
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kekulé diradicaloids derived from a classical N-heterocyclic carbene†
Dennis
Rottschäfer
 a,
Beate
Neumann
a,
Hans-Georg
Stammler
a,
Diego M.
Andrada
b and
Rajendra S.
Ghadwal
*a
a,
Beate
Neumann
a,
Hans-Georg
Stammler
a,
Diego M.
Andrada
b and
Rajendra S.
Ghadwal
*a
aAnorganische Molekülchemie und Katalyse, Lehrstuhl für Anorganische Chemie und Strukturchemie, Centrum für Molekulare Materialien, Fakultät für Chemie, Universität Bielefeld, Universitätsstr. 25, D-33615 Bielefeld, Germany. E-mail: rghadwal@uni-bielefeld.de; Web: http://www.ghadwalgroup.de Fax: +49 521 106 6026; Tel: +49 521 106 6167
bAllgemeine und Anorganische Chemie, Universität des Saarlandes, Campus C4.1, D-66123 Saarbrücken, Germany
First published on 24th April 2018
Abstract
The direct double carbenylation of 1,4-diiodobenzene and 4,4′-dibromobiphenyl with a classical N-heterocyclic carbene, SIPr (1) (SIPr = :C{N(2,6-iPr2C6H3)}2CH2CH2), by means of nickel catalysis gives rise to 1,3-imidazolinium salts [(SIPr)(C6H4)(SIPr)](I)2 (2) and [(SIPr)(C6H4)2(SIPr)](Br)2 (3) as off-white solids. Two-electron reduction of 2 and 3 with KC8 cleanly yields Kekulé diradicaloid compounds [(SIPr)(C6H4)(SIPr)] (4) and [(SIPr)(C6H4)2(SIPr)] (5), respectively, as crystalline solids. Structural parameters and DFT as well as CASSCF calculations suggest the closed-shell singlet ground state for 4 and 5. Calculations reveal a very low singlet–triplet energy gap ΔES–T for 5 (10.7 kcal mol−1), while ΔES–T for 4 (29.1 kcal mol−1) is rather large.
Introduction
Molecules containing two unpaired electrons in two nearly degenerate molecular orbitals are called diradicals, which may have parallel (triplet) or antiparallel (singlet) spins.1 Depending on the interaction between unpaired electrons, singlet diradicals are further classified as open-shell (OS) and closed-shell (CS) singlets. Diradicaloids are molecules with partial singlet diradical nature in their ground state.2 In this context, Thiele's (TH)3 and Chichibabin's (CH)4 hydrocarbons I (Fig. 1), reported shortly after Gomberg's discovery5 of the Ph3C˙ radical in 1900, are noteworthy examples. They can be described either as an OS diradical (Ia) featuring a phenylene or diphenylene linker or a CS p-quinodimethane (Ib). Since their first isolation, the ground state spin state of I has been a subject of intense experimental and theoretical investigations,6 leading to a very controversial discussion, the so-called “diradical paradox”.7 In 1986, Montgomery et al.8 determined the solid-state molecular structures of these highly reactive compounds by single crystal X-ray diffraction. Currently, CH is generally described as a diradicaloid (the resonance hybrid of 1a and 1b) with a significant diradical character, whereas TH is considered as a p-quinodimethane. | ||
| Fig. 1 Benzenoid (Ia) and quinoid (Ib) resonance forms of Thiele's (n = 1) and Chichibabin's (n = 2) HCs and analogues dicationic bis(triarylamino) (II) and benzannulated (III) derivatives. Singlet carbenes derived cumulene/diradicaloid compounds (IV) and radicals (V). NHC derived diradicaloids VI. | ||
Stable diradicaloids delocalized over π-conjugated systems are appealing synthetic targets because of their unique physical properties and potential applications in nonlinear optics, molecular electronics, and organic spintronics.9 Therefore, different synthetic strategies have been developed over the past few years to thermodynamically and/or kinetically stabilize various analogues of TH and CH. Bis(triarylamine) dications II are isoelectronic to I, in which the Ph2C˙ moiety of I is replaced by an Ar2N+˙ unit.10 The benzannulation approach has also been successfully employed to isolate stable polycyclic hydrocarbons such as III.11
In recent years, various paramagnetic compounds featuring main-group elements or transition metals have been stabilized by the use of Bertrand's cyclic alkyl amino carbenes (CAACs).12 Remarkably, during the preparation of this manuscript for submission, we came across three consecutive reports dealing with the synthesis of extended cumulenes IV supported by CAACs13 or a classical N-heterocyclic carbene (NHC).14 Very recently, we reported stable crystalline radicals V derived from classical NHCs.15 While these reports nicely emphasize the relevance of singlet carbenes in the synthesis of carbon-centered π-conjugated systems and radicals, synthetic access to diradicaloids VI, which are NHC-analogues of I, remained a challenge. Herein, we report the synthesis and characterization of two crystalline Kekulé diradicaloid compounds [(SIPr)(C6H4)(SIPr)] (4) and [(SIPr)(C6H4)2(SIPr)] (5) derived from a classical NHC, SIPr (1) (SIPr = :C{N(2,6-iPr2C6H3)}2CH2CH2).
