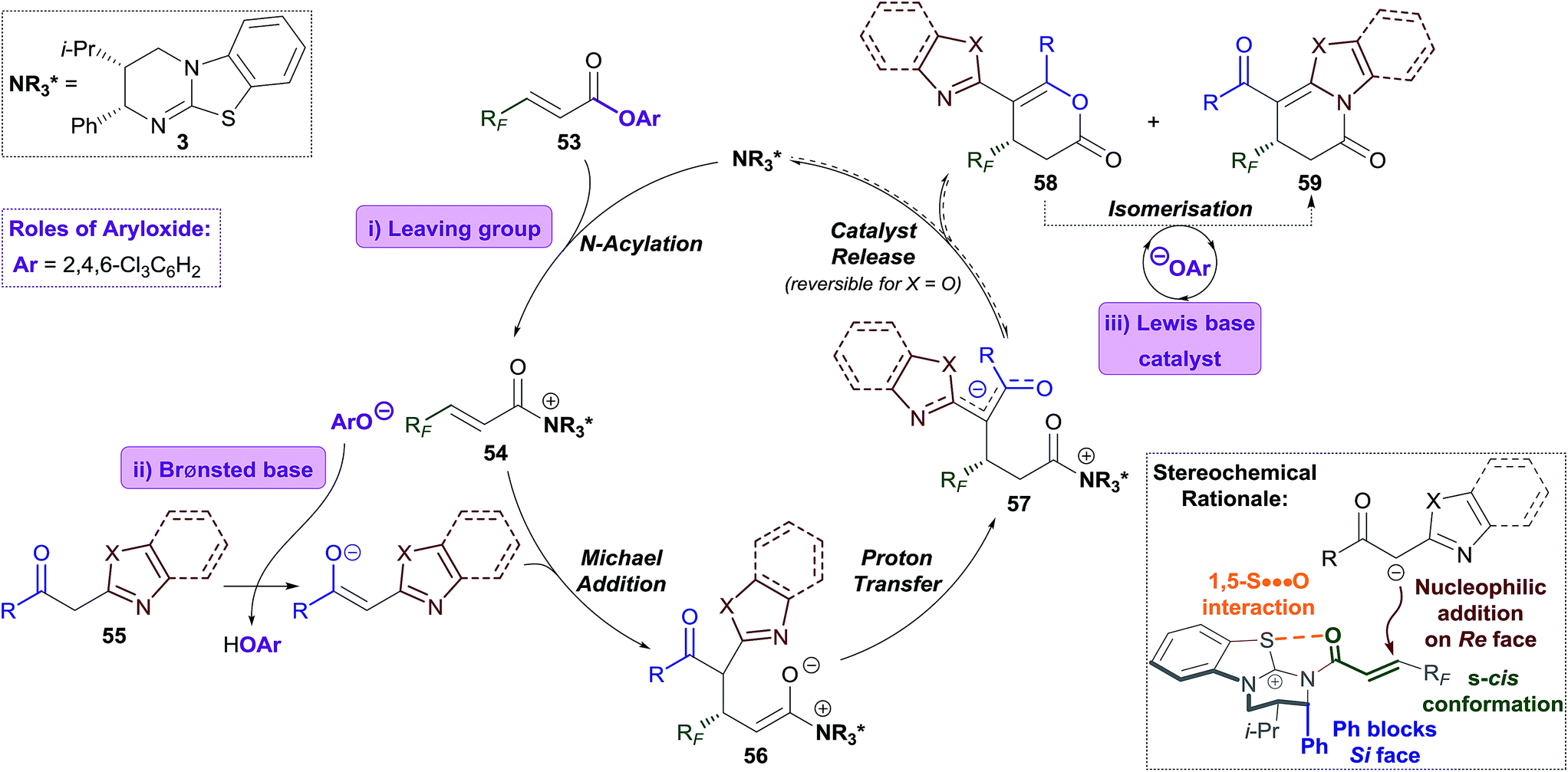 | ||
| Scheme 5 Proposed mechanism and stereochemical rationale. | ||
2.6 Proposed mechanism
The Michael addition–annulation process is proposed to begin with N-acylation of HyperBTM 3 by α,β-unsaturated TCP ester 53 to give α,β-unsaturated acyl isothiouronium trichlorophenoxide ion pair 54 (Scheme 5).9c Deprotonation of the 2-acylbenzazole pro-nucleophile 55 by trichlorophenoxide provides trichlorophenol and a stabilised enolate, which undergoes Michael addition to α,β-unsaturated acyl isothiouronium 54 to give ammonium enolate 56. Proton transfer, likely facilitated by trichlorophenol, gives zwitterionic intermediate 57, which may undergo cyclisation through oxygen or nitrogen, regenerating the catalyst and giving dihydropyranone 58 or dihydropyridinone 59, respectively. Trichlorophenoxide present in the reaction can facilitate isomerisation of dihydropyranone 58 to give the thermodynamically-favoured dihydropyridinone 59. This isomerisation is most facile for benzothiazole-derived dihydropyranones (X = S), and presumably takes place through nucleophilic ring-opening of the dihydropyranone 58, followed by lactamisation (see Fig. 1c). In the optimised protocol this isomerisation step was most efficiently catalysed by the addition of less sterically-hindered aryloxides, such as 3,5-bis(trifluoromethyl)phenoxide or 3,4,5-trifluorophenoxide. For benzoxazole-derived dihydropyranones, a second isomerisation pathway is possible, which is catalysed by HyperBTM 3 operating as a Lewis base, and trichlorophenol acting as a Brønsted acid.The stereochemical outcome of the reaction can be rationalised by the α,β-unsaturated acyl isothiouronium 54 adopting an s-cis conformation, with a syn-coplanar non-covalent 1,5-S⋯O interaction between the acyl O and catalyst S providing a conformational lock.6c,13b,14a,e,i,26 Michael addition of the acylbenzazole-derived enolate to α,β-unsaturated acyl isothiouronium 54 then takes place anti- to the stereodirecting pseudo-axial phenyl substituent of the isothiourea catalyst (Scheme 5, bottom).
3. Conclusions
The isothiourea-catalysed enantioselective synthesis of a range of polyfluorinated dihydropyranone and dihydropyridinone products was achieved via a Michael addition–annulation process using α,β-unsaturated acyl ammonium catalysis (29 examples, up to 98%, >99:1 er). β-Fluoroalkyl-substituted α,β-unsaturated trichlorophenyl esters were used as the α,β-unsaturated acyl ammonium precursors, and