Results and discussion
The direct two-fold carbenylation of 1,4-diiodobenzene and 4,4′-dibromobiphenyl with SIPr (1) under Ni-catalysis gave the starting materials 2 and 3, respectively (Scheme 1).16 Compounds 2 and 3 are off-white solids and have been characterized by NMR spectroscopy and mass spectrometry. They exhibited characteristic 1H and 13C NMR resonances expected for C2-arylated 1,3-imidazolinium salts.15,16 Molecular structures of 2 and 3 have been determined by single crystal X-ray diffraction studies (Fig. 2). The metric parameters of 2 and 3 (Table 1) are fully consistent with those of reported mono-C2-arylated 1,3-imidazolinium species.15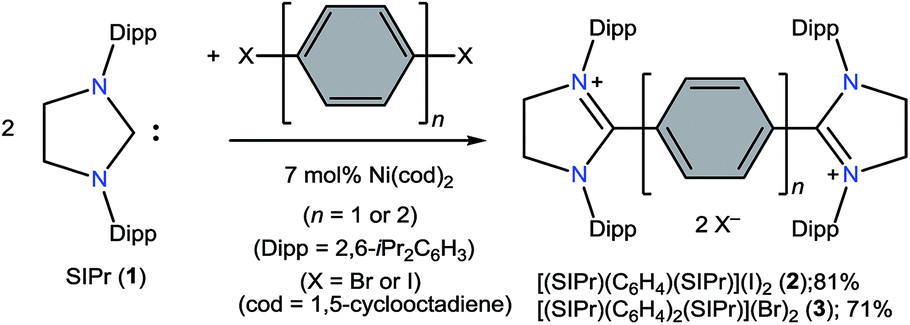 | ||
| Scheme 1 Synthesis of C2-arylated bis(1,3-imidazolinium) salts 2 and 3. | ||
 | ||
| Fig. 2 Solid-state molecular structures of 2 (left) and 3. Hydrogen atoms and counter anions have been omitted for clarity. Selected bond lengths and angles for 2 and 3 are given in Table 1. | ||
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | N–Cα | Cα–Ci | Ci–Co | Co–Co′ | N–C–N | Ci′– Ci′ | ΣN | BLAc |
| a The values for N–Cα, Cα–Ci, Ci–Co, Co–Co′, and N–C–N (except Co–Co′ of 2, Cα–Ci and N–C–N of 2, 3, and 5) are averaged and given without estimated standard deviations (esd's). b Optimized structures and further detail are provided in the ESI (see Fig. S6–S9). c BLA = Ci–Co − Co–Co′. | ||||||||
| 2 | 1.326 | 1.478(2) | 1.397 | 1.390(2) | 113.2(1) | — | 356.4 | — |
| TH | — | 1.381 | 1.449 | 1.346 | — | — | — | 0.10 |
| 4 | 1.404 | 1.371 | 1.451 | 1.349 | 108.2 | — | 349.4 | 0.10 |
| CS | 1.413 | 1.390 | 1.461 | 1.362 | 105.8 | — | 355.1 | 0.10 |
| T | 1.407 | 1.445 | 1.421 | 1.391 | 109.0 | 353.6 | 0.03 | |
| 3 | 1.331 | 1.466(3) | 1.395 | 1.383 | 111.5(2) | 1.494(4) | 355.5 | — |
| CH | — | 1.415 | 1.424 | 1.372 | — | 1.448 | — | 0.05 |
| 5 | 1.390 | 1.386(2) | 1.443 | 1.359 | 107.9(1) | 1.408(2) | 347.3 | 0.08 |
| CS | 1.403 | 1.398 | 1.450 | 1.367 | 107.0 | 1.415 | 352.9 | 0.08 |
| T | 1.409 | 1.424 | 1.433 | 1.387 | 107.6 | 1.481 | 351.4 | 0.04 |
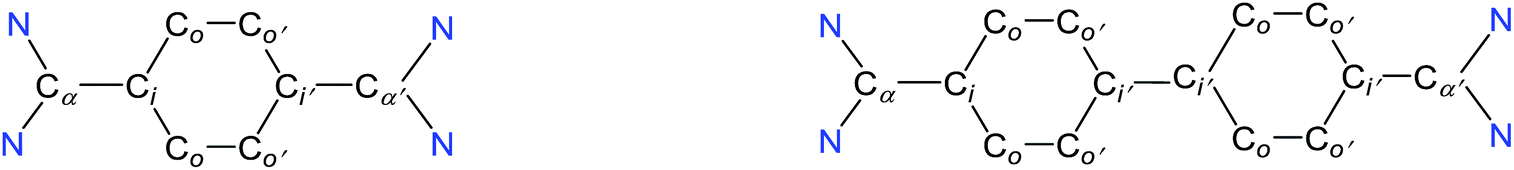
Having the desired compounds in hand, we analyzed the electrochemical properties of 2 and 3 by cyclic voltammetry. Cyclic voltammograms (CVs) of 2 and 3 (Fig. 3) showed two sequential quasi-reversible redox processes, suggesting that both imidazolinium fragments are electrochemically coupled. The first reduction at E1/2 = −0.80 V (2) and E1/2 = −1.04 V (3) is most probably to form a radical cation, which undergoes second reduction at E1/2 = −0.95 V (2) and −1.28 V (3) to give the corresponding neutral species. Note, the reduction potential for 2 and 3 is significantly lower compared to the related C2-protonated imidazolinium salt (SIPr)HCl (ca. −2.3 V).17 Indeed, chemical reduction of 2 and 3 with KC8 at −78 °C in THF immediately resulted in the formation of a highly colored solution and a black precipitate (graphite) (Scheme 2). Upon usual workup, compound 4 (green) and 5 (blue) were isolated as air sensitive solids in an almost quantitative yield. Compounds 4 and 5 are indefinitely stable in solution as well as in solid state under an inert gas atmosphere. Single crystals of 4 and 5 suitable for X-ray diffraction were grown by storing a saturated n-hexane solution of each at −35 °C for 20 h.
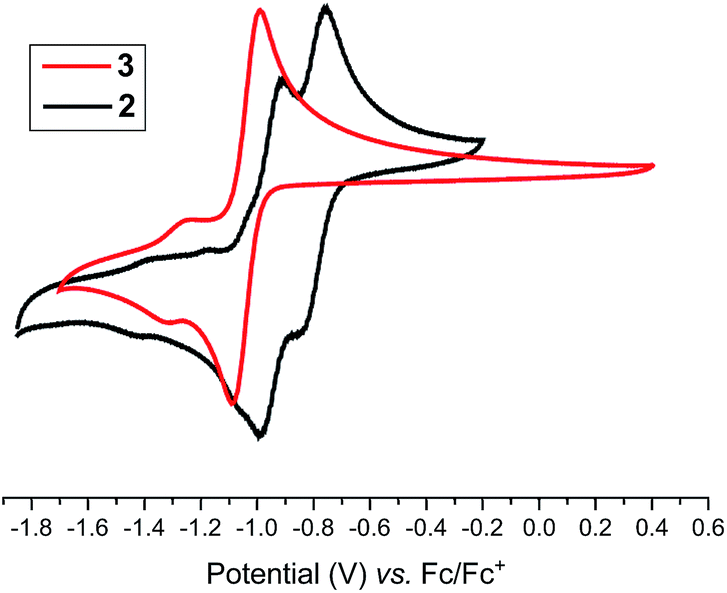 | ||
| Fig. 3 Cyclic voltammograms of 2 and 3 (CH3CN/nBu4NPF6, 100 mV s−1, vs. Fc+/Fc). | ||
 | ||
| Scheme 2 KC8 reduction of 2 and 3 to 4 and 5. | ||
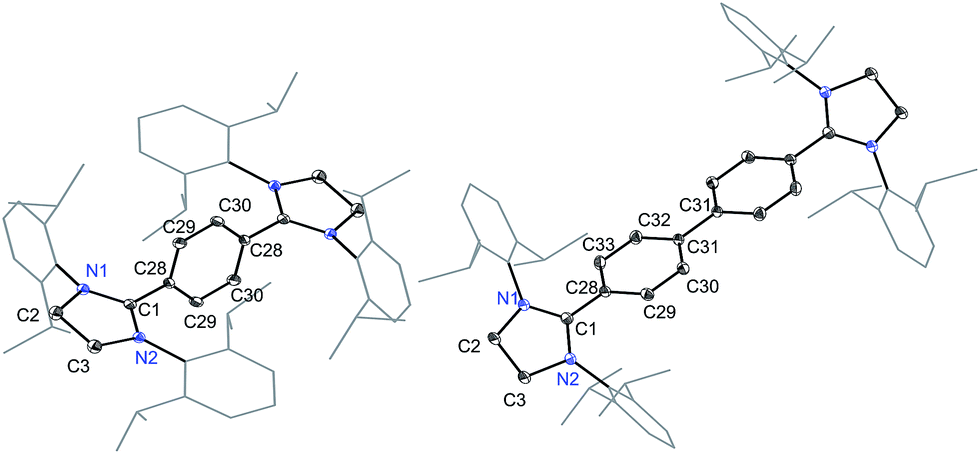
The solid-state molecular structures of 4 and 5 are shown in Fig. 4. The structural parameters of 4, 5 and their precursors 2, 3 along with TH and CH are given in Table 1. The Cα–N (av. 1.328 Å), Cα–Ci (av. 1.472 Å), and Ci–Co/Co–Co′ (av. 1.391 Å) bond lengths of 2 and 3 are in accordance with those of mono C2-arylated imidazolinium salts.15 The Cα–N bond length of 4 (av. 1.404 Å) and 5 (1.390 Å) are considerably longer, whereas Cα–Ci bond lengths of 4 (av. 1.371 Å) and 5 (1.386(2) Å) are significantly shorter compared to those of 2 and 3. The Cα–Ci bond lengths of 4 and 5 are although longer than those in typical olefins (1.33–1.34 Å) and that of (IPr)CH2 (1.332(4) Å)18 but compare well with that of the vinylsilane IPrCH(SiHCl2) (1.379(2) Å)19 in which a considerable π-electron density has transferred to the silicon atom. In addition, the Ci–Co (1.442(2)–1.448(3) Å) and Co–Co′ (1.347(4)–1.360(2) Å) bond lengths of 4 and 5 are clearly distinguishable (i.e., no longer benzenoid). While the bond length alternation (BLA) in 4 (0.10 Å) is similar to TH (0.10 Å), the same in 5 (0.08 Å) is intermediate of 4 and CH (0.05 Å).8 In addition, the Ci–Ci′ bond length of 5 (1.408 Å) is shorter compared to that of CH (1.448 Å). This indicates a higher quinoidal contribution to the resonance structure of 5 compared to CH, which is however lower in comparison with 4 and TH. This becomes also evident when the planarity of imidazolidine rings is compared with that of the bridging phenyl or diphenylene ring. The imidazolidine rings are puckered and feature the plane twist angle of 56.42(5)° in 2 and av. 8.28(10)° in 4 from the bridging phenyl ring, indicating a significant contribution of the quinoid resonance form in the later. As expected, the plane twist angle in 5 (17.56(5)°) is also smaller compared to that of 3 (46.41(9)°), but it is almost twice that of the 4 (8.28(10)°). This also emphasizes the diminished quinoidal contribution to the resonance structure of 5 compared to 4.