a range of 2-acyl(benz)azoles used as the nucleophilic reaction partner. Significantly, the trichlorophenoxide leaving group was shown to play a variety of other roles in the reaction, including acting as (i) a Brønsted base, circumventing the need for the addition of an auxiliary base; and (ii) a Lewis base, catalysing the isomerisation of dihydropyranone products into thermodynamically-favoured dihydropyridinones. The isomerisation process was most efficient using less sterically-hindered aryloxide catalysts bearing electron-withdrawing groups, such as 3,5-bis(trifluoromethyl)phenoxide, 3,4,5-trifluorophenoxide or para-nitrophenoxide. These findings led to the development of a sequential Michael addition–annulation–isomerisation protocol for the synthesis of a range of benzothiazole-derived dihydropyridinone products as the only constitutional isomer in excellent yield and enantiocontrol. The method could also be applied when using 2-acylbenzoxazole pro-nucleophiles, with the selective formation of either dihydropyranones or dihydropyridinones achieved by including or omitting the isomerisation step. The aryloxide-promoted isomerisation protocol was further applied to our previously-reported Michael addition–annulation process using homoanhydrides, demonstrating the wide applicability of the method. Finally, the phenol derivative produced upon protonation of the aryloxide during the reaction was shown to act as a Brønsted acid, which promoted an isothiourea-catalysed kinetic resolution of benzoxazole-derived dihydropyranones. Overall, this work provides a concise and efficient method for the synthesis of polyfluorinated heterocyclic products in high yield and enantioselectivity. Identification of the multiple roles of the aryloxide leaving group in this process should inform future work in this area and provide inspiration for new reaction design.27
Conflicts of interest
No conflicts of interest to declare.Acknowledgements
The research leading to these results has received funding from the ER0043 under the European Union's Seventh Framework Programme (FP7/2007–2013)/E.R.C. grant agreement 279850. A. D. S. thanks the Royal Society for a Wolfson Research Merit Award. We thank the EPSRC UK National Mass Spectrometry Facility at Swansea University.Notes and references
- (a) S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560–1638 CrossRef PubMed; (b) E. Vedejs and S. E. Denmark, Lewis Base Catalysis in Organic Synthesis, Wiley, Weinheim, 2016 Search PubMed.