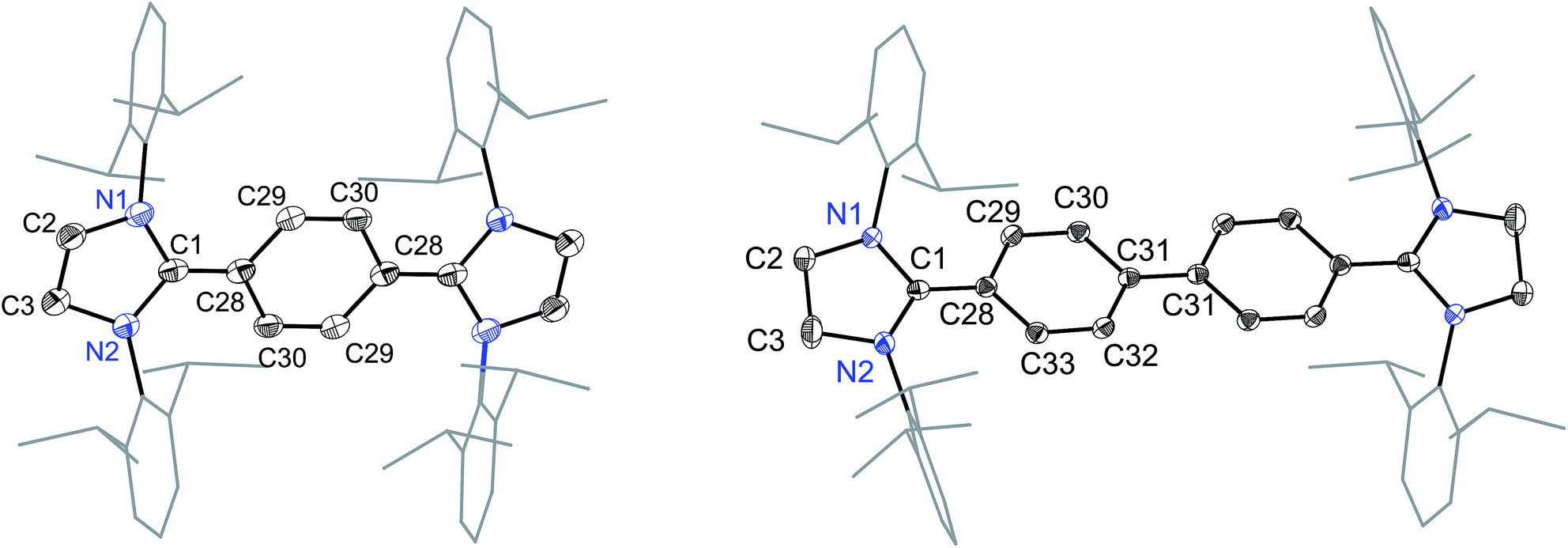 | ||
| Fig. 4 Solid-state molecular structures of [(SIPr)(C6H4)(SIPr)] (4) (left) and [(SIPr)(C6H4)2(SIPr)] (5) (right). Hydrogen atoms have been omitted for clarity. Selected bond lengths and angles for 4 and 5 are given in Table 1. | ||
In order to gain deeper insight into the electronic structures of 4 and 5, DFT calculations were performed at the B3LYP/def2-SVP and BH&HLYP/def2-SVP level of theory (see ESI† for details). The frontier molecular orbitals (FMOs) of 4 and 5 are shown in Fig. 5. Geometry optimizations were performed considering three electronic states namely, closed-shell singlet (CS), open-shell singlet (OS), and triplet (T) states (Table 1). As can be seen, the calculated CS singlet state structures are in a better agreement with X-ray structures. The pyramidalization of the nitrogen atom of imidazolidine ring can also be accounted for the quinoidal character of 4 and 5. The nitrogen atoms in 2 and 3 are almost planar (the sum of the angles at the nitrogen atom ΣN = 355–356°), whereas one of the nitrogen atoms of NHC units in 4 (ΣN = 349.4°) and 5 (ΣN = 347.3°) is pyramidalized. This becomes also obvious when ΣN of 4 and 5 calculated for CS and T states are compared. The OS calculations were carried out within the Noodleman's broken-symmetry (BS) formalism.20 All BS computations showed no spin-contamination collapsing on the CS species. In the case of 5, a broken symmetry singlet wave function −2.3 kcal mol−1 lower compared to the CS singlet species is recognized (Table 2).
 | ||
| Fig. 5 Molecular orbitals (isovalue 0.05) of the CS singlet state species of 4 (top) and 5 (bottom) calculated at B3LYP/def2-TZVPP level of theory. Hydrogens have been omitted for clarity. | ||
| Method | Compound | CS | OS | T | y |
|---|---|---|---|---|---|
| B3LYP/def2-TZVPP | 4 | 0.0 | 0.0 | +29.1 | 0.00 |
| 5 | 0.0 | 0.0 | +10.7 | 0.43 | |
| BH&HLYP/def2-TZVPP | 4 | 0.0 | 0.0 | +27.8 | 0.0 |
| 5 | 0.0 | −2.3 | +4.8 | 0.35 |
An improvement of the electronic energies of each species was performed at B3LYP/def2-TZVPP, PBE0/def2-TZVPP, BH&HLYP/def2-TZVPP, and M06-2X/def2-TZVPP levels of theory (see the ESI†). All these methods predicted that for 4 the triplet form is 29.1 kcal mol−1 (B3LYP), 28.7 kcal mol−1 (PBE0), 27.8 kcal mol−1 (BH&HLYP), or 32.0 kcal mol−1 (M06-2X) higher in energy (Table T3 in the ESI†). The computed singlet–triplet energy difference ΔES–T for 5 is 10.7 kcal mol−1 (B3LYP), 9.5 kcal mol−1 (PBE0), 4.8 kcal mol−1 (BH&HLYP), or 9.8 kcal mol−1 (M06-2X) (Table T4 in the ESI†). It should however be noted that among all the functionals used, BH&HLYP was the only one that recognized a singlet broken symmetry state (Table 2). We also calculated the diradical character (y) of 4 and 5 as described by Nakano (y = 0 for the closed-shell and y = 1 for the pure singlet diradical).21 Compound 4 is a perfectly closed-shell species while compound 5 has 43% diradical character (Table 2). The estimated values are however lower than those reported for the TH (0.31) and CH (0.72) HCs.6c
We also performed CASSCF(2,2)+NEVPT2/def2-SVP calculations on model systems 4Me and 5Me, where the 2,6-iPr2C6H3 groups of compounds 4 and 5 were replaced by methyl groups (See the ESI, Tables T5–T7†). These calculation led to a CI (configuration interaction) vector having coefficients of 0.96 for the closed-shell 2,0 configuration, 0.0 for the 1,1 configuration, and 0.3 for the 0,2 configuration. The diradical index computed according to Neese et al.22 suggest that 4Me and 5Me are closed-shell species with 7.0% and 7.8% diradicaloid character, respectively. Indeed, the occupation of HOMO is in both case 1.92 electrons, while LUMO has 0.07 electrons.
From the above discussion, it becomes clear that the closed-shell singlet is the ground state for 4 and 5. Indeed, a carefully prepared sample of 4 was EPR silent and exhibit well resolved 1H and 13C NMR resonances in the expected region (see Plots P8–P10 in the ESI†). Under similar conditions, the sample prepared by dissolving crystals of 5 exhibit a doublet EPR signal (Fig. S5 in the ESI†), which corresponds to a mono-radical species. This observation is in line with the baffling controversy over the magnetic properties of CH, the so-called “diradical paradox”.7a Similar observations have been also made in many other closed-shell compounds reported recently.11a,14,23 Baumgarten6c addressed this issue in a very recent report that concludes that the weak intensity triplet expected for CH can be masked under even ≈0.1% of mono-radical impurity.
Compound 4 showed an intense absorption with maximum (λmax) at 431 nm along with a weak absorption at 619 nm (Fig. S3†). Compound 5 exhibited λmax at 618 nm with a shoulder at 580 nm (Fig. S4†). We carried out time-dependent TD-PCM(THF)-B3LYP/def2-SVP calculations for the assignment of the UV-visible spectral bands. The results are summarized in Tables T8 and T9 (see the ESI†). The results for 4 in the closed-shell configuration gave three main excitations at 256 nm, 350 nm, and 539 nm with the oscillator strengths of 0.092, 1.386, and 0.066, respectively. The main absorption corresponds to the HOMO → LUMO + 8 transition (Fig. S13†). For compound 5, the absorption bands are red shifted and the oscillator strengths are stronger. The main absorption is due to the HOMO → LUMO transition and occurs at 441 nm with the oscillator strength of 3.296 (Fig. 5).
Experimental
Materials and methods