- (a) L. C. Morrill and A. D. Smith, Chem. Soc. Rev., 2014, 43, 6214–6226 RSC; (b) P. Chauhan and D. Enders, Angew. Chem., Int. Ed., 2014, 53, 1485–1487 CrossRef PubMed.
- L. Hao, Y. Du, H. Lv, X. Chen, H. Jiang, Y. Shao and Y. R. Chi, Org. Lett., 2012, 14, 2154–2157 CrossRef PubMed.
- For a review see ref. 2b. For selected examples see: (a) Z. Fu, J. Xu, T. Zhu, W. W. Y. Leong and Y. R. Chi, Nat. Chem., 2013, 5, 835–839 CrossRef PubMed; (b) J. Xu, Z. Jin and Y. R. Chi, Org. Lett., 2013, 15, 5028–5031 CrossRef PubMed; (c) J. Cheng, Z. Huang and Y. R. Chi, Angew. Chem., Int. Ed., 2013, 52, 8592–8596 CrossRef PubMed; (d) Z. Fu, X. Wu and Y. R. Chi, Org. Chem. Front., 2016, 3, 145–149 RSC; (e) H. Wang, X. Chen, Y. Li, J. Wang, S. Wu, W. Xue, S. Yang and Y. R. Chi, Org. Lett., 2018, 20, 333–336 CrossRef PubMed.
- She reported that an N-hydroxyphthalimide leaving group could be exploited in a subsequent N- to C-sulfonyl transfer of N-tosyl dihydropyridinone products: R. Han, L. He, L. Liu, X. Xie and X. She, Chem.–Asian J., 2016, 11, 193–197 CrossRef PubMed.
- (a) T. H. West, D. S. B. Daniels, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2014, 136, 4476–4479 CrossRef PubMed; (b) T. H. West, D. M. Walden, J. E. Taylor, A. C. Brueckner, R. C. Johnson, P. H.-Y. Cheong, G. C. Lloyd-Jones and A. D. Smith, J. Am. Chem. Soc., 2017, 139, 4366–4375 CrossRef PubMed; (c) T. H. West, S. S. M. Spoehrle and A. D. Smith, Tetrahedron, 2017, 73, 4138–4149 CrossRef; (d) S. S. M. Spoehrle, T. H. West, J. E. Taylor, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2017, 139, 11895–11902 CrossRef PubMed; (e) K. Kasten, A. M. Z. Slawin and A. D. Smith, Org. Lett., 2017, 19, 5182–5185 CrossRef PubMed.
- Chi also reported the use of aryl esters in tertiary amine catalysis in 2014, albeit not in an enantioselective transformation, see: L. Hao, X. Chen, S. Chen, K. Jiang, J. Torres and Y. R. Chi, Org. Chem. Front., 2014, 1, 148–150 RSC.
- For a review of aryloxide-promoted catalyst turnover see: (a) W. C. Hartley, T. J. C. O'Riordan and A. D. Smith, Synthesis, 2017, 49, 3303–3310 CrossRef; for specific examples see: (b) H. Wack, A. E. Taggi, A. M. Hafez, W. J. Drury and T. Lectka, J. Am. Chem. Soc., 2001, 123, 1531 CrossRef PubMed; (c) A. M. Hafez, A. E. Taggi, H. Wack, J. Esterbrook and T. Lectka, Org. Lett., 2001, 3, 2049 CrossRef PubMed; (d) Y. Kawanaka, E. M. Phillips and K. A. Scheidt, J. Am. Chem. Soc., 2009, 131, 18028 CrossRef PubMed; (e) J. Douglas, K. B. Ling, C. Concellón, G. Churchill, A. M. Z. Slawin and A. D. Smith, Eur. J. Org. Chem., 2010, 2010, 5863–5869 CrossRef; (f) S. Y. Lee, S. Neufeind and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 8899 CrossRef PubMed.