All syntheses and manipulations were performed under an inert gas (Ar or N2) atmosphere using standard Schlenk techniques and an MBraun LABmaster Pro glovebox. THF, n-hexane, and o-xylene were dried over NaK, distilled prior to use, and stored over 3 Å molecular sieve. Melting points were measured using a Büchi B-545 melting point apparatus. NMR spectra were recorded using a Bruker Avance III 500 or a Bruker Avance III 500HD NMR spectrometer. Chemical shifts are given in δ ppm and are referenced to the solvent residual peaks:24 DMSO-d6 (1H, δ = 2.50 ppm and 13C, δ = 39.52 ppm), C6D6 (1H, δ = 7.16 ppm and 13C, δ = 128.06 ppm). ESI mass spectra were recorded with a Bruker Esquire 3000 spectrometer. UV-visible spectra were recorded using a ThermoFisher Evolution 300 spectrophotometer.Synthetic procedures of 2–5
Compounds 2 and 3 were prepared by the direct C2-arylation of SIPr (1) with 1,4-diiodobenzene and 4,4′-dibromobiphenyl under nickel catalysis.16,25[(SIPr)C6H4(SIPr)](I)2 (2)
To a Schlenk flask containing SIPr (1) (1.76 g, 4.50 mmol), 1,4-diiodobenzene (0.70 g, 2.21 mmol), and Ni(cod)2 (87 mg, 0.32 mmol) was added 40 mL o-xylene. The resulting reaction mixture was heated under reflux for 4 h and then brought to room temperature. Filtration through a G4 porosity frit afforded an off-white solid, which was washed with (2 × 10 mL) toluene and dried under vacuum. Compound 2 was obtained as a white solid in 81% (1.90 g) yield. Mp: 348–350 °C (dec.). Elemental analysis (%) calcd for 2, C60H80I2N4 (1111): C 64.86; H 7.26; N 5.04; found: C 64.54, H 7.22, N 4.79. 1H NMR (500 MHz, DMSO-d6, 25 °C): δ = 0.68 (d, J = 6.7 Hz, 24H, CH(CH3)2), 1.23 (d, J = 6.5 Hz, 24H, CH(CH3)2), 2.87 (sept, J = 6.8 Hz, 8H, CH(CH3)2), 4.55 (s, 8H, NCH2), 6.83 (s, 4H, C6H4), 7.22 (d, J = 7.8 Hz, 8H, m-C6H3), 7.42 (t, J = 7.8 Hz, 4H, p-C6H3) ppm. 13C NMR (125 MHz, DMSO-d6, 25 °C): δ = 22.5, 26.0, 28.2, 53.6, 124.8, 125.3, 125.5, 125.7, 128.7, 129.1, 129.4, 131.4, 136.0, 145.1, 146.1, 164.2 ppm. MS (ESI pos.): m/z = 428.67 [2 − 2I]++.[(SIPr)(C6H4)2(SIPr)](Br)2 (3)
Compound 3 was prepared by adopting a similar method as descried for 2 using SIPr (1) (1.50 g, 3.84 mmol), 4,4′-dibromobiphenyl (0.59 g, 1.89 mmol), and Ni(cod)2 (75 mg, 0.25 mmol). Yield: 1.47 g, 71%. Mp: 358–360 °C. Elemental analysis (%) calcd for 3, C66H84Br2N4 (1093): C 72.51; H 7.75; N 5.12; found: C 71.75, H 7.47, N 4.88. 1H NMR (500 MHz, DMSO-d6, 25 °C): δ = 0.96 (d, J = 6.7 Hz, 24H, CH(CH3)2), 1.31 (d, J = 6.5 Hz, 24H, CH(CH3)2), 3.06 (sept, J = 6.6 Hz, 8H, CH(CH3)2), 4.56 (s, 8H, NCH2), 6.97 (d, J = 8.6 Hz, 4H, C6H4), 7.34 (d, J = 7.7 Hz, 8H, m-C6H3), 7.46 (t, J = 7.7 Hz, 4H, p-C6H3), 7.72 (d, J = 8.5 Hz, 4H, C6H4) ppm. 13C NMR (125 MHz, DMSO-d6, 25 °C): δ = 22.8, 25.7, 28.40, 53.5, 125.6, 127.3, 129.6, 130.6, 131.2, 145.3, 165.3 ppm. MS (ESI pos.): m/z = 466.4 [3 − 2Br]++.[(SIPr)C6H4(SIPr)] (4)
To a Schlenk flask containing 2 (250 mg, 0.25 mmol) and KC8 (74 mg, 0.55 mmol) was added 15 mL pre-cooled (−78 °C) THF, which immediately led to the formation of a green suspension. The reaction mixture was brought to room temperature and further stirred for 2 h. Filtration through a plug of Celite afforded a green solution. The volatiles were removed under vacuum and the remaining green residue was extracted with n-hexane and stored at −24 °C to obtain 4 as a crystalline solid. Yield: 204 mg, 95%; mp 181 °C (dec.). Elemental analysis (%) calcd for 4, C60H80N4 (857): C 84.06; H 9.41; N 6.54; found: C 83.36, H 9.13, N 6.32. UV-vis THF, λ (nm) (ε (M−1 cm−1)): 399 (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 906), 431 (17389), 578 (2020), 619 (2791), 908 (486). MS (ESI pos.): m/z = 428.3 [4 + 2H]+. 1H NMR (500 MHz, C6D6, 25 °C): δ = 1.21 (d, J = 6.8 Hz, 24H, HC(CH3)2), 1.24 (d, J = 6.8 Hz, 24H, HC(CH3)2), 3.37 (s, 8H, NCH2), 3.49 (sept, J = 6.8 Hz, 8H, HC(CH3)2), 5.04 (s, 4H, C6H4), 7.02 (d, J = 7.5 Hz, 8H, m-C6H3), 7.10 (t, J = 7.4 Hz, 4H, p-C6H3) ppm.13C NMR (125 MHz, C6D6, 25 °C): δ = 23.7, 25.2 (HC(CH3)2), 28.5 (HC(CH3)2), 53.0 (CH2N), 117.6 (C6H4), 124.3 (p-C6H3), 127.4 (m-C6H3), 128.1, 128.4, 134.9, 141.5, 147.1 (o-/ipso-C6H3,p-C6H4) ppm.
906), 431 (17389), 578 (2020), 619 (2791), 908 (486). MS (ESI pos.): m/z = 428.3 [4 + 2H]+. 1H NMR (500 MHz, C6D6, 25 °C): δ = 1.21 (d, J = 6.8 Hz, 24H, HC(CH3)2), 1.24 (d, J = 6.8 Hz, 24H, HC(CH3)2), 3.37 (s, 8H, NCH2), 3.49 (sept, J = 6.8 Hz, 8H, HC(CH3)2), 5.04 (s, 4H, C6H4), 7.02 (d, J = 7.5 Hz, 8H, m-C6H3), 7.10 (t, J = 7.4 Hz, 4H, p-C6H3) ppm.13C NMR (125 MHz, C6D6, 25 °C): δ = 23.7, 25.2 (HC(CH3)2), 28.5 (HC(CH3)2), 53.0 (CH2N), 117.6 (C6H4), 124.3 (p-C6H3), 127.4 (m-C6H3), 128.1, 128.4, 134.9, 141.5, 147.1 (o-/ipso-C6H3,p-C6H4) ppm.
[(SIPr)(C6H4)2(SIPr)] (5)
Compound 5 was prepared by employing a similar method as described above for 4 using 3 (800 mg, 0.73 mmol) and KC8 (198 mg, 1.46 mmol) in THF (20 mL) as a deep blue solid. Yield: 670 mg, 98%; mp 201 °C (dec.). Elemental analysis (%) calcd for 5, C66H84N4 (933): C 84.93; H 9.07; N 6.00; found: C 84.55, H 8.77, N 5.68. UV-vis THF, λ (nm) (ε (M−1 cm−1)): 580 (14860), 618 (20940), 911 (520). MS (ESI pos.): m/z = 466.3 [5 + 2H]+.
X-ray crystallography
Solid-state molecular structures of 2 (Fig. S6†), 3 (Fig. S7†), and crystallographic details of 2–5 (Tables T1 and T2) are given in the ESI.† CCDC 1826567 (2), 1826568 (3), 1826569 (4), and 1826570 (5) contain the supplementary crystallographic data for this paper.†Computational calculations
Geometry optimizations were performed using Gaussian 09 (ref. 26) together with TurboMole V6.5.27 All geometry optimizations were computed using the functional B3LYP28 and BH&HLYP29 in combination with the def2-SVP basis set.30 The stationary points were located with the Berny algorithm31 using redundant internal coordinates. Analytical Hessians were computed to determine the nature of stationary points. The improvements in the electronic energies were carried out by computing single points on the B3LYP/def2-SVP geometries at the B3LYP/def2-TZVPP, BH&HLYP/def2-TZVPP, PBE0/def2-TZVPP32 and M06-2X/def2-TZVPP33 levels of theory. Further calculation details on optimized molecular structures, relative free energies, frontier molecular orbital analyses, and UV-visible spectra are provided in the ESI.†Conclusions
In conclusion, we have presented the synthesis and characterization of the first Kekulé diradicaloid compounds 4 and 5 derived from a classical NHC 1. Compounds 4 and 5 may be considered as NHC analogues of Thiele's and Chichibabin's hydrocarbons. Remarkably, the precursor compounds 2 and 3 are readily accessible by the double carbenylation of para-dihaloarenes with a commercially available NHC 1 using Ni-catalysis. Experimental and computational studies fully corroborate the closed-shell singlet ground state of 4 and 5. The results demonstrate that by using appropriate linkers, synthetic access to non-Kekulé diradicals and poly-radicals derived from NHCs seems feasible and therefore is worth pursuing.Conflicts of interest
There are no conflicts to declareAcknowledgements
We gratefully acknowledge the support from the Deutsche Forschungsgemeinschaft and thank Professor Norbert W. Mitzel for his generous support and encouragement. We also thank Dr Maurice van Gastel for analysing compounds 4 and 5 by EPR spectroscopy.References
- (a) W. T. Borden in The Foundations of Physical Organic Chemistry: Fifty Years of the James Flack Norris Award, American Chemical Society, 2015, vol. 1209, ch. 11, pp. 251–303 Search PubMed; (b) M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed; (c) L. Salem and C. Rowland, Angew. Chem., Int. Ed., 1972, 11, 92–111 CrossRef CAS.
- (a) Y. Jung and M. Head-Gordon, ChemPhysChem, 2003, 4, 522–525 CrossRef CAS PubMed; (b) M. Seierstad, C. R. Kinsinger and C. J. Cramer, Angew. Chem., Int. Ed., 2002, 41, 3894–3896 CrossRef CAS; (c) M. Abe, J. Ye and M. Mishima, Chem. Soc. Rev., 2012, 41, 3808–3820 RSC; (d) H. Grützmacher and F. Breher, Angew. Chem., Int. Ed., 2002, 41, 4006–4011 CrossRef; (e) F. Breher, Coord. Chem. Rev., 2007, 251, 1007–1043 CrossRef CAS.
- J. Thiele and H. Balhorn, Ber. Dtsch. Chem. Ges., 1904, 37, 1463–1470 CrossRef.
- A. E. Tschitschibabin, Ber. Dtsch. Chem. Ges., 1907, 40, 1810–1819 CrossRef CAS.
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef.
- (a) F. Popp, F. Bickelhaupt and C. Maclean, Chem. Phys. Lett., 1978, 55, 327–330 CrossRef CAS; (b) W. T. Borden, Diradicals, Wiley-Interscience, New York, 1982 Search PubMed; (c) P. Ravat and M. Baumgarten, Phys. Chem. Chem. Phys., 2015, 17, 983–991 RSC; (d) A. Konishi and T. Kubo, Top. Curr. Chem., 2017, 375, 83 CrossRef PubMed; (e) T. Kubo, Chem. Lett., 2015, 44, 111–122 CrossRef.
- (a) Y. Kanzaki, D. Shiomi, K. Sato and T. Takui, J. Phys. Chem. B, 2012, 116, 1053–1059 CrossRef CAS PubMed; (b) H. M. McConnell, J. Chem. Phys., 1960, 33, 1868–1869 CrossRef CAS.