- (a) K. J. Schwarz, J. L. Amos, J. C. Klein, D. T. Do and T. N. Snaddon, J. Am. Chem. Soc., 2016, 138, 5214–5217 CrossRef PubMed; (b) X. Jiang, J. J. Beiger and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 87–90 CrossRef PubMed; (c) A. Matviitsuk, M. D. Greenhalgh, D.-J. Barrios Antúnez, A. M. Z. Slawin and A. D. Smith, Angew. Chem., Int. Ed., 2017, 56, 12282–12287 CrossRef PubMed; (d) J. N. Arokianathar, A. B. Frost, A. M. Z. Slawin, D. Stead and A. D. Smith, ACS Catal., 2018, 8, 1153–1160 CrossRef.
- S. Vellalath and D. Romo, Angew. Chem., Int. Ed., 2016, 55, 13934–13943 CrossRef PubMed.
- For a review on the related area of α,β-unsaturated acyl azolium catalysis see: C. Zhang, J. F. Hooper and D. W. Lupton, ACS Catal., 2017, 7, 2583–2596 CrossRef.
- E. Bappert, P. Müller and G. C. Fu, Chem. Commun., 2006, 2604–2606 RSC.
- (a) S. Pandiancherri, S. J. Ryan and D. W. Lupton, Org. Biomol. Chem., 2012, 10, 7903–7911 RSC; (b) E. R. T. Robinson, C. Fallan, C. Simal, A. M. Z. Slawin and A. D. Smith, Chem. Sci., 2013, 4, 2193–2200 RSC; (c) S. Vellalath, K. N. Van and D. Romo, Angew. Chem., Int. Ed., 2013, 52, 13688–13693 CrossRef PubMed; (d) G. Liu, M. E. Shirley, K. N. Van, R. L. McFarlin and D. Romo, Nat. Chem., 2013, 5, 1049–1057 CrossRef PubMed.
- See ref. 9c, and: (a) M. E. Abbasov, B. M. Hudson, D. J. Tantillo and D. Romo, J. Am. Chem. Soc., 2014, 136, 4492–4495 CrossRef PubMed; (b) S. Goudedranche, X. Bugaut, T. Constantieux, D. Bonne and J. Rodriguez, Chem.–Eur. J., 2014, 20, 410–415 CrossRef PubMed; (c) Y. Fukata, T. Omamura, K. Asano and S. Matsubara, Org. Lett., 2014, 16, 2184–2187 CrossRef PubMed; (d) Y. Fukata, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2015, 137, 5320–5323 CrossRef PubMed; (e) E. R. T. Robinson, D. M. Walden, C. Fallan, M. D. Greenhalgh, P. H.-Y. Cheong and A. D. Smith, Chem. Sci., 2016, 7, 6919–6927 RSC; (f) A. Matviitsuk, J. E. Taylor, D. B. Cordes, A. M. Z. Slawin and A. D. Smith, Chem.–Eur. J., 2016, 22, 17748–17757 CrossRef PubMed; (g) N. A. Ahlemeyer and V. B. Birman, Org. Lett., 2016, 18, 3454–3457 CrossRef PubMed; (h) E. R. T. Robinson, A. B. Frost, P. Elías-Rodríguez and A. D. Smith, Synthesis, 2017, 49, 409–423 Search PubMed; (i) M. E. Abbasov, B. M. Hudson, D. J. Tantillo and D. Romo, Chem. Sci., 2017, 8, 1511–1524 RSC; (j) Y. Fukata, K. Yao, R. Miyaji, K. Asano and S. Matsubara, J. Org. Chem., 2017, 82, 12655–12668 CrossRef PubMed; (k) J. Cao, K. Sun, S. Dong, T. Lu, Y. Dong and D. Du, Org. Lett., 2017, 19, 6724–6727 CrossRef PubMed; (l) N. A. Ahlemeyer, E. V. Streff, P. Muthupandi and V. B. Birman, Org. Lett., 2017, 19, 6486–6489 CrossRef PubMed; (m) K. N. Van and D. Romo, J. Org. Chem., 2018, 83, 632–643 CrossRef PubMed.
- For a review on isothiourea catalysis see: (a) J. Merad, J.-M. Pons, O. Chuzel and C. Bressy, Eur. J. Org. Chem., 2016, 2016, 5589–5610 CrossRef; for examples see ref. 6, 9, 13, 14a,d–j,l,m, 23b, 24, 26a and: (b) V. B. Birman and X. Li, Org. Lett., 2006, 8, 1351–1354 CrossRef PubMed; (c) C. Joannesse, C. P. Johnson, C. Concellon, C. Simal, D. Philp and A. D. Smith, Angew. Chem., Int. Ed., 2009, 48, 8914–8918 CrossRef PubMed; (d) C. A. Leverett, V. C. Purohit and D. Romo, Angew. Chem., Int. Ed., 2010, 49, 9479–9483 CrossRef PubMed; (e) D. Belmessieri, L. C. Morill, C. Simal, A. M. Z. Slawin and A. D. Smith, J. Am. Chem. Soc., 2011, 133, 2714–2720 CrossRef PubMed.
- (a) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Chem. Rev., 2011, 111, 455–529 CrossRef PubMed; (b) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826–870 CrossRef PubMed; (c) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef PubMed.
- See ESI for full details.†.
- The absolute configuration of recovered 18 was assigned as (S) by X-ray crystallographic analysis (CCDC 1827462†). The absolute configuration of all other benzothiazole-derived products was assigned by analogy.
- (a) J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465–7478 RSC; (b) J. A. Erickson and J. I. McLoughlin, J. Org. Chem., 1995, 60, 1626–1631 CrossRef; (c) Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, J. Med. Chem., 2017, 60, 797–804 CrossRef PubMed.
- The absolute configuration of recovered 41 was assigned as (S) by X-ray crystallographic analysis (CCDC 1827463†). The absolute configuration of all other benzoxazole-derived products was assigned by analogy.
- For a review on the use of ammonium aryloxide salts as Brønsted and Lewis base catalysts, see: (a) J. Godemert, S. Oudeyer and V. Levacher, ChemCatChem, 2016, 8, 74–85 CrossRef.
- H. B. Kagan and J. C. Fiaud, in Topics in Stereochemistry, ed. E. L. Eliel and S. H. Wilen, John Wiley & Sons, 1988, vol. 18, pp. 249–330 Search PubMed.
- (a) A. Wittkopp and P. R. Schreiner, Chem.–Eur. J., 2003, 9, 407–414 CrossRef PubMed; (b) Z. Zhang, Z. Bao and H. Xing, Org. Biomol. Chem., 2014, 12, 3151–3162 RSC.
- (a) J. Liang, J. Craig and G. C. Fu, J. Org. Chem., 1998, 63, 3154–3155 CrossRef; (b) X. Yang, G. Lu and V. B. Birman, Org. Lett., 2010, 12, 892–895 CrossRef PubMed.
- Birman has reported the use of an isothiourea catalyst and benzoic acid for the kinetic resolution of β-lactams and an oxazinone through ring-opening/alcoholysis. Birman proposed nucleophilic addition of the isothiourea catalyst as the selectivity-determining step. The sense of enantiodiscrimination reported is the same as that observed in our current study, see: V. D. Bumbu and V. B. Birman, J. Am. Chem. Soc., 2011, 133, 13902–13905 CrossRef PubMed.
- (a) P. Liu, X. Yang, V. B. Birman and K. N. Houk, Org. Lett., 2012, 14, 3288–3291 CrossRef PubMed; (b) D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef PubMed; (c) B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef PubMed; (d) X. Zhang, Z. Gong, J. Li and T. Lu, J. Chem. Inf. Model., 2015, 55, 2138–2153 CrossRef PubMed.
- The research data underpinning this publication can be found at DOI: 10.17630/2dec867e-570d-4bcc-99ba-2cefe7479357.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, product characterisation data (mpt, NMR, IR, HRMS, [α]D, HPLC), traces (NMR, HPLC) and X-ray crystallographic data. CCDC 1827462 and 1827463. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01324a |
| This journal is © The Royal Society of Chemistry 2018 |