- L. K. Montgomery, J. C. Huffman, E. A. Jurczak and M. P. Grendze, J. Am. Chem. Soc., 1986, 108, 6004–6011 CrossRef CAS PubMed.
- (a) K. Kamada, K. Ohta, T. Kubo, A. Shimizu, Y. Morita, K. Nakasuji, R. Kishi, S. Ohta, S.-i. Furukawa, H. Takahashi and M. Nakano, Angew. Chem., Int. Ed., 2007, 46, 3544–3546 CrossRef CAS PubMed; (b) Z. Sun, Q. Ye, C. Chi and J. Wu, Chem. Soc. Rev., 2012, 41, 7857–7889 RSC; (c) Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197–204 CrossRef CAS PubMed; (d) M. Nakano and B. Champagne, J. Phys. Chem. Lett., 2015, 6, 3236–3256 CrossRef CAS.
- G. Tan and X. Wang, Acc. Chem. Res., 2017, 50, 1997–2006 CrossRef CAS PubMed.
- (a) Z. Zeng, X. Shi, C. Chi, J. T. Lopez Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC; (b) G. Li, H. Phan, T. S. Herng, T. Y. Gopalakrishna, C. Liu, W. Zeng, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 5012–5016 CrossRef CAS PubMed; (c) J. Liu, J. Ma, K. Zhang, P. Ravat, P. Machata, S. Avdoshenko, F. Hennersdorf, H. Komber, W. Pisula, J. J. Weigand, A. A. Popov, R. Berger, K. Müllen and X. Feng, J. Am. Chem. Soc., 2017, 139, 7513–7521 CrossRef CAS PubMed.
- (a) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed; (b) K. C. Mondal, S. Roy and H. W. Roesky, Chem. Soc. Rev., 2016, 45, 1080–1111 RSC; (c) S. Roy, K. C. Mondal and H. W. Roesky, Acc. Chem. Res., 2016, 49, 357–369 CrossRef CAS PubMed; (d) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed; (e) B. Rao, H. Tang, X. Zeng, L. Liu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2015, 54, 14915–14919 CrossRef CAS PubMed; (f) C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC.
- (a) M. M. Hansmann, M. Melaimi, D. Munz and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2546–2554 CrossRef CAS PubMed; (b) M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2206–2213 CrossRef CAS PubMed.
- B. M. Barry, R. G. Soper, J. Hurmalainen, A. Mansikkamaki, K. N. Robertson, W. L. McClennan, A. J. Veinot, T. L. Roemmele, U. Werner-Zwanziger, R. T. Boere, H. M. Tuononen, J. A. C. Clyburne and J. D. Masuda, Angew. Chem., Int. Ed., 2018, 57, 749–754 CrossRef CAS PubMed.
- D. Rottschäfer, B. Neumann, H.-G. Stammler, M. van Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 4765–4768 CrossRef PubMed.
- N. K. T. Ho, B. Neumann, H. G. Stammler, V. H. Menezes da Silva, D. G. Watanabe, A. A. C. Braga and R. S. Ghadwal, Dalton Trans., 2017, 46, 12027–12031 RSC.
- B. Gorodetsky, T. Ramnial, N. R. Branda and J. A. C. Clyburne, Chem. Commun., 2004, 1972–1973 RSC.
- S. M. Ibrahim Al-Rafia, A. C. Malcolm, S. K. Liew, M. J. Ferguson, R. McDonald and E. Rivard, Chem. Commun., 2011, 47, 6987–6989 RSC.
- R. S. Ghadwal, S. O. Reichmann, F. Engelhardt, D. M. Andrada and G. Frenking, Chem. Commun., 2013, 49, 9440–9442 RSC.
- L. Noodleman, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef CAS.
- K. Kamada, K. Ohta, A. Shimizu, T. Kubo, R. Kishi, H. Takahashi, E. Botek, B. Champagne and M. Nakano, J. Phys. Chem. Lett., 2010, 1, 937–940 CrossRef CAS.
- (a) V. Bachler, G. Olbrich, F. Neese and K. Wieghardt, Inorg. Chem., 2002, 41, 4179–4193 CrossRef CAS PubMed; (b) D. Herebian, E. Bothe, F. Neese, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 2003, 125, 9116–9128 CrossRef CAS PubMed; (c) F. Neese, J. Phys. Chem. Solids, 2004, 65, 781–785 CrossRef CAS; (d) D. Herebian, K. E. Wieghardt and F. Neese, J. Am. Chem. Soc., 2003, 125, 10997–11005 CrossRef CAS PubMed.
- (a) J. Wang, X. Xu, H. Phan, T. S. Herng, T. Y. Gopalakrishna, G. Li, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2017, 56, 14154–14158 CrossRef CAS PubMed; (b) Y. Su, X. Wang, Y. Li, Y. Song, Y. Sui and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 1634–1637 CrossRef CAS PubMed; (c) Y. Su, X. Wang, X. Zheng, Z. Zhang, Y. Song, Y. Sui, Y. Li and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 2857–2861 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- R. S. Ghadwal, S. O. Reichmann and R. Herbst-Irmer, Chem.–Eur. J., 2015, 21, 4247–4251 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. Bruschi, P. Fantucci, M. Pizzotti and C. Rovizzi, J. Mol. Catal. A: Chem., 2003, 204, 793–803 CrossRef.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- C. Y. Peng, P. Y. Ayala, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 1996, 17, 49–56 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1826567–1826570. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01209a |
| This journal is © The Royal Society of Chemistry 2018